

Abstract
Amphotericin B (AMB) is one of the most effective antifungal agents; however, its use is often limited by the occurrence of adverse events, especially nephrotoxicity. The present study was designed to determine the possible mechanisms underlying the nephrotoxic action of AMB. The exposure of a porcine proximal renal tubular cell line (LLC-PK1 cells) to AMB caused cell injury, as assessed by mitochondrial enzyme activity, the leakage of lactate dehydrogenase, and tissue ATP depletion. Propidium iodide uptake was enhanced, while terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling was not affected by AMB, suggesting a lack of involvement of apoptosis in AMB-induced cell injury. The cell injury was inhibited by the depletion of membrane cholesterol with methyl-β-cyclodextrin, which lowered the extracellular Na+ concentration or the chelation of intracellular Ca2+. The rise in the intracellular Ca2+ concentration may be mediated through the activation of the ryanodine receptor (RyR) on the endoplasmic reticulum and the mitochondrial Na+-Ca2+ exchanger, since cell injury was attenuated by dantrolene (an RyR antagonist) and CGP37157 (an Na+-Ca2+ exchanger inhibitor). Moreover, AMB-induced cell injury was reversed by PD169316 (a p38 mitogen-activated protein [MAP] kinase inhibitor), c-Jun N-terminal kinase inhibitor II, and PD98059 (a MEK1/2 inhibitor). The phosphorylations of these MAP kinases were enhanced by AMB in a calcium-independent manner, suggesting the involvement of MAP kinases in AMB-induced cell injury. These findings suggest that Na+ entry through membrane pores formed by the association of AMB with membrane cholesterol leads to the activation of MAP kinases and the elevation of the intracellular Ca2+ concentration, leading to renal tubular cell injury.
Amphotericin B (AMB), a polyene macrolide antibiotic, is still the most effective drug for the treatment of systemic fungal infections in patients with cancer, AIDS, and organ transplantation. The antifungal action of this agent has been considered to be mediated through ion-permeable pores that are formed by the association of AMB with ergosterol in the fungal cell membranes (1, 6). However, the clinical use of this antifungal agent is often limited by its severe nephrotoxicity (7, 20, 36). The incidence of AMB-induced nephrotoxicity has been reported to be between 49% and 65% (7). Wingard et al. (36) reported on 239 patients with a depressed immune response who received AMB for suspected or proven aspergillosis and found that 53% of the patients showed a doubled serum creatinine level after the injection of AMB; the value exceeded 2.5 mg/dl in 29% of the patients, and dialysis was required in 15% of the patients. Unfortunately, there have been few effective means of prevention of AMB-induced nephrotoxicity or therapy for AMB-induced nephrotoxicity, except for aggressive hydration and correction of electrolyte levels (7, 22). Recently, new lipid formulations of AMB have been developed to reduce the nephrotoxicity of conventional AMB (Fungizone). There are currently three lipid formulations on the market worldwide: liposomal AMB (Ambisome), AMB lipid complex (Abelcet), and AMB colloidal dispersion (Amphotec). Torrado et al. (32) have summarized the data on the incidence of nephrotoxicity induced by lipid formulations of AMB and showed that treatment with liposomal AMB and AMB colloidal dispersion resulted in lower rates of nephrotoxicity than treatment with conventional AMB did. Although the incidence of nephrotoxicity is evidently lower in patients treated with liposomal AMB (14.6%) than in patients treated with AMB (33.2%), the liposomal formulation is still considered to be nephrotoxic (13). The etiology of the nephrotoxicity of AMB remains to be clarified; however, vasoconstriction and the direct toxic action of AMB on renal tubular epithelial cells have been postulated to be the major causes of AMB-induced nephrotoxicity (7, 20). Varlam et al. (33) have reported that AMB causes concentration-dependent apoptosis in the porcine renal proximal tubular epithelial cell line LLC-PK1 and medullary interstitial cells in vitro, while it induces apoptosis in rat renal tubular epithelial cells in vivo in a dose-dependent fashion. Therefore, in the present study, we determined the possible mechanisms underlying the toxic action of AMB on LLC-PK1 cells.
MATERIALS AND METHODS
Chemicals.
AMB was obtained from Bristol-Myers Squibb K.K. (Tokyo, Japan). Methyl-β-cyclodextrin (Mβ-CD), cholesterol, EGTA, ruthenium red (RR), and xestospongin C (Xesto) were obtained from Sigma-Aldrich Co. (St. Louis, MO). 1,2-Bis(ο-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA-AM), CGP37157 (CGP), leupeptin, PD169316, c-Jun N-terminal kinase (JNK) inhibitor II (JNKI), a JNKI negative control, and PD98059 were purchased from Calbiochem (San Diego, CA). Dantrolene sodium (Dant) was obtained from Wako Pure Chemicals (Osaka, Japan).
Cell culture.
Cells of a porcine renal tubular epithelial cell line, LLC-PK1 cells, were obtained from the American Type Culture Collection (Manassas, VA). The cells were grown in a 75-cm2 flask (Corning Incorporated, Corning, NY) and were maintained in medium 199 (MP Biomedicals, Inc., Irvine, CA) supplemented with 10% fetal bovine serum (Cansera International Inc., Canada) and 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco BRL, Grand Island, NY) in an atmosphere of 5% CO2-95% air at 37°C. For the experiments, the cells were seeded on 24-well plates (Nalge Nunc International, Rochester, NY) at a density of 1.0 × 104 cells/cm2 and were cultured at 37°C for 24 h, and then the medium was washed twice with serum-free Krebs-Ringer solution and incubated in serum-free Krebs-Ringer solution in the presence of AMB and other agents at 37°C in 5% CO2-95% air.
WST-8 assay.
Cell viability was assessed by measuring the mitochondrial activity that reduced 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt (WST-8) to formazan, as described previously (18). Briefly, after treatment with AMB, the cells were washed with phosphate-buffered saline (PBS). The cells were then incubated with 210 μl serum-free medium and 10 μl WST-8 assay solution (Cell Counting Kit-8; Dojindo, Kumamoto, Japan) for 1 h at 37°C in humidified air supplemented with 5% CO2. The incubation medium was carefully taken and transferred to 96-well flat-bottom plastic plates (Nalge Nunc International). The amount of formazan formed was measured from the absorbance at 450 nm with a reference wavelength of 620 nm by using a microplate reader (Immuno-Mini NJ-2300; Inter Medical, Tokyo, Japan).
Leakage of LDH.
Cells were seeded at a density of 3 × 104 cells/well onto 24-well plastic plates (Nalge Nunc International) and were used for experiments on the following day, by which time they had reached 70 to 80% confluence. The leakage of lactate dehydrogenase (LDH) was expressed as the amount of LDH released into the medium as a percentage of the total amount of LDH released 24 h after exposure to AMB. LDH activity was determined with an LDH assay kit (Takara Biochemicals, Osaka, Japan).
Propidium iodide staining.
Propidium iodide staining was carried out with a commercial apoptosis assay kit (Mebcyto apoptosis kit; Medical & Biological Lab Co., Ltd., Nagoya, Japan), as described previously (37). In brief, cells were cultured on eight-chamber plastic slides (Iwaki, Tokyo, Japan) at 2.0 × 104 cells/chamber. At 24 h after the cells were seeded, the cells were incubated with AMB (15 μg/ml) for 24 h. The cells were visualized with a fluorescent microscope (BX51; Olympus, Tokyo, Japan).
TUNEL.
Cells were cultured on eight-chamber plastic slides (Iwaki) at 2.0 × 104 cells/chamber. At 24 h after the cells were seeded, the cells were incubated with AMB (15 μg/ml) for 24 h. The cells were then washed with PBS and fixed for 30 min at room temperature with 4% (wt/vol) paraformaldehyde in PBS. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was carried out with an in situ apoptosis detection kit (Takara Biochemicals), according to the manufacturer's instructions. The stained cells were visualized with a microscope (Olympus).
Measurement of cellular ATP content.
The cells were seeded at a density of 6 × 104 cells/well onto six-well plastic plates (Nalge Nunc International). After exposure to AMB, the cells were homogenized with 0.4 N perchloric acid. The homogenates were centrifuged at 10,000 × g for 20 min, and the resultant supernatant was neutralized to pH 6.0 with 10% K2CO3 solution. The ATP content was determined by high-performance liquid chromatography (HPLC) with spectrophotometric detection, according to the method described previously (28). In brief, a portion of the neutralized supernatant was injected directly into an HPLC system. The system was composed of a pump (LC-6A; Shimadzu Co., Kyoto, Japan), a reversed-phase separation column (250 by 4.6 mm; Wakosil II 5C18 RS; Wako Pure Chemical), a UV spectrophotomonitor (SPD-6A; Shimadzu), and a chromatorecorder (Chromatopac C-R4A; Shimadzu). The column temperature was set at 35°C. The mobile phase was 0.2 M ammonium dihydrogen phosphate (pH 4.1) containing 1.5% acetonitrile and tetrabutylammonium hydrogen sulfate (40 mg/liter) (Sigma) as an ion-pair agent. The flow rate was 1.0 ml/min. ATP was detected at an optical density of 260 nm. The cell pellet was dissolved in 0.5 ml of 1 N NaOH, and the protein content was determined by the method of Bradford (4) and by using bovine serum albumin as the standard.
Cholesterol depletion and repletion.
Cholesterol depletion and repletion were performed with Mβ-CD, as described previously (21). Cholesterol depletion was carried out by incubating the cells with Mβ-CD for 1 h at 37°C, and the cells were then washed with PBS before the experiments were performed. Cholesterol repletion was carried out by incubating cholesterol-depleted cells with serum-free medium containing a 4% (vol/vol) cholesterol-Mβ-CD stock solution for 1 h at 37°C. A stock solution consisting of a mixture of cholesterol and Mβ-CD was prepared by adding 100 μl of cholesterol (20 mg/ml in ethanol) to 10 ml of a 5 mM Mβ-CD solution and mixing the ingredients at 40°C.
Western blot analysis.
Cells were cultured on 24-well plastic plates (Nalge Nunc International) at 2.0 × 104 cells/well. At 24 h after the cells were seeded, the cells were incubated with Krebs-Ringer buffer containing AMB (15 μg/ml). The cells were then washed three times with ice-cold PBS and lysed in 100 μl of ice-cold lysis buffer (2 mM EGTA, 2 mM dithiothreitol, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin). Protein samples were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and were then transferred to a nitrocellulose membrane by the semidry method. The membrane was incubated with Tris-buffered saline containing Tween 20 (20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 0.2% Tween 20) and 5% nonfat milk. After the membranes were blocked, they were incubated with specific antibodies raised against phospho-p38 mitogen-activated protein kinase (MAPK) (Thr180/Thr182), total p38, phospho-p44/42 extracellular signal-regulated kinase (ERK), total ERK, phospho-JNK (Thr183/Tyr185), and total JNK (Cell Signaling Technology, Inc., Beverly, MA). Secondary antibodies, conjugated with horseradish peroxidase (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), were detected by use of the Western Lightning kit (Perkin-Elmer Life Sciences, Boston, MA). The optical density of the film was scanned and measured with Scion Image software (Scion Corp., Frederick, MD).
Mitochondrial membrane potential as measured by JC-1 staining.
Changes in mitochondrial membrane potentials were assessed by using the JC-1 stain, as described previously (38). In brief, cells were cultured on eight-chamber plastic slides (Iwaki) at 2.0 × 104 cells/chamber. At 24 h after the cells were seeded, the cells were incubated with Krebs-Ringer buffer containing AMB (15 μg/ml) for 3 h. CGP (10 μM), RR (10 μM), JNKI (10 μM), a JNKI negative control (10 μM), and PD98059 (10 μM) were added 1 h before AMB treatment and were included throughout experiment. The cells were then washed with PBS and incubated with the Krebs-Ringer buffer containing JC-1 stain (Wako Pure Chemical) for 15 min. Fluorescence images were observed with a fluorescent microscope (BX51; Olympus).
Statistical analysis.
Data are expressed as the means ± standard error of the means (SEMs) and were statistically analyzed by one-way analysis of variance, followed by Dunnett's test for multiple comparisons or by Student's t test for comparisons between two groups. Statistical significance was defined as a P value of <0.05.
RESULTS
AMB-induced injury in renal tubular LLC-PK1 cells.
As shown in Fig. 1A, AMB (1 to 20 μg/ml, 24 h) caused concentration-dependent cell injury, as determined by the WST-8 assay and from the amount of LDH leakage. Significant effects were observed at AMB concentrations of ≥5 μg/ml. The toxic effect of AMB was time dependent, in which significant injury was already observed at 3 h after exposure to 20 μg/ml AMB and was more marked at 24 h (Fig. 1A). On the other hand, AMB (15 μg/ml, 24 h) caused increases in the numbers of propidium iodide-stained cells but no marked change in the number of TUNEL-positive cells (Fig. 1B).
FIG. 1.

Concentration-dependent and time-dependent cell injury to LLC-PK1 cells induced by AMB, as assessed by the WST-8 assay or determination of the level of LDH leakage (A) and staining with the representative stains propidium iodide (PI) and TUNEL (B). Cells were exposed to various concentrations of AMB for 24 h. The data represent the means ± SEMs of four to eight experiments. **, P < 0.01 versus the results for the control group.
Depletion of cellular ATP after exposure to AMB.
As shown in Fig. 2, AMB (1 to 10 μg/ml, 24 h) caused a marked decrease in the cellular ATP content in a concentration-dependent manner. The ATP-depleting effect of AMB (10 μg/ml) was time dependent, and a significant decrease was observed at 6 h after exposure to AMB.
FIG. 2.

Concentration-dependent and time-dependent decreases in tissue ATP content after exposure of LLC-PK1 cells to AMB. Cells were exposed to various concentrations of AMB for 24 h. The data represent the means ± SEMs of four to eight experiments. **, P < 0.01 versus the results for the control group.
Reversal by membrane cholesterol depletion of AMB-induced cell injury in LLC-PK1 cells.
To determine the role of membrane cholesterol in the toxic effect of AMB in LLC-PK1 cells, we examined the effect of the depletion of membrane cholesterol by Mβ-CD on the toxic effect of AMB. As shown in Fig. 3, the AMB-induced loss of cell viability was prevented by the depletion of membrane cholesterol. The inhibitory effect of Mβ-CD on AMB-induced cell toxicity was reversed by cholesterol repletion.
FIG. 3.
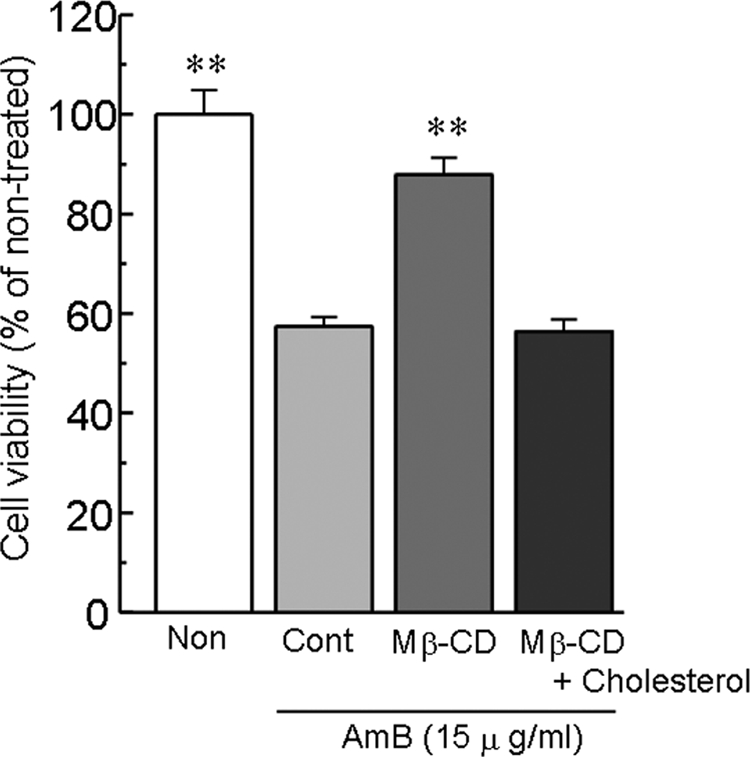
Inhibition by Mβ-CD of AMB-induced loss of cell viability in LLC-PK1 cells and its reversal of the inhibition by cholesterol. Cholesterol depletion was carried out by incubating the cells with Mβ-CD for 1 h at 37°C. Cholesterol repletion was carried out by incubating cholesterol-depleted cells with serum-free medium containing a 4% (vol/vol) cholesterol-Mβ-CD stock solution for 1 h at 37°C. The cells were then washed with PBS and incubated with AMB for 6 h. Cell viability was assessed by the WST-8 assay. The data represent the means ± SEMs of four to six experiments. **, P < 0.01 versus the results for the control (Cont) group.
Roles of Na+ and Ca2+ in AMB-induced LLC-PK1 cell injury.
To determine whether Na+ and Ca2+ contribute to AMB-induced renal cell injury, the effects of Krebs-Ringer solution with a low Na+ concentration (70 mM Na+) and the chelation of extracellular and intracellular Ca2+ with EGTA and the cell-permeant compound BAPTA-AM, respectively, on the loss of cell viability induced by AMB were investigated. The AMB-induced cell injury was attenuated by lowering the extracellular Na+ concentration or the chelation of intracellular but not extracellular Ca2+ (Fig. 4).
FIG. 4.

Inhibition of the AMB-induced loss of cell viability in LLC-PK1 cells by lowering the extracellular Na+ concentration (A) or the chelation of intracellular Ca2+ with BAPTA-AM (B). Cells were exposed to AMB (15 μg/ml) for 6 h in normal Krebs-Ringer buffer, 70 mM Na+-containing buffer, or Ca2+-free and EGTA (1 mM)-containing buffer. BATPTA-AM was included at a concentration of 2 μM. Cell viability was assessed by the WST-8 assay. The data represent the means ± SEMs of four to six experiments. **, P < 0.01 versus the results for the respective AMB-free group; ††, P < 0.01 versus the results for the group treated with BAPTA-AM.
Effects of Ca2+ modulators in the ER and mitochondria on AMB-induced LLC-PK1 cell injury.
To elucidate the roles of the endoplasmic reticulum (ER) and the mitochondria in AMB-induced renal cell injury, the effects of various agents that affect Ca2+ mobilization in the ER and the mitochondria on the AMB-induced loss of cell viability were examined. Cells were treated with Krebs-Ringer buffer containing AMB (15 μg/ml) for 6 h in the absence or the presence of Dant, a ryanodine receptor (RyR) blocker; Xesto, an inositol=1,4,5=triphosphate (IP3) receptor antagonist; CGP, an inhibitor of the mitochondrial Na+-Ca2+ exchanger; or RR, an inhibitor of the mitochondrial Ca2+ uniporter. As shown in Fig. 5A, Dant (30 μM) inhibited AMB-induced cell injury, whereas Xesto (1 μM) was not effective. On the other hand, CGP (10 μM) and RR (10 μM) reversed the cell injury induced by AMB (Fig. 5B).
FIG. 5.

Effects of Ca2+ modulators of ER (A) and mitochondria (B) on AMB-induced loss of cell viability in LLC-PK1 cells. Cells were exposed to AMB (15 μg/ml) for 6 h in Krebs-Ringer buffer, and their viability was assessed by the WST-8 assay. The RyR antagonist Dant, the inhibitor of the mitochondrial Na+-Ca2+ exchanger CGP (10 μM), and the inhibitor of mitochondrial Ca2+ uniporter RR (10 μM) were included 1 h before AMB treatment, while the IP3 receptor antagonist Xesto (1 μM) was added 20 min before AMB treatment; the inhibitors and the antagonists were included throughout the experiment. The data represent the means ± SEMs of four experiments. *, P < 0.05 versus the results for the control group; **, P < 0.01 versus the results for the control group. Non, nontreated; Cont, control.
Involvement of the MAPK pathway in AMB-induced LLC-PK1 cell injury.
To determine the role of the MAPK pathway in AMB-induced cell injury, the effects of various MAPK inhibitors on the AMB-induced loss of cell viability were examined. As shown in Fig. 6, the cell injury induced by AMB was slightly but significantly reversed by JNKI (10 μM) or the p38 MAPK inhibitor PD169316 (10 μM) and was markedly attenuated by the ERK inhibitor PD98059 (10 μM). Subsequently, we investigated by Western blot analysis whether MAPK is activated by phosphorylation after exposure to AMB. As shown in Fig. 7, AMB markedly enhanced the phosphorylation of JNK (54 and 46 kDa) and p38, while the activation of ERK was slight and transient. On the other hand, the AMB-induced phosphorylation of JNK, p38, and ERK was not reversed by BAPTA-AM (data not shown).
FIG. 6.
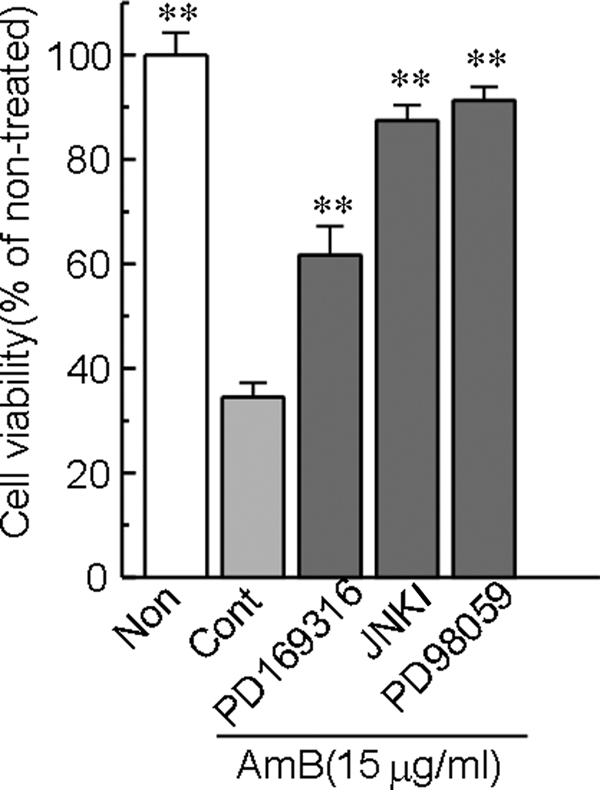
Effects of MAPK inhibitors on the AMB-induced loss of cell viability in LLC-PK1 cells. Cells were exposed to 15 μg/ml AMB for 6 h in Krebs-Ringer buffer. The p38 MAPK inhibitor PD169316 (10 μM), JNKI (10 μM), and the ERK inhibitor PD98059 (10 μM) were added 1 h before AMB treatment and were included throughout the experiment. Cell viability was assessed by the WST-8 assay. The data represent the means ± SEMs of five to six experiments. **, P < 0.01 versus the results for the control group. Non, nontreated; Cont, control.
FIG. 7.

Time courses of phospho-JNK, phospho-p38, and phospho-ERK activation induced by AMB in LLC-PK1 cells. Cells were stimulated with AMB (15 μg/ml) for the indicated times. Phospho-JNK, phospho-p38, and phospho-ERK activation was determined by Western blot analysis with specific antibodies, as described in Materials and Methods. The results are shown as the means ± SEMs from three independent experiments.
Involvement of MAPK in depolarization of mitochondrial membranes induced by AMB.
Finally, we determined the changes in mitochondrial membrane potential with the JC-1 stain after exposure to AMB. As shown in Fig. 8, a red/orange fluorescence was predominant in nontreated control cells, indicating that JC-1 existed in the aggregated form in mitochondrial membranes at resting potential. In AMB-treated cells, a number of cells revealed a green fluorescence, indicating the existence of free JC-1 at the depolarized mitochondrial membrane potential. Interestingly, AMB-induced mitochondrial membrane depolarization (green fluorescence) was almost completely reversed by CGP and RR. Moreover, the membrane depolarization induced by AMB was inhibited by JNKI and PD98059.
FIG. 8.

AMB-induced depolarization of mitochondrial membranes as assessed with the JC-1 stain. Cells were exposed to AMB (15 μg/ml) for 3 h in Krebs-Ringer buffer, and cell viability was assessed by the WST-8 assay. CGP (10 μM), RR (10 μM), JNK (10 μM), a JNK negative control (10 μM), and PD98059 (10 μM) were added 1 h before AMB treatment and were included throughout experiment. Cells were visualized under a fluorescent microscope.
DISCUSSION
AMB is one of the oldest antifungal agents but remains the “gold standard” for the treatment of life-threatening fungal infections. However, its use is often limited by serious nephrotoxicity (7, 20, 34). Although the precise modes of nephrotoxic action of AMB remain to be clarified, several mechanisms underlying the nephrotoxicity of AMB have been postulated and include vasoconstriction and/or a direct toxic action on renal tubular epithelial cells (7, 20). In the present study, we focused on the direct toxic action of AMB on cells of the cultured renal tubular cell line LLC-PK1.
The exposure of LLC-PK1 cells to AMB caused cell injury, as assessed by a WST-8 staining assay and measurement of the leakage of LDH, in concentration- and time-dependent manners. A number of reports in the literature have indicated that AMB causes toxic action on renal cells at concentrations ranging from 5 to 20 μg/ml (2, 10, 19). Consistent with these findings, in the present study, AMB at this concentration range produced a significant loss of cell viability or LDH leakage in LLC-PK1 cells. In addition, AMB caused a concentration-dependent decrease in the cellular ATP content, although the minimal concentration that showed a significant effect was lower (≥1 μg/ml) than that observed in the cell toxicity assay. In subsequent experiments, to induce moderate (50 to 60%) and consistent cell injury, the concentration of AMB and the time of exposure were chosen to be 15 μg/ml and 6 h, respectively.
In the clinical setting, the dose of conventional AMB is usually 0.4 to 0.7 mg/kg of body weight/day, which results in plasma concentrations of 0.5 to 2.0 μg/ml (11, 16). It has been reported that the nephrotoxicity appears after treatment with high doses of AMB (≥1.5 mg/kg/day), indicating that the plasma concentration is estimated to be approximately 4 μg/ml (12), a concentration quite similar to the cytotoxic concentration (5 μg/ml) observed in the present experiments.
It has been considered that membrane leakage due to the formation of pores by the association of AMB with membrane cholesterol contributes to the nephrotoxic action of AMB (7, 20). In the present study, we confirmed the involvement of membrane pore formation in AMB-induced LLC-PK1 cell injury, in which cell injury was inhibited by the depletion of membrane cholesterol with Mβ-CD and the inhibitory effect of Mβ-CD was reversed by cholesterol repletion. Moreover, the AMB-induced cell injury was prevented by the lowering of the extracellular Na+ concentration or the chelation of intracellular Ca2+ with BAPTA-AM but not by the chelation of extracellular Ca2+ with EGTA. It has been demonstrated that the membrane pores formed by the complex of AMB and cholesterol preferably increase the permeation of Na+ (15, 24). Therefore, it is suggested that the influx of Na+ through the pores formed by the association of AMB with membrane cholesterol may lead to the necrosis found in renal tubular cells.
On the other hand, the RyR blocker Dant but not the IP3 receptor antagonist Xesto inhibited AMB-induced cell injury. In addition, both CGP (a mitochondrial Na+-Ca2+ exchanger inhibitor) and RR (a mitochondrial Ca2+ uniporter inhibitor) reversed the cell injury induced by AMB. Although RR is a potent inhibitor of mitochondrial Ca2+ uptake, this compound also inhibits intracellular calcium release channels, such as RyR (17). The ER and mitochondria have been shown to play important roles in Ca2+ signaling and cell death (9, 14, 34). It has also been reported that IP3 receptor- and RyR-mediated Ca2+ signaling can trigger mitochondrial membrane permeabilization, leading to cell death (3, 14). Moreover, Nikolaeva et al. (26) have shown in rat optic nerves that elevation of the intracellular Ca2+ concentration induced by chemical ischemia with NaN3, an inhibitor of mitochondrial complex IV, is inhibited when Na+ influx is reduced by the replacement of Na+ with N-methyl-d-glucamine or Li+. In addition, the intracellular Ca2+ concentration is reduced by CGP, as well as ryanodine, which is also known to inhibit RyR at high concentrations (26). Taken together, our present findings suggests that the influx of Na+ through membrane pores formed by the complex of AMB and cholesterol causes a rise in the intracellular Ca2+ concentration by stimulating RyR on the ER and the mitochondrial Na+-Ca2+ exchanger.
More importantly, we have reported here for the first time that the activation of the MAPK pathway contributes to AMB-induced renal tubular cell injury. MAPKs, such as p38, JNK, and ERK, have been reported to play important roles in the regulation of cell survival in a variety of cell types, including renal tubular cells (8, 31, 35). In the present study, MAPK inhibitors such as JNKI and PD98059 reversed the mitochondrial membrane depolarization as well as the cell injury induced by AMB. Moreover, AMB increased the amounts of the phosphorylated forms of JNK (54 and 46 kDa), p38, and ERK. It has been reported that Ca2+ release from the ER is implicated in the activation of MAPK (27, 29). However, in the present study, the AMB-induced phosphorylation of JNK, p38, and ERK was not affected by the removal of intracellular Ca2+ with BAPTA-AM (data not shown), suggesting that the phosphorylation of MAPKs is not dependent on the intracellular Ca2+ concentration. On the other hand, Taniguchi et al. (30) have shown in the smooth muscle cells of spontaneously hypertensive rats that angiotensin II induces a rise in the intracellular Ca2+ concentration by the ERK-dependent activation of the Na+-Ca2+ exchanger. In addition, Muchekehu and Harvey (23) have reported that 17β-estradiol increases the intracellular Ca2+ concentration in the presence of thapsigargin through the activation of RyRs, which is prevented by inhibitors of protein kinase Cδ delta and protein kinase A or PD98059, indicating a role for the protein kinase Cδ, protein kinase A, and ERK signaling pathway in the activation of RyRs. Considering the fact that both BAPTA-AM and MAPK inhibitors markedly attenuated AMB-induced renal tubular cell injury, it seems likely that the phosphorylation of MAPKs increases the intracellular Ca2+ concentration by stimulating RyRs and the mitochondrial Na+-Ca2+ exchanger, which leads to cell injury. It has recently been reported that hypotonic stress stimulates renal epithelial Na+ transport by activating MAPKs such as p38, JNK, and ERK (5, 25). Therefore, it is likely that the activation of MAPKs also contributes at least in part to AMB-induced Na+ entry into cells. Further studies are needed to clarify the mechanisms underlying the AMB-induced MAPK activation and other intracellular signals that lead to renal tubular cell death.
Conclusion.
We found in the present study that AMB causes necrotic cell death in LLC-PK1 cells, in which Na+ influx through ion-permeable pores formed by the association with membrane cholesterol and the subsequent elevation of the intracellular Ca2+ concentration through the activation of RyR on the ER and the mitochondrial Na+-Ca2+ exchanger and by the activation of MAPKs may be involved. The depolarization of mitochondrial membranes was also evident after exposure to AMB, which was mediated by the activation of the Na+-Ca2+ exchanger as well as MAPKs.
Acknowledgments
This research was supported in part by Grant-in-Aid for Scientific Research (grant C20928015) from the Ministry of Education, Science, Sport and Culture of Japan.
Footnotes
Published ahead of print on 12 January 2009.
REFERENCES
- 1.Andreoli, T. E. 1973. On the anatomy of amphotericin B-cholesterol pores in lipid bilayer membranes. Kidney Int. 4:337-345. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett, K., E. Yau, S. C. Hartsel, A. Hamer, G. Tsai, D. Bizzotto, and K. M. Wasan. 2004. Effect of heat-treated amphotericin B on renal and fungal cytotoxicity. Antimicrob. Agents Chemother. 48:333-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernardi, P., A. Krauskopf, E. Basso, V. Petronilli, E. Blachly-Dyson, F. Di Lisa, and M. A. Forte. 2006. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273:2077-2099. [DOI] [PubMed] [Google Scholar]
- 4.Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Chiri, S., S. Bogliolo, J. Ehrenfeld, and B. Ciapa. 2004. Activation of extracellular signal-regulated kinase ERK after hypo-osmotic stress in renal epithelial A6 cells. Biochim. Biophys. Acta 1664:224-229. [DOI] [PubMed] [Google Scholar]
- 6.De Kruijiff, B., and R. A. Demel. 1974. Polyene antibiotic-sterol interactions in membranes of Acholeplasma laidlawii cells and lecithin liposomes. Biochim. Biophys. Acta 339:57-70. [DOI] [PubMed] [Google Scholar]
- 7.Deray, G. 2002. Amphotericin B nephrotoxicity. J. Antimicrob. Chemother. 49(Suppl. 1):37-41. [DOI] [PubMed] [Google Scholar]
- 8.di Mari, J. F., R. Davis, and R. L. Safirstein. 1999. MAPK activation determines renal epithelial cell survival during oxidative injury. Am. J. Physiol. 277:F195-F203. [DOI] [PubMed] [Google Scholar]
- 9.Duchen, M. R. 2000. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 529:57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaboriau, F., M. Chéron, C. Petit, and J. Bolard. 1997. Heat-induced superaggregation of amphotericin B reduces its in vitro toxicity: a new way to improve its therapeutic index. Antimicrob. Agents Chemother. 41:2345-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallis, H. A., R. H. Drew, and W. W. Pickard. 1990. Amphotericin B: 30 years of clinical experience. Rev. Infect. Dis. 12:308-329. [DOI] [PubMed] [Google Scholar]
- 12.Gates, C., and R. J. Pinney. 1993. Amphotericin B and its delivery by liposomal and lipid formulations. J. Clin. Pharm. Ther. 18:147-153. [DOI] [PubMed] [Google Scholar]
- 13.Girois, S. B., F. Chapuis, E. Decullier, and B. G. Revol. 2006. Adverse effects of antifungal therapies in invasive fungal infections: review and meta-analysis. Eur. J. Clin. Microbiol. Infect. Dis. 25:138-149. [DOI] [PubMed] [Google Scholar]
- 14.Hajnóczky, G., G. Csordás, S. Das, C. Garcia-Perez, M. Saotome, S. Sinha Roy, and M. Yi. 2006. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40:553-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartsel, S. C., S. K. Benz, W. Ayenew, and J. Bolard. 1994. Na+, K+ and Cl− selectivity of the permeability pathways induced through sterol-containing membrane vesicles by amphotericin B and other polyene antibiotics. Eur. Biophys. J. 23:125-132. [DOI] [PubMed] [Google Scholar]
- 16.Heinemann, V., B. Kähny, U. Jehn, D. Mühlbayer, A. Debus, K. Wachholz, D. Bosse, H. J. Kolb, and W. Wilmanns. 1997. Serum pharmacology of amphotericin B applied in lipid emulsions. Antimicrob. Agents Chemother. 41:728-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hymel, L., H. Schindler, M. Inui, and S. Fleischer. 1988. Reconstitution of purified cardiac muscle calcium release channel (ryanodine receptor) in planar bilayers. Biochem. Biophys. Res. Commun. 152:308-314. [DOI] [PubMed] [Google Scholar]
- 18.Isobe, I., M. Michikawa, and K. Yanagisawa. 1999. Enhancement of MTT, a tetrazolium salt, exocytosis by amyloid beta-protein and chloroquine in cultured rat astrocytes. Neurosci. Lett. 266:129-132. [DOI] [PubMed] [Google Scholar]
- 19.Joly, V., L. Saint-Julien, C. Carbon, and P. Yeni. 1990. Interactions of free and liposomal amphotericin B with renal proximal tubular cells in primary culture. J. Pharmacol. Exp. Ther. 255:17-22. [PubMed] [Google Scholar]
- 20.Lemke, A., A. F. Kiderlen, and O. Kayser. 2005. Amphotericin B. Appl. Microbiol. Biotechnol. 68:151-162. [DOI] [PubMed] [Google Scholar]
- 21.Liu, J., M. Liang, L. Liu, D. Malhotra, Z. Xie, and J. I. Shapiro. 2005. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 67:1844-1854. [DOI] [PubMed] [Google Scholar]
- 22.Mayer, J., M. Doubek, J. Doubek, D. Horký, P. Scheer, and M. Stepánek. 2002. Reduced nephrotoxicity of conventional amphotericin B therapy after minimal nephroprotective measures: animal experiments and clinical study. J. Infect. Dis. 186:379-388. [DOI] [PubMed] [Google Scholar]
- 23.Muchekehu, R. W., and B. J. Harvey. 2008. 17beta-estradiol rapidly mobilizes intracellular calcium from ryanodine-receptor-gated stores via a PKC-PKA-Erk-dependent pathway in the human eccrine sweat gland cell line NCL-SG3. Cell Calcium 44:276-288. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen, R. 1977. Effect of the polyene antibiotic filipin on the permeability of the inward- and the outward-facing membranes of the isolated from skin (Rana temporaria). Acta Physiol. Scand. 99:399-411. [DOI] [PubMed] [Google Scholar]
- 25.Niisato, N., A. Taruno, and Y. Marunaka. 2007. Involvement of p38 MAPK in hypotonic stress-induced stimulation of beta- and gamma-ENaC expression in renal epithelium. Biochem. Biophys. Res. Commun. 358:819-824. [DOI] [PubMed] [Google Scholar]
- 26.Nikolaeva, M. A., B. Mukherjee, and P. K. Stys. 2005. Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia. J. Neurosci. 25:9960-9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh-hashi, K., M. Kaneyama, Y. Hirata, and K. Kiuchi. 2006. ER calcium discharge stimulates GDNF gene expression through MAPK-dependent and-independent pathways in rat C6 glioblastoma cells. Neurosci. Lett. 405:100-105. [DOI] [PubMed] [Google Scholar]
- 28.Shino, Y., Y. Itoh, T. Kubota, T. Yano, T. Sendo, and R. Oishi. 2003. Role of poly(ADP-ribose) polymerase in cisplatin-induced injury in LLC-PK1 cells. Free Radic. Biol. Med. 35:966-977. [DOI] [PubMed] [Google Scholar]
- 29.Szanda, G., P. Koncz, A. Rajki, and A. Spät. 2008. Participation of p38 MAPK and a novel-type protein kinase C in the control of mitochondrial Ca2+ uptake. Cell Calcium 43:250-259. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi, S., K. Furukawa, S. Sasamura, Y. Ohizumi, K. Seya, and S. Motomura. 2004. Gene expression and functional activity of sodium/calcium exchanger enhanced in vascular smooth muscle cells of spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 43:629-637. [DOI] [PubMed] [Google Scholar]
- 31.Tian, W., Z. Zhang, and D. M. Cohen. 2000. MAPK signaling and the kidney. Am. J. Physiol. Renal Physiol. 279:F593-F604. [DOI] [PubMed] [Google Scholar]
- 32.Torrado, J. J., R. Espada, M. P. Ballesteros, and S. Torrado-Santiago. 2008. Amphotericin B formulations and drug targeting. J. Pharm. Sci. 97:2405-2425. [DOI] [PubMed] [Google Scholar]
- 33.Varlam, D. E., M. M. Siddiq, L. A. Parton, and H. Rüssmann. 2001. Apoptosis contributes to amphotericin B-induced nephrotoxicity. Antimicrob. Agents Chemother. 45:679-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verkhratsky, A., and E. C. Toescu. 2003. Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J. Cell. Mol. Med. 7:351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang, J., H. Huang, P. Liu, F. Tang, J. Qin, W. Huang, F. Chen, F. Guo, W. Liu, and B. Yang. 2006. Inhibition of phosphorylation of p38 MAPK involved in the protection of nephropathy by emodin in diabetic rats. Eur. J. Pharmacol. 553:297-303. [DOI] [PubMed] [Google Scholar]
- 36.Wingard, J. R., P. Kubilis, L. Lee, G. Yee, M. White, L. Walshe, R. Bowden, E. Anaissie, J. Hiemenz, and J. Lister. 1999. Clinical significance of nephrotoxicity in patients treated with amphotericin B for suspected or proven aspergillosis. Clin. Infect. Dis. 29:1402-1407. [DOI] [PubMed] [Google Scholar]
- 37.Yano, T., Y. Itoh, T. Sendo, T. Kubota, and R. Oishi. 2003. Cyclic AMP reverses radiocontrast media-induced apoptosis in LLC-PK1 cells by activating A kinase/PI3 kinase. Kidney Int. 64:2052-2063. [DOI] [PubMed] [Google Scholar]
- 38.Yano, T., Y. Itoh, M. Yamada, N. Egashira, and R. Oishi. 2008. Combined treatment with l-carnitine and a pan-caspase inhibitor effectively reverses amiodarone-induced injury in cultured human lung epithelial cells. Apoptosis 13:543-552. [DOI] [PubMed] [Google Scholar]