

Abstract
Ketolides represent the latest generation of macrolide antibiotics, displaying improved activities against some erythromycin-resistant strains, while maintaining their activity against erythromycin-susceptible ones. In this study, we present a new ketolide, K-1325, that carries an alkyl-aryl side chain at C-13 of the lactone ring. According to our genetic and biochemical studies, K-1325 binds within the nascent polypeptide exit tunnel, at a site previously described as the primary attachment site of all macrolide antibiotics. Compared with telithromycin, K-1325 displays enhanced antimicrobial activity against wild-type Escherichia coli strains, as well as against strains bearing the U2609C mutation in 23S rRNA. Chemical protection experiments showed that the alkyl-aryl side chain of K-1325 interacts specifically with helix 35 of 23S rRNA, a fact leading to an increased affinity of U2609C mutant ribosomes for the drug and rationalizing the enhanced effectiveness of this new ketolide.
The macrolide class of antibiotics represents a large family of protein synthesis inhibitors that has received a lot of attention because many are already used in human medicine (17, 24). Each family member is characterized by the presence of a lactone ring to which distinctive side chain residues are attached. Erythromycin (Fig. 1) was the first member of the macrolide family introduced into therapeutic use, followed by clarithromycin and azithromycin (Fig. 1) (17). Structural studies have revealed that all of the above-mentioned macrolides bind within a hydrophobic crevice of the nascent polypeptide exit tunnel (3, 13, 31, 37), consistent with the long-standing view that macrolides act by hindering the progression of the nascent peptide chain through the ribosomal tunnel, which eventually leads to peptidyl-tRNA “drop-off” (19, 20).
FIG. 1.

Chemical structures of erythromycin, clarithromycin, azithromycin, telithromycin, cethromycin, and Kosan-1325 (K-1325).
The rapid and sudden increase in macrolide resistance in the 1980s and 1990s (29) led to the development of improved compounds, termed ketolides (1, 22). Telithromycin and cethromycin (ABT-773) (Fig. 1) represent two of the most distinguished ketolide members. Both are semisynthetic derivatives of erythromycin, having a keto group at the C-3 position of the lactone ring, instead of l-cladinose. In addition to this structural modification, ketolides possess a carbamate group fused at positions 11 and 12 of the lactone ring as well as a heteroaromatic side chain that is linked via a flexible alkyl linker to the lactone ring at various positions: C-6 for cethromycin and the C-11-C-12 position for telithromycin (22). Cethromycin and telithromycin are effective against some pathogens that are resistant to erythromycin due to efflux or inducible erm methyltransferase mechanisms (17). However, with a few exceptions (33), both lack activity against constitutive resistance. Moreover, both drugs are capable of inducing erm genes, although at a lower rate and in a narrow range of their concentrations (2). It might be kept in mind that there are species-specific interactions of macrolides with the ribosomes, and similar mutations in different bacteria may confer somewhat different resistance profiles (38, 39).
It is believed that the success of the ketolide class to overcome macrolide resistance in these cases is due to the aromatic side chains, which enhance ribosome binding and help evade the efflux systems. Mutational and footprinting studies have indicated a possible interaction of the aryl-alkyl side chains of telithromycin and cethromycin with the loop of helix 35 (H35) that forms the wall of the exit tunnel opposite to A2058 (9, 11, 14, 40). Although direct contact of the side chain with H35 is not evident in crystallographic structures of ketolides complexed to the Deinococcus radiodurans ribosomes, the side chain does extend to the corresponding region of domain II and is located in close proximity to the U790 region of the 23S rRNA (Escherichia coli numbering is used throughout) (3, 31).
The success of telithromycin and cethromycin in treating infections caused by macrolide-resistant streptococcal and staphylococcal strains has triggered an intensive search for new and improved ketolide derivatives (10, 12, 15, 41, 42). Within the framework of these efforts, Kosan Bioscience Inc. has identified a ketolide carrying an extended group at the C-13 position (named K-1325; Fig. 1). This compound exhibits increased activity against some macrolide-resistant strains as well as an improved pharmacokinetic profile (Kosan Bioscience Inc., unpublished data). Here, we investigate the interaction of K-1325 with wild-type and macrolide-resistant ribosomes. Our footprinting and competition assays indicate that only one molecule of K-1325 interacts with the ribosome at the classic macrolide binding site. K-1325 binds twofold tighter to wild-type E. coli ribosomes than does erythromycin, it is better than erythromycin at displacing tylosin from the ribosome, and it is a more effective inhibitor of in vitro translation on the macrolide-resistant A2058G ribosomes. Finally, chemical protection assays indicate that the heterocyclic side chain of K-1325 interacts with domain II of the 23S rRNA, analogous to the activity of telithromycin; however, the differences in footprinting suggest that K-1325 adopts a mode of interaction distinct from that of telithromycin.
MATERIALS AND METHODS
Materials.
Puromycin dihydrochloride (disodium salt), erythromycin, GTP, ATP, and tRNA from E. coli strain W were purchased from Sigma (St. Louis, MO). Poly(U) and poly(A) were purchased from Fluka and Roche, respectively. l-[2,3,4,5,6-3H]phenylalanine, l-[4,5-3H]lysine, and [γ-32P]ATP were purchased from Amersham Biosciences Inc. (Piscataway, NJ). [14C]-erythromycin was purchased from Moravek Biochemicals (Brea, CA). Telithromycin was a gift from Aventis-Pharma to D. L. Kalpaxis. Avian myeloblastosis virus-reverse transcriptase was from Roche Diagnostics (Mannheim, Germany). Cellulose nitrate filters (type HA; 24-mm diameter, 0.45-μm pore size) were from Millipore Corporation (Bedford, MA). Glass fiber filters were from Schleider and Schuell (Dassel, Germany). Ketolide K-1325 was provided by Kosan Bioscience Inc.
Biochemical preparations.
70S ribosomes, either tight-coupled or reassociated, were prepared from E. coli K-12 cells as described previously (4), and they were kept in buffer containing 20 mM HEPES/KOH, pH 7.6, 50 mM CH3COONH4, 6 mM (CH3COO)2Mg, and 4 mM β-mercaptoethanol. The S-100 fraction was prepared as previously described (28), and it was treated with DE-52 cellulose in order to become free of tRNAs and most RNases. Heteropolymeric mRNA (MF-mRNA) encoding formylmethionyl (fMet)-Phe was prepared with runoff transcription (30), and elongation factor G was isolated from E. coli as previously described (8). Ac[3H]Phe-tRNA was prepared using specific tRNA under standard conditions (28) and was freed of uncharged tRNA by reverse-phase high-performance liquid chromatography on a Nucleosil column by using a programmed binary gradient of buffers 1 (20 mM ammonium acetate, pH 5.0, 10 mM magnesium acetate, 400 mM NaCl) and 2 (60% [vol/vol] methanol in buffer 1).
Preparation of defined ribosomal complexes.
All ribosomal complexes were prepared as previously reported (8). Standard ionic conditions were used, namely, 20 mM HEPES-KOH, pH 7.6, 4.5 mM magnesium acetate, 150 mM ammonium acetate, 2 mM spermidine, 0.05 mM spermine, and 4 mM β-mercaptoethanol (called buffer A), and were kept constant throughout all the steps of complex formation. Briefly, tight-coupled or reassociated 70S ribosomes at a final concentration of 0.3 μM were incubated in the presence of the appropriate mRNA (2.0 μΜ) and the charged tRNA (0.5 μM) at 37°C for 15 min unless otherwise indicated. Measurements of tRNA binding and titration of tRNA binding sites were made as described previously (4). All determinations were performed in duplicate with a deviation from the average of less than 10%.
Puromycin reaction.
The reaction between defined ribosomal complexes and excess of puromycin was carried out at 37°C for 2 min. Usually, the reaction volume was 20 μl and puromycin (in buffer A) was added to a final concentration of 1 mM. The reaction was stopped by the addition of an equal volume of 0.3 M sodium acetate, pH 5.5, saturated with MgSO4. Extraction with 1 ml of ethyl acetate followed, and the radioactivity contained in 700 μl of the organic phase was counted by liquid scintillation.
Poly(U)-dependent poly(Phe) synthesis.
The poly(Phe) assay was carried out in buffer A. Tight-coupled 70S ribosomes (0.5 μΜ) were preincubated for 10 min at 37°C with each antibiotic at the appropriate concentration, in a mixture of 15 μl also containing 25 μg poly(U) mRNA, [3H]phenylalanine (5 nmol, 50 dpm/pmol), 1 A260 unit tRNA bulk (E. coli), 3 mM ATP, 1.5 mM GTP, 5 mM acetyl phosphate, and an optimized amount of S-100 fraction. After 30 min of incubation at 37°C, hot trichloroacetic acid precipitation followed, and polypeptides were isolated on glass fiber filters (5). As a positive control, edeine was used at the appropriate concentration. The remaining radioactivity on the filters was measured in a liquid scintillation counter.
Poly(A)-dependent poly(Lys) synthesis.
The poly(Lys) assay was performed as described for the poly(Phe) assay, except that poly(A) replaced poly(U) and [3H]lysine (3 nmol, 500 dpm/pmol) replaced phenylalanine. Incubation took place at 37°C for 1 h. The trichloroacetic acid solution used for polylysine peptide precipitation was treated with 0.25% sodium wolframate before use.
Competition of [14C]-erythromycin binding.
Tight-coupled 70S ribosomes (0.20 μΜ final concentration) were incubated in buffer A with [14C]-erythromycin (150 dpm/pmol) in the appropriate concentration. After incubation for 10 min at 37°C, the mixture was diluted with 3 ml of cold buffer A and was filtered through a 25-mm-diameter cellulose nitrate membrane filter (Millipore; 0.45-μm pore size). The filter was immediately washed twice with 3 ml of cold buffer A, and the bound radioactivity was determined by measuring the radioactivity bound on the filter. Next, the binding of [14C]-erythromycin was studied in competition with cold erythromycin or K-1325, by adding a constant [14C]-erythromycin concentration (0.6 μM) and increasing the concentration of nonradioactive compounds.
Effects of 23S rRNA mutations on antibiotic effect.
E. coli TA531 cells that lack chromosomal rrn alleles but contain plasmids expressing either wild-type or mutated 23S rRNA were used to determine the antibiotic effect on the growth of wild-type or mutated cells and to prepare ribosomes for cell-free assays. Wild-type, A2058G mutant, and U2609C mutant cells contained plasmids pKK3535 (6), pSTL102 (35), and pKK3535(U2609C) (11), respectively. All strains were kindly provided by A. S. Mankin (University of Illinois).
Antibiotic probing and chemical modification.
Aliquots of E. coli 70S ribosomes, 50 pmol per tube, were initially activated for 5 min at 37°C in buffer containing 20 mM HEPES/KOH, pH 7.8, 20 mM MgCl2, and 300 mM KCl. Next, antibiotics were added in the appropriate concentration up to the final volume of 100 μl in the same buffer, and the reaction mixtures were incubated for 10 min at 37°C. After cooling on ice, chemical modification of ribosomes was carried out by using DMS (dimethyl sulfate) or CMCT [1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluene] as described previously (34). The ribosomes were then ethanol precipitated, and the pellets were resuspended in 50 μl of buffer containing 10 mM Tris/HCl, pH 7.5, 100 mM NH4Cl, 5 mM EDTA, and 0.5% sodium dodecyl sulfate. rRNA was isolated by extraction with equal volumes of phenol, phenol-chloroform, and chloroform, followed by ethanol precipitation. Finally, rRNA was resuspended in water.
Primer extension.
The modifications in 23S rRNA were monitored by primer extension analysis using avian myeloblastosis virus-reverse transcriptase and 5′-labeled primer. The primers used were complementary to nucleotides 888 to 906, 2105 to 2121, and 2677 to 2694. The cDNA products were separated by electrophoresis on 6% polyacrylamide sequencing gels. Gels were scanned with PhosphorImager-type Fujifilm (FLA-3000; Berthold) and analyzed with ImageQuant software AIDA (Raytest). The positions of the stops in cDNA synthesis were identified by reference to dideoxy sequencing reactions on 23S rRNA, run in parallel (34). The experiments were repeated up to four times.
MIC determination.
For MIC determinations, cells were grown overnight in Luria-Bertani medium which always contained 100 μg ampicillin per ml. The cultures were diluted into fresh Luria-Bertani medium and were again grown. Exponential-phase cultures were then diluted to a final A600 of 0.050 and incubated with each one of the antibiotics at 37°C for 15 h without shaking and for 3 h with shaking. The MIC was recorded as the lowest concentration of the drug that caused culture growth arrest.
RESULTS
The new ketolide K-1325 was first tested for inhibition of growth of E. coli TA531 cells that lack chromosomal rrn alleles but contain plasmids expressing either wild-type or mutated 23S rRNA. As seen in Table 1, K-1325 is much more effective than erythromycin or telithromycin against E. coli cells expressing wild-type rRNA, whereas the A2058G mutation renders E. coli cells resistant to all macrolides tested, in particular erythromycin. In contrast, the U2609C mutation, which also renders E. coli cells resistant to telithromycin (11, 41), does not affect the MIC of K-1325 recorded in cells producing wild-type rRNA.
TABLE 1.
Effects of rRNA mutations on the susceptibility of E. coli to macrolide antibiotics
| Mutationa | MIC (μg/ml)
|
||
|---|---|---|---|
| Erythromycin | Telithromycin | K-1325 | |
| Wild type | 128 | 16 | 8 |
| A2058G | >500 | >150 | >250 |
| U2609C | 128 | 64 | 8 |
Wild-type or mutant RNAs were expressed in the TA531 strain of E. coli (39) from plasmid-borne genes that were the single source of rRNA in the cell. Wild-type cells contained plasmid pKK3535 (6), the A2058G mutant contained plasmid pSTL102 (35), and the U2609C mutant contained plasmid pKK3535(2609C) (11).
Next, we explored the binding position of K-1325 in the ribosome competition and chemical footprinting experiments. Figure 2A reveals that the binding of [14C]-erythromycin to ribosomes is progressively reduced, as the concentration of K-1325 increases. For comparison, the corresponding plot obtained using nonradioactive erythromycin instead of K-1325 is also presented. The plots shown in Fig. 2A follow the equation.
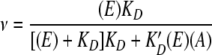 |
(1) |
where ν is the saturation ratio that is equal to the number of [14C]-erythromycin molecules bound per ribosome, E and K′D are the concentration and dissociation constants of [14C]-erythromycin, and A and KD are the concentration and dissociation constants of the competitor (nonradioactive erythromycin or K-1325, respectively). Equation 1 implies that
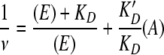 |
(2) |
By fixing the data to equation 2, the KD and K′D values for K-1325 and erythromycin were determined as 7.5 nM and 15 nM, respectively. Equation 2 predicts that one molecule of ligand is bound per ribosome. Consequently, the fact that the data for K-1325 fit so perfectly to equation 2 (Fig. 2B) strongly suggests that only one molecule of K-1325 is bound per ribosome.
FIG. 2.

Competition of [14C]-erythromycin binding to vacant ribosomes by nonradioactive erythromycin or K-1325. (A) Plot of ν (pmol of bound [14C]-erythromycin per pmol of ribosomes) versus the concentration of K-1325 (▿) or nonradioactive erythromycin (•). The concentration of ribosomes was multiplied by a 0.72 factor, which represents the active fraction of tight ribosomes capable of binding erythromycin. (B) Plot of 1/ν versus the concentration of K-1325 (▿) or erythromycin (•).
To investigate the mode of binding of K-1325 to the ribosome, we chemically probed the 23S rRNA in the presence and absence of K-1325. Since the “classical” ribosomal binding site for macrolides is known from X-ray crystallography studies (3, 13, 32, 37), we used primer extension to monitor the interaction of the drug with domains V and II of the 23S rRNA. Additionally, we used E. coli 70S ribosomes containing wild-type 23S rRNA as well as ribosomes bearing the resistance mutation U2609C (11, 41). In domain V of the 23S rRNA, nucleotides A2058 and A2059 are strongly protected from DMS modification by K-1325 as well as by erythromycin (Fig. 3A). In contrast to erythromycin and independently of the U2609C mutation, an additional novel observation was that the presence of K-1325 induced an enhancement in the modification of nucleotide A2062 of the 23S rRNA (Fig. 3A). In Fig. 3A, we can also see the difference between K-1325 and erythromycin with respect to protection of U2609: while the presence of erythromycin results in a mild protection of U2609, no protection by K-1325 is observed regardless of whether wild-type or mutant ribosomes are used.
FIG. 3.

Modification of 23S rRNA accessibility toward chemical modifiers DMS and CMCT by bound antibiotics. 70S ribosomes, either wild type (wt) or U2609C mutant, complexed or not complexed with antibiotics, were probed with DMS or CMCT, and modification sites of 23S rRNA were detected by primer extension analysis. Modifications in domain V of 23S rRNA (A) and in domain II of 23S rRNA (B) are shown.
Next, we monitored the modification of nucleotides within domain II of the 23S rRNA, which also comprises part of the classical macrolide binding site. As shown in Fig. 3B, nucleotide A752 is strongly protected by K-1325 from DMS, whereas erythromycin enhanced modification of A752, in agreement with previous studies (14, 27). In addition, binding of K-1325 decreased the accessibility of A789 to DMS and U790 to CMCT, whereas erythromycin failed to impart the same protections (Fig. 3B). Collectively, these results suggest that K-1325 directly interacts with domain II of the 23S rRNA, which is located deeper in the ribosomal tunnel, unlike erythromycin, which appears to indirectly induce conformational change within this region.
To investigate the effect of K-1325 on protein synthesis, in vitro translation assays polymerizing phenylalanine and lysine were performed in the presence or in the absence of K-1325. For comparison, parallel experiments were carried out in the presence of erythromycin or edeine. Figure 4A shows that poly(U)-dependent synthesis of poly(Phe) is not inhibited by K-1325 or by erythromycin. In contrast, poly(A)-dependent synthesis of poly(Lys) is strongly blocked by the presence of either K-1325 or erythromycin (Fig. 4B). The initiation inhibitor edeine inhibited both poly(Phe) and poly(Lys) synthesis, as reported previously (8). The differential effects of macrolides on poly(Phe) and poly(Lys) synthesis have been observed previously (16, 36). One possible explanation for the lack of inhibitory effect of macrolides on poly(Phe) synthesis is that the highly hydrophobic nascent poly(Phe) chain may displace K-1325 from its binding site on 50S subunits as it passes through the tunnel. However, a second and more likely hypothesis is that the nascent poly(Phe) chain does not pass through the tunnel during its synthesis (26), and therefore the synthesis of this polypeptide is not inhibited by erythromycin or K-1325. Indeed, it is possible to synthesize long poly(Phe) chains while erythromycin remains bound to the ribosome (23).
FIG. 4.

Effect of K-1325 on poly(Phe) and poly(Lys) synthesis. (A) Poly(Phe) synthesis in the absence or in the presence of erythromycin and K-1325. Edeine was used as a positive control. The concentration of each antibiotic was 20 μM, and the reaction mixture was incubated at 37°C for 15 min. (B) Poly(Lys) synthesis in the absence or in the presence of erythromycin, K-1325, and edeine. Experimental conditions and antibiotic concentrations were the same as before, except that the incubation time was 1 h. (C) Poly(Lys) assay as described for panel B, except that the ribosomes used carry the A2058G mutation.
The poly(Lys) assay was also performed with ribosomes carrying the A2058G mutation. As shown in Fig. 4C, only a small inhibition was observed, even at high concentrations (50 μM) of K-1325, a fact supporting the notion that such ribosomes exhibit low affinity for this ketolide. In contrast, ribosomes carrying the U2609C mutation behaved similarly to wild-type ribosomes (data not shown), indicating that this mutation does not affect the K-1325 binding.
To verify the above results, we also applied another experimental approach. We have previously demonstrated that 16-membered macrolides, like tylosin, inhibit AcPhe-puromycin synthesis, whereas 14-membered macrolides, such as erythromycin, do not (7, 16). This is due to the fact that the C-5-disaccharide of tylosin reaches into the peptidyl transferase (PTase) center of the ribosome, whereas the C-5-monosaccharide of 14-membered macrolides is too short (3, 13, 32, 37). However, since erythromycin still competes with tylosin for binding on the 50S subunit, erythromycin restores AcPhe-puromycin reactivity by chasing tylosin from the ribosome. Therefore, Pi ribosomal complexes were prepared from wild-type and mutant ribosomes by binding AcPhe-tRNA to the P-site in the presence of MF-mRNA. The activity of the Pi complex in reacting with puromycin was used to monitor the competition between tylosin and K-1325 or erythromycin for binding to the large ribosomal subunit. As seen in Fig. 5A, when Pi complex was reacted with 1 mM puromycin, most of the AcPhe-tRNA bound to the complex was converted to AcPhe-puromycin, and this was not affected by erythromycin or K-1325. In contrast, the presence of 5 μM tylosin almost completely abolished AcPhe-puromycin synthesis. Preincubation of the Pi complex with erythromycin (10 μM) or K-1325 (10 μM) relieved the inhibitory effect exerted by tylosin on AcPhe-puromycin synthesis (Fig. 5A). This result indicates that the affinity of K-1325 for wild-type ribosomes is similar to that of erythromycin. Additionally, when the same assay was performed using U2609C mutant ribosomes, both erythromycin and K-1325 were effective at relieving the inhibitory effect exerted by tylosin (Fig. 5B). This suggests that little or no difference in affinity between erythromycin and K-1325 is seen on the U2609C mutant ribosomes. In contrast, when the same assay was performed using A2058G mutant ribosomes, although tylosin had a lower inhibitory effect on AcPhe-puromycin formation, K-1325 exerted a higher protection effect than erythromycin, indicating a better affinity for the A2058G mutant ribosomes (Fig. 5C).
FIG. 5.

Puromycin reaction with wild-type and mutant ribosomes, in the absence or in the presence of antibiotics. Pi ribosomal complex, containing Ac[3H]Phe-tRNA at the P-site of MF-mRNA-programmed 70S ribosomes free of unbound donor, was reacted with 1 mM puromycin at 37°C for 2 min. The reaction took place in the absence of antibiotic (first bar) and in the presence of 10 μM erythromycin (second bar), K-1325 (third bar), and 5 μΜ tylosin (fourth bar). Next, Pi complex was first exposed to erythromycin or K-1325 (final concentration, 10 μM) at 37°C for 2 min, and then tylosin was added (final concentration, 5 μM). (A) Wild-type ribosomes. (B) Ribosomes carrying the U2609C mutation. (C) Ribosomes carrying the A2058G mutation.
Further kinetic studies revealed that K-1325, like erythromycin (7, 19), acts as a slow-binding, slowly dissociating inhibitor (21) (data not shown). Using a previously established method (16), we found that the dissociation rate constant for K-1325 from the ribosomal Pi complex is equal to 0.003 min−1 (Fig. 6), i.e., about one order of magnitude lower than that of erythromycin, supporting the notion that the binding of K-1325 to wild-type ribosomes is tighter than that with erythromycin.
FIG. 6.

Determination of the dissociation rate constant (k7) of the complex between antibiotic K-1325 and the Pi ribosomal complex. Drug-ribosome complex was prepared in the presence of 2 × 10−6 M antibiotic and absorbed on a cellulose nitrate filter. Then, it was exposed to 1 × 10−5 M tylosin for various time intervals, after which the remaining activity was titrated with puromycin. The k7 value was the estimate from the slope of the linear time plot.
DISCUSSION
After the successful introduction of telithromycin into the market, many research labs tried to develop other novel versions of ketolides to combat the rising tide of multidrug-resistant bacteria. One of these compounds is the new ketolide K-1325, developed by Kosan Bioscience Inc., which carries an aryl-alkyl side chain extending from the C-13 position of the lactone ring. The purpose of the present study was to characterize the interaction of this novel ketolide with E. coli ribosomes.
As expected, our competition (Fig. 2) and footprinting (Fig. 3) results indicate that like other macrolide antibiotics, K-1325 binds within the exit tunnel located on the large ribosomal subunit. This is consistent with the lack of inhibitory effect of K-1325 on the binding of tRNAs to the ribosome, Elongation factor G-dependent translocation, and the PTase activity of the ribosome (data not shown). Specifically, we show that K-1325 has the capability to compete with erythromycin for binding to ribosomes as well as to reduce the inhibitory effect of tylosin on peptide bond formation. Indeed, K-1325 and erythromycin have no effect on the puromycin reaction, whereas tylosin strongly inhibits this reaction (Fig. 5). This is consistent with biochemical and crystallographic studies revealing that the C-5-disaccharides of 16-membered macrolides reach into the PTase center to perturb the PTase activity, whereas the single C-5-monosaccharide of K-1325, erythromycin, and telithromycin is too short to extend into the PTase center (13, 18, 27). Interestingly, structural studies of azithromycin bound to the D. radiodurans 50S subunit reveal that in addition to the drug being bound at the classical site, a second molecule is present deeper in the tunnel (31). In contrast, our results indicate that such an interaction with two molecules of the drug bound per ribosome does not seem to exist for K-1325. This conclusion is derived from Fig. 2B and 6, where the linearity of both diagrams supports the notion that only one molecule of K-1325 binds per ribosome.
According to the chemical protection data, K-1325, like erythromycin (9, 11, 41), strongly protects A2058 and A2059 from DMS modification. This is consistent with X-ray crystallography structures showing that the desosamine sugar of macrolides contacts these bases (Fig. 7A) and can even form hydrogen bond interactions (3, 13, 32, 37). The importance of this contact is emphasized by the reduced inhibitory effect that erythromycin and K-1325 have on poly(Lys) synthesis when A2058G mutant ribosomes are used (Fig. 4C). That K-1325 is slightly more effective than erythromycin is in agreement with the strong resistance of A2058G mutant strains to erythromycin but the relatively higher susceptibility to telithromycin and K-1325 (Table 1) (41).
FIG. 7.

Relative positions of K-1325 (yellow) compared to tylosin (cyan) and telithromycin (purple) bound to the H. marismortui large ribosomal subunit (H50S) and of telithromycin (green) bound to the D. radiodurans large ribosomal subunit (D50S). Note that in H50S, the aryl-alkyl side chain of telithromycin stacks upon U2609, whereas in the D50S structure, the same side chain penetrates deeper into the tunnel and stacks on U790 within domain II of the 23S rRNA. (B) Details as in panel A, except illustrating the ribosomal environment of the tunnel with a surface representation. E. coli numbering for all bases is used in both panels. Brown color represents the exit route of the nascent peptide. This figure utilizes PDB1K9M (tylosin-H50S) (13), 1YIJ (telithromycin-H50S) (37), and telithromycin-D50S (39) and was created with PyMol (http://www.pymol.org).
The tighter binding of K-1325 is most likely due to the fact that it carries an alkyl-aryl side chain which enables additional contacts with domain II of 23S rRNA to be established. Indeed, we found that K-1325 strongly protects A752 in the loop of H35 of domain II of the 23S rRNA in E. coli ribosomes from DMS modification (Fig. 3B), as has been reported previously for telithromycin (11). In contrast, erythromycin, which lacks such an extension, enhances the accessibility of A752 to DMS, suggesting that although erythromycin does not directly interact with this region (32, 37), drug binding induces conformational changes there. Curiously, direct contact of the C-11-C-12 side chain with A752 in the loop of H35 is not evident in crystallographic structures of telithromycin in complex with the large subunit of D. radiodurans ribosomes (3, 39); however, the heterocyclic side chain is seen to approach a groove formed by nucleotides A751 and A789-U790 and to establish stacking interactions with the base of U790 (Fig. 7A). In this respect, it is relevant that K-1325, but not erythromycin, protects A788 and U790 from chemical attack by DMS and CMCT, respectively (Fig. 3B).
The mutation U2609C confers resistance to telithromycin but does not alter the susceptibility to erythromycin (Table 1) (41). An advantage of K-1325 over telithromycin is that the U2609C mutation does not influence the low MIC (8 μg/ml) of K-1325 compared to that of wild-type E. coli strains (Table 1). Consistent with this, K-1325 does not protect position U2609 in wild-type or U2609C mutant ribosomes (Fig. 3A), whereas modest protection by telithromycin has been observed previously (11, 41). Crystallographic studies of telithromycin complexed with the large subunit of Haloarcula marismortui indeed show that the alkyl-aryl side chain folds back over the lactone ring and stacks upon U2609 (HmC2644) (Fig. 7A) (37). However, such an interaction was not observed in the telithromycin complex with the D. radiodurans large ribosomal subunit (3, 39). Erythromycin strongly protects U2609 in wild-type and mutant U2609C ribosomes, consistent with the competition results (Fig. 5B), illustrating that the U2609C mutation does not perturb the binding of erythromycin. This is in excellent agreement with resistance and antibiotic binding data published previously (11), as well as a more recent study showing that the U2609C mutation does not significantly change the kinetics of erythromycin binding to ribosomes (25).
Collectively, these results suggest that although K-1325 interacts with domain II of the 23S rRNA like other ketolides, such as telithromycin, the differences in footprinting patterns indicate that the mode of interaction differs. The side chain of K-1325 extends from the C-13 of the lactone ring rather than C-11-C-12 as for telithromycin, and this would favor direct interaction of the K-1325 side chain with A752, rather than with U790, as shown in Fig. 7. Moreover, in E. coli, U790 is positioned as in H. marismortui rather than as in D. radiodurans, and stacking on U790 would require conformational flipping-out of the base. This may suggest that in E. coli the side chain of telithromycin adopts a similar position as that seen in H. marismortui. In fact, the weak protection of U790 by K-1325 observed here for E. coli could even result from stabilizing U790 in a flipped-in position rather than by direct interaction. However, to fully understand how the side chain of K-1325 interacts with the E. coli ribosomes, a crystal structure of the K-1325-E. coli ribosome complex will be necessary.
In conclusion, our results suggest that K-1325 is a potent inhibitor, displaying better MICs than erythromycin and telithromycin against wild-type and U2609C mutant E. coli strains. We note, however, that K-1325, like erythromycin and telithromycin, exhibits reduced effectiveness against the mutant A2058G E. coli strains. Binding studies indicate that K-1325 has a twofold higher affinity for the ribosome than does erythromycin, consistent with its better ability to compete with tylosin for ribosome binding. Footprinting studies indicate that the tighter binding of K-1325 results from additional interactions that the alkyl-aryl side chain establishes with nucleotides located in domain II of 23S rRNA. The distinct interactions of K-1325, compared to those of telithromycin, may explain the superior resistance profile of K-1325 compared to that for telithromycin for U2609C mutant E. coli cells. Finally, our results emphasize the importance of the position of the lactone ring in which the alkyl-aryl side chain is attached, as it governs the mode of interaction with the ribosome, which in turn dictates the affinity and susceptibility to macrolide resistance mutations in the ribosome.
Acknowledgments
We thank Kosan Bioscience Inc. for providing the new ketolide K-1325 and partial financial support, the Deutsche Forschungsgemeinschaft for financial support (WI3285/1-1 to D.N.W.), and Alexander Mankin for kindly providing the plasmids expressing either wild or mutated 23S rRNA.
Footnotes
Published ahead of print on 21 January 2009.
REFERENCES
- 1.Ackermann, G., and A. C. Rodloff. 2003. Drugs of the 21st century: telithromycin (HMR 3647)—the first ketolide. J. Antimicrob. Chemother. 51:497-511. [DOI] [PubMed] [Google Scholar]
- 2.Bailey, M., T. Chettiath, and A. Mankin. 2008. Induction of erm(C) expression by noninducing antibiotics. Antimicrob. Agents Chemother. 52:866-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berisio, R., J. Harms, F. Schluenzen, R. Zarivach, H. A. S. Hansen, P. Fucini, and A. Yonath. 2003. Structural insight into the antibiotic action of telithromycin against resistant mutants. J. Bacteriol. 185:4276-4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaha, G., U. Stelzl, C. M. T. Spahn, R. K. Agrawal, J. Frank, and K. H. Nierhaus. 2000. Preparation of functional ribosomal complexes and the effect of buffer conditions on tRNA positions observed by cryoelectron microscopy. Methods Enzymol. 317:292-309. [DOI] [PubMed] [Google Scholar]
- 5.Bommer, U., N. Burkhardt, R. Junemann, C. M. T. Spahn, F. J. Triana-Alonso, and K. H. Nierhaus. 1996. Ribosomes and polysomes, p. 271-301. In J. Graham and D Rickwoods (ed.), Subcellular fractionation. A practical approach. Oxford University Press, New York, NY.
- 6.Brosius, J., A. Ullrich, M. A. Raker, A. Gray, T. J. Dull, R. R. Gutell, and H. F. Noller. 1981. Construction and fine mapping of recombinant plasmids containing the rrnB ribosomal RNA operon of E. coli. Plasmid 6:112-118. [DOI] [PubMed] [Google Scholar]
- 7.Dinos, G., and D. Kalpaxis. 2000. Studies on the interaction between a ribosomal complex active in peptide bond formation and the macrolide antibiotics tylosin and erythromycin. Biochemistry 39:11621-11628. [DOI] [PubMed] [Google Scholar]
- 8.Dinos, G., D. N. Wilson, Y. Teraoka, W. Szaflarski, P. Fucini, D. Kalpaxis, and K. H. Nierhaus. 2004. Dissecting the ribosomal inhibition mechanisms of edeine and pactamycin: the universally conserved residues G693 and C795 regulate P-site tRNA binding. Mol. Cell 13:113-124. [DOI] [PubMed] [Google Scholar]
- 9.Douthwaite, S., L. H. Hansen, and P. Mouvais. 2000. Macrolide-ketolide inhibition of MLS-resistant ribosomes is improved by alternative drug interaction with domain II of 23S rRNA. Mol. Microbiol. 36:183-193. [DOI] [PubMed] [Google Scholar]
- 10.Fu, H., S. Marquez, X. Gu, L. Katz, and D. C. Myles. 2006. Synthesis and in vitro antibiotic activity of 16-membered 9-O-arylalkyloxime macrolides. Bioorg. Med. Chem. Lett. 16:1259-1266. [DOI] [PubMed] [Google Scholar]
- 11.Garza-Ramos, G., I. Xiong, P. Zhong, and A. Mankin. 2001. Binding site of macrolide antibiotics on the ribosome: new resistance mutation identifies a specific interaction of ketolides with rRNA. J. Bacteriol. 183:6898-6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant, E., D. Guiadeen, D. Abbanat, B. D. Foleno, K. Bush, and M. J. Macielag. 2006. Synthesis and antibacterial activity of 6-O-heteroarylcarbamoyl-11,12-lactoketolides. Bioorg. Med. Chem. Lett. 16:1929-1933. [DOI] [PubMed] [Google Scholar]
- 13.Hansen, J. L., J. A. Ippolito, N. Ban, P. Nissen, P. B. Moore, and T. A. Steitz. 2002. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell 10:117-128. [DOI] [PubMed] [Google Scholar]
- 14.Hansen, L. H., P. Mauvais, and S. Douthwaite. 1999. The macrolide-ketolide antibiotic binding site is formed by structures in domains II and V of 23S ribosomal RNA. Mol. Microbiol. 31:623-631. [DOI] [PubMed] [Google Scholar]
- 15.Kaneto, P., K. Romero, B. Li, and R. Buzon. 2007. Novel tethers in ketolide antibiotics. Bioorg. Med. Chem. Lett. 17:5049-5053. [DOI] [PubMed] [Google Scholar]
- 16.Karahalios, P., D. L. Kalpaxis, H. Fu, L. Katz, D. N. Wilson, and G. P. Dinos. 2006. On the mechanism of action of 9-O-arylalkyloxime derivatives of 6-O-mycaminosyltylonolide, a new class of 16-membered macrolide antibiotics. Mol. Pharmacol. 70:1271-1280. [DOI] [PubMed] [Google Scholar]
- 17.Katz, L., and G. W. Ashley. 2005. Translation and protein synthesis: macrolides. Chem. Rev. 105:499-527. [DOI] [PubMed] [Google Scholar]
- 18.Kirillov, S. V., B. T. Porse, and R. A. Garrett. 1999. Peptidyl transferase antibiotics perturb the relative positioning of the 3′-terminal adenosine of P/P′-site-bound tRNA and 23S rRNA in the ribosome. RNA 5:1003-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lovmar, M., T. Tenson, and M. Ehrenberg. 2004. Kinetics of macrolide action: the josamycin and erythromycin cases. J. Biol. Chem. 279:53506-53515. [DOI] [PubMed] [Google Scholar]
- 20.Menninger, J. R., and D. P. Otto. 1982. Erythromycin, carbomycin, and spiramycin inhibit protein synthesis by stimulating the dissociation of peptidyl-tRNA from ribosomes. Antimicrob. Agents Chemother. 21:811-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison, J. F., and C. Walsh. 1988. The behavior and significance of slow-binding inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61:201-301. [DOI] [PubMed] [Google Scholar]
- 22.Nilius, A. M., and Z. Ma. 2002. Ketolides: the future of macrolides? Curr. Opin. Pharmacol. 2:493-500. [DOI] [PubMed] [Google Scholar]
- 23.Odom, O. W., W. D. Picking, T. Tsalkova, and B. Hardesty. 1991. The synthesis of polyphenylalanine on ribosomes to which erythromycin is bound. Eur. J. Biochem. 198:713-722. [DOI] [PubMed] [Google Scholar]
- 24.Omura, S. 2002. Macrolide antibiotics: chemistry, biology and practice. Academic Press, Orlando, FL.
- 25.Petropoulos, A. D., E. C. Kouvela, G. Dinos, and D. L. Kalpaxis. 2008. Stepwise binding of tylosin and erythromycin to Escherichia coli ribosomes, characterized by kinetic and footprinting analysis. J. Biol. Chem. 283:4756-4765. [DOI] [PubMed] [Google Scholar]
- 26.Picking, W. D., O. W. Odom, T. Tsalkova, I. Serdyuk, and B. Hardesty. 1991. The conformation of nascent polylysine and polyphenylalanine peptides on ribosomes. J. Biol. Chem. 266:1534-1542. [PubMed] [Google Scholar]
- 27.Poulsen, S. M., C. Kofoed, and B. Vester. 2000. Inhibition of the ribosomal peptidyl transferase reaction by the mycarose moiety of the antibiotics carbomycin, spiramycin and tylosin. J. Mol. Biol. 304:471-481. [DOI] [PubMed] [Google Scholar]
- 28.Rheinberger, H. J., U. Geigenmuller, M. Wedde, and K. H. Nierhaus. 1988. Parameters for the preparation of Escherichia coli ribosomes and ribosomal subunits active in tRNA binding. Methods Enzymol. 164:658-670. [DOI] [PubMed] [Google Scholar]
- 29.Sahm, D. F., J. A. Karlowsky, L. J. Kelly, I. A. Critchley, M. E. Jones, C. Thornsberry, Y. Mauriz, and J. Kahn. 2001. Need for annual surveillance of antimicrobial resistance in Streptococcus pneumoniae in the United States: 2-year longitudinal analysis. Antimicrob. Agents. Chemother. 45:1037-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer, M. A., A. O. Tastan, S. Patzke, G. Blaha, C. M. Spahn, D. N. Wilson, and K. H. Nierhaus. 2002. Codon-anticodon interaction at the P site is a prerequisite for tRNA interaction in the small ribosomal subunit. J. Biol. Chem. 277:19095-19105. [DOI] [PubMed] [Google Scholar]
- 31.Schlunzen, F., J. Harms, F. Franceschi, H. Hansen, H. Bartels, R. Zarivach, and A. Yonath. 2003. Structural basis for the antibiotic activity of ketolides and azalides. Structure 11:329-338. [DOI] [PubMed] [Google Scholar]
- 32.Schlunzen, F., R. Zarivach, A. Basham, A. Tocilj, R. Albrecht, A. Yonath, and F. Franceschi. 2001. Structural basis for the interaction of antibiotics with the peptidyl transferase center in eubacteria. Nature 413:814-821. [DOI] [PubMed] [Google Scholar]
- 33.Shortridge, V. D., P. Zhong, Z. Cao, J. M. Beyer, L. S. Almer, N. C. Ramer, S. Z. Doktor, and R. K. Flamm. 2002. Comparison of in vitro activities of ABT-773 and telithromycin against macrolide-susceptible and -resistant streptococci and staphylococci. Antimicrob. Agents Chemother. 46:783-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stern, S., D. Moazed, and H. Noller. 1988. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 164:481-489. [DOI] [PubMed] [Google Scholar]
- 35.Triman, K., E. Becker, C. Dammel, J. Katz, H. Mori, S. Douthwaite, C. Yapijakis, S. Yoast, and H. F. Noller. 1989. Isolation of temperature-sensitive mutants of 16 S rRNA in Escherichia coli. J. Mol. Biol. 209:645-653. [DOI] [PubMed] [Google Scholar]
- 36.Tsagkalia, A., F. Leontiadou, M. A. Xaplanteri, G. Papadopoulos, D. L. Kalpaxis, and T. Choli-Papadopoulou. 2005. Ribosomes containing mutants of L4 ribosomal protein from Thermus thermophilus display multiple defects in ribosomal functions and sensitivity against erythromycin. RNA 11:1633-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu, D., G. Blaha, P. B. Moore, and T. A. Steitz. 2005. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 121:257-270. [DOI] [PubMed] [Google Scholar]
- 38.Vester, B., and S. Douthwaite. 2001. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 45:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson, D. N., J. M. Harms, K. H. Nierhaus, F. Schluenzen, and P. Fucini. 2005. Species-specific antibiotic-ribosome interactions: implications for drug development. Biol. Chem. 386:1239-1252. [DOI] [PubMed] [Google Scholar]
- 40.Xiong, L., S. Shah, P. Mauvais, and A. S. Mankin. 1999. A ketolide resistance mutation in domain II of 23S rRNA reveals the proximity of hairpin 35 to the peptidyl transferase centre. Mol. Microbiol. 31:633-639. [DOI] [PubMed] [Google Scholar]
- 41.Xiong, L., Y. Korkhin, and A. S. Mankin. 2005. Binding site of the bridged macrolides in the Escherichia coli ribosome. Antimicrob. Agents Chemother. 49:281-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu, B., B. A. Marinelli, D. Abbanat, B. D. Foleno, K. Bush, and M. J. Macielag. 2007. Synthesis and antibacterial activity of 3-keto-6-O-carbamoyl-11,12-cyclic thiocarbamate erythromycin A derivatives. Bioorg. Med. Chem. Lett. 17:3900-3904. [DOI] [PubMed] [Google Scholar]