

Abstract
Moxifloxacin (MXF) is a fluoroquinolone antibiotic that is effective against respiratory infections. However, the mechanisms of MXF lung diffusion are unknown. Active transport in other tissues has been suggested for several members of the fluoroquinolone family. In this study, transport of MXF was systematically investigated across a Calu-3 lung epithelial cell model. MXF showed polarized transport, with the secretory permeability being twice as high as the absorptive permeability. The secretory permeability was concentration dependent (apparent Pmax = 13.6 × 10−6 cm·s−1; apparent Km = 147 μM), suggesting saturated transport at concentrations higher than 350 μg/ml. The P-glycoprotein inhibitor PSC-833 inhibited MXF transport in both directions, whereas probenecid, a multidrug resistance-related protein inhibitor, appeared to have no effect in the Calu-3 model. Moreover, rifampin, a known inducer of efflux transport proteins, upregulated the expression of P-glycoprotein in Calu-3 cells and enhanced MXF active transport. In conclusion, this study clearly indicates that MXF is subject to P-glycoprotein-mediated active transport in the Calu-3 model. This P-glycoprotein-dependent secretion may lead to higher MXF epithelial lining fluid concentrations than those in plasma. Furthermore, drug-drug interactions may be expected when MXF is combined with other P-glycoprotein substrates or modulators.
Fluoroquinolones (FQs) are one of the main classes of antimicrobial agents, with a broad spectrum of activity and a concentration-dependent killing effect (25, 29). Moxifloxacin (MXF) is an FQ that has been shown to be effective against respiratory pathogens, including Haemophilus influenzae, Moraxella catarrhalis, and penicillin-resistant strains of Streptococcus pneumoniae (3, 24). The use of MXF is recommended as therapy for patients with community-acquired pneumonia, exacerbations of chronic bronchitis, chronic obstructive pulmonary disease, or tuberculosis (21). Pharmacokinetics of FQs have been studied in different tissues, such as the liver, kidney, and intestinal tract, and it appears that multiple transporters contribute to drug disposition (2). Active transport has been suggested for many FQs, involving mainly the ATP-binding cassette (ABC) transporter family (P-glycoprotein [P-gp] and multidrug resistance-related proteins [MRPs]). However, there is a lack of information regarding the mechanisms of MXF lung distribution. To our knowledge, cellular transport of MXF through the lung tissue is still unclear, and an understanding of its mechanisms would help to better predict its pulmonary disposition, which is directly linked to the bacterial killing effect. Moreover, when MXF is used as a second-line therapy or in combination with drugs such as rifampin (22), drug-drug interaction can occur, as presented in some rifampin-MXF interaction studies (23, 30). In this particular case, MXF was recommended for the treatment of multidrug-resistant tuberculosis and a decrease in MXF plasma concentration was observed in association with rifampin. Although MXF phase II metabolism induction was suggested, drug-drug interaction could also occur through a P-gp-dependent secretion pathway, since rifampin is best known as a strong inducer of P-gp (20). P-gp has been identified in the lung (14), and therefore a rifampin-MXF interaction could also affect MXF pulmonary distribution. The aim of this study was to evaluate the transport mechanisms of MXF by using the Calu-3 cell model, which has been reported to express tight junctions, P-gp, and MRP1 efflux proteins (11, 12).
MATERIALS AND METHODS
Materials.
Sodium fluorescein (SF), probenecid, verapamil-HCl, and HEPES were purchased from Sigma-Aldrich. Hanks' balanced salt solution and phosphate-buffered saline (PBS), pH 7.4, were purchased from Gibco BRL Life Technology (Grand Island, NY). MXF-HCl (>98.5% pure) and PSC-833 were kindly supplied by Bayer Healthcare (Leverkusen, Germany) and Novartis (Basel, Switzerland), respectively. Transwell clear polyester membranes with a 4.67-cm2 area and a pore size of 0.4 μm were obtained from Corning Costar (Cambridge, MA). Cell culture media and supplements were purchased from PAN Biotech GmbH (Aidenbach, Germany). Tetrabutylammonium perchlorate and acid citric monohydrate were obtained from Fluka (Buchs, Switzerland). High-performance liquid chromatography-grade acetonitrile was purchased from VWR International (Fontenay sous Bois, France). All other reagents were analytical grade.
Calu-3 cell culture.
Calu-3 cells were purchased from the American Type Culture Collection (Rockville, MD) and maintained as a monolayer culture in plastic tissue culture flasks of 75 cm2 (Nunc, Roskilde, Denmark). The cells were cultured in Dulbecco's modified Eagle's medium-Ham's F12 medium (1:1) supplemented with l-glutamine (2 mM), fetal calf serum (10% [vol/vol]), and gentamicin (50 μg/ml) and incubated at 37°C. The atmosphere was kept at 90 to 95% relative humidity with 5% (vol/vol) CO2 in air. Upon reaching 80 to 90% confluence, the cells were detached with approximately 3 ml of 0.05% trypsin-0.02% EDTA solution in PBS (PAN Biotech GmbH, Aidenbach, Germany) and plated into new flasks at a split ratio of 1:3. The medium was renewed every 2 to 3 days. The Calu-3 cells at passages 22 to 30 were seeded at a density of 5 × 105 cells/cm2 onto transparent tissue culture inserts, which were placed in six-well plates and incubated as previously described. The volume in the apical (AP) compartment (1.5 ml) was removed 24 h after incubation, and the cells were cultured under air-interface conditions for 15 days. The growth medium in the basolateral (BL) compartment (2.6 ml) was replaced with fresh medium every other day (10). At this point, the presence of functional tight junctions was evidenced by the fact that medium from the BL side ceased to diffuse to the AP chamber (27).
Concentration effect.
Transport experiments were conducted in the BL-to-AP direction. Solutions of MXF from 10 to 800 μM (i.e., 4.4 to 350 μg/ml MXF-HCl) were prepared in triplicate in transport medium (TM) (Hanks' buffered salt solution buffered with 10 mM HEPES and adjusted to pH 7.4 with 1 M sodium hydroxide). On the study day, Dulbecco's modified Eagle's medium-Ham's F12 medium was withdrawn from the chamber. The Calu-3 monolayers were quickly rinsed with PBS and incubated for 30 min in TM. Following the equilibration period, the drug-free TM on the BL side was replaced with fresh TM containing MXF and the plates were returned to the incubator. Subsequently, sample aliquots (200 μl) were taken from the AP compartment at 60, 120, and 180 min, and the same volume was replaced with fresh TM. Following the transport study, a control for monolayer integrity was performed using SF (10). Briefly, the monolayers were rinsed with PBS, fresh TM was added to the BL chamber, and a solution of SF in TM (10 μg/ml) was poured into the AP side. The inserts were incubated, and samples (200 μl) were taken after 60 min. The threshold apparent permeability (Papp) of 0.7 × 10−6 cm·s−1 for SF was retained for the tight junction integrity rejection parameter for all experiments. This corresponds to <1% of the initial amount in the AP compartment.
Inhibition studies.
Transport experiments were conducted in both the AP-to-BL and BL-to-AP directions. Solutions of MXF (50 μM) were prepared in TM in the presence of the following efflux pump inhibitors at the indicated concentrations: verapamil, 100 μM; probenecid, 100 μM; and PSC-833, 3 μM. Cells were preincubated for 30 min with the TM containing the respective inhibitor. The volume of the donor compartment was then replaced with test solution (MXF plus inhibitor). Samples from the acceptor compartment were taken and replenished with fresh inhibitory solution in TM as previously described. Control experiments using MXF (50 μM) in TM were conducted in parallel. The monolayer integrity test was performed as mentioned above.
Influence of rifampin on MXF transport.
Calu-3 cells were seeded on semipermeable membranes and cultured for 15 days as already described, but in the presence or absence of rifampin. Fresh solutions of rifampin at 10 μM and 25 μM in culture medium were used. On the study day, the cell monolayers were rinsed three times with TM for 10 min to ensure elimination of rifampin, and transport experiments with MXF were conducted as mentioned earlier.
MXF and SF assay.
Samples containing MXF were directly analyzed by reversed-phase high-performance liquid chromatography with fluorescence detection (λex = 285 nm; λem = 490 nm), as previously described for other FQs (16, 17). Samples for standard curves were prepared in TM, and curves were linear within the range of validation (0.06 to 22.5 μg/ml), with coefficients of correlation of >0.995 using 1/X2 as a weighing factor, where X was the MXF concentration (μg/ml). The chromatographic system consisted of a Waters 717 autosampler (Waters Millipore, Milford, MA), a Lachrom 7110 solvent delivery pump (Merck, Darmstadt, Germany), and a Waters 474 fluorescence detector (Waters Millipore). The temperature of the autosampler was set at 4°C throughout the analysis. Chromatography was carried out using a Nucleosil ODS-5 C18 (250 mm by 4.6-mm internal diameter, with 5-μm pore size; Interchrom, Montluçon, France) analytical column protected by an Interchrom C18 guard column (10 mm by 4-mm internal diameter, with a 5-μm pore size). Following injection (50 μl), samples were recorded and analyzed by a Kromasystem integrator (Bio-Tek, St. Quentin en Yvelines, France). The mobile phase consisted of aqueous citric acid solution (0.1 M) containing 20% acetonitrile (vol/vol) and 10 mM tetrabutylammonium perchlorate. The flow rate was 1 ml/min, and under these conditions, the retention time of MXF was about 4 min. The intra- and interday precisions for MXF were satisfactory, with coefficients of variance of between 0.3 and 6.7%. Similarly, the accuracy of the assay was between 94 and 114% at three different concentrations (0.1, 1, and 10 μg/ml). SF concentrations were measured using a Packard Fluorocount fluorescence microplate reader (model AF 1001). The excitation and emission wavelengths were set to 490 nm and 530 nm, respectively.
Data analysis.
Papp values were obtained according to equation 1, as follows:
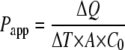 |
(1) |
where ΔQ/ΔT is the amount of drug (Q) appearing in the acceptor compartment as a function of time, obtained from the slope of the linear portion of the amount transported-versus-time plot; A is the surface area of the semipermeable membrane; and C0 is the initial concentration of MXF or SF in the donor compartment. The efflux ratio (ER) was determined by the quotient of the Papp in the secretory (BL to AP) direction over the Papp in the absorptive (AP to BL) direction.
For the concentration dependency study, a variable-slope sigmoidal dose-response equation was used to fit the data for simultaneous passive diffusion and active transport, as follows:
 |
(2) |
where Papp is the observed apparent permeability, Papp Min and Papp Max are the minimal and maximal permeability values, respectively, Km is the drug concentration associated with the response halfway between the minimal and maximal permeability values, C0 is the initial MXF concentration in the donor compartment, and λ is the Hill factor. The first and second terms of the equation represent the passive and active permeabilities, respectively. Model parameter values as well as corresponding confidence intervals were determined using GraphPad Prism, version 4.00, for Windows (GraphPad Software Inc., San Diego, CA).
Western blotting.
Cells treated or not with 10 or 25 μM rifampin for 15 days were detached after a 30-min incubation in PBS at 37°C. Ten million cells for each condition were collected and lysed with lysis buffer (10 mM Tris, pH 7.4, 1% NP-40, 0.5% deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 20 μM leupeptin, 0.8 μM aprotinin, 10 μM pepstatin) for 20 min on ice. Lysates were clarified by centrifugation at 15,000 × g for 5 min at 4°C, and the supernatants were kept at −80°C until use. Samples (30 μg) were resuspended in Laemmli sample buffer and fractionated in a 7% gradient sodium dodecyl sulfate-polyacrylamide gel, followed by Western blotting. For Western blots, proteins were transferred from gels to nitrocellulose membranes (0.20-μm pore size; Sartorius, Göttingen, Germany), using a Miniprotean electroblotter (Bio-Rad Laboratories, Hercules, CA). Immunoblots were washed three times with Tris-buffered saline with 0.1% Tween 20 (TBS-Tween) and incubated overnight at 4°C with 25 μg of anti-P-gp (mouse monoclonal C219; Abcam, Inc., Cambridge, MA) or anti-actin (rabbit polyclonal; Sigma) in TBS-Tween. Following three washes with TBS-Tween, membranes were incubated for 1 h at 4°C with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G and horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (GE Healthcare-Amersham, Chalfont St. Giles, United Kingdom) in TBS-Tween with 5% nonfat dried milk. The membranes were washed three times for 5 min per wash with TBS-Tween, and bound antibodies were detected using enhanced luminol and oxiding reagent as specified by the manufacturer (ECL+ kit; GE Healthcare-Amersham). Band intensities were analyzed with Scion Image for Windows (Scion Corporation, Frederick, MD).
Statistical analysis.
All values are presented as means ± standard errors of the means (SEM). The statistical evaluation of the data was performed with one-way or two-way analysis of variance (ANOVA) followed by Bonferroni's post hoc tests, using GraphPad Prism, version 4.00, for Windows. P values of <0.05 were considered statistically significant.
RESULTS
Concentration effect.
Permeability in the secretory direction decreased as the BL concentrations of MXF increased (Fig. 1). This result suggests an active mediated transport of MXF through the Calu-3 cell monolayer. Accordingly, a model with a variable-slope sigmoidal dose response for simultaneous passive diffusion and active transport successfully described the data, with Papp Max and Papp Min estimated to be 13.6 × 10−6 ± 0.3 × 10−6 cm·s−1 and 7.9 × 10−6 ± 0.4 × 10−6 cm·s−1, respectively. The apparent Km was 64.6 μg/ml (i.e., 147.5 μM), and the Hill factor was −2.11. The 95% confidence intervals for the apparent Km and Hill factor were 49.9 to 83.7 μg/ml and −3.7 to −1.1, respectively, and those for Papp Min and Papp Max were 7 × 10−6 to 8.82 × 10−6 cm·s−1 and 12.96 × 10−6 to 14.24 × 10−6 cm·s−1, respectively. A concentration below the apparent Km (50 μM MXF, i.e., 21.9 μg/ml MXF-HCl) was chosen for the next transport experiments.
FIG. 1.

Concentration dependence of Papp in the secretory direction (BL-AP) of MXF-HCl across Calu-3 monolayers. The graph shows the total Papp, reflecting active transport and passive diffusion. Data are expressed as means ± SEM (n = 3). A sigmoidal dose-response equation was used to fit the data (R2 = 0.92).
Inhibition studies.
The amount of MXF transported over time and the permeability of the Calu-3 monolayer in the absence and presence of efflux pump inhibitors are presented in Fig. 2 and Table 1, respectively. In all cases, the amount of MXF transported over time increased linearly with time for up to 180 min. The amount of MXF transported after 180 min across the Calu-3 monolayers was twice as high in the secretory direction as in the absorptive direction (2.49 ± 0.07 μg·cm−2 versus 1.22 ± 0.10 μg·cm−2) in the absence of efflux pump inhibitors (Fig. 2). This polarized transport was abolished in the presence of verapamil (100 μM) and PSC-833 (3 μM), whereas no effect was observed in the presence of probenecid (100 μM). The permeability of MXF in the absence of inhibitors (control) was also twice as high in the secretory direction as in the absorptive direction (Table 1). The ER was equal to 2, confirming the presence of active transport for MXF. The presence of verapamil or PSC-833 increased, by 53% to 69%, and reduced, by 30%, the absorptive and secretory permeabilities, respectively. The ERs for both inhibitors were close to 1 (Table 1). No difference in MXF transport was observed in the presence or absence of probenecid.
FIG. 2.

Amounts of MXF transported across Calu-3 monolayers over time in the absence (control) and presence of verapamil (100 μM), PSC-833 (3 μM), or probenecid (100 μM). The concentration of MXF in the donor compartment was 50 μM (21.9 μg/ml MXF). Data are expressed as means ± SEM (n = 4 to 6). Two-way ANOVA and Bonferroni's post hoc test were used to compare BL-to-AP transport against AP-to-BL transport. *, P < 0.001.
TABLE 1.
Papp of MXF (50 μM) in the presence of different inhibitors or inducers of transport proteins in the AP-to-BL and BL-to-AP directions, together with the ER across Calu-3 monolayers
| Inhibitor or inducer | Affected transporter |
Papp (10−6 cm·s−1)a
|
ER | |
|---|---|---|---|---|
| AP-to-BL direction | BL-to-AP direction | |||
| Control | 5.25 ± 0.18 | 10.53 ± 0.10 | 2.0 | |
| Verapamil | P-gp, MRP | 8.05 ± 0.27*** | 8.07 ± 0.05** | 1.0 |
| PSC-833 | P-gp, not MRP | 8.87 ± 0.06*** | 7.74 ± 0.02** | 0.9 |
| Probenecid | MRP, not P-gp | 4.42 ± 0.03 (NS) | 9.58 ± 0.37 (NS) | 2.2 |
| Rifampinb | ||||
| 10 μM | P-gp, MRP | 4.35 ± 0.33* | 11.86 ± 0.13** | 2.7 |
| 25 μM | P-gp, MRP | 4.08 ± 0.30** | 12.48 ± 0.18** | 3.1 |
Two-way ANOVA and Bonferroni's post hoc test were used to compare data against the control data. ***, P < 0.001; **, P < 0.01; *, P < 0.05; NS, P > 0.05.
Cells were pretreated for 15 days.
Influence of rifampin on P-gp expression and MXF transport.
Upregulation of P-gp was obtained with 10 and 25 μM rifampin treatment. The 25 μM concentration was chosen as the highest concentration that did not affect cell growth (15). The results in Fig. 3 show a unique, 170-kDa band revealed by the C219 anti-P-gp antibody. The presence of 10 and 25 μM rifampin increased the C219 band's relative intensity by 30% and 92%, respectively. In parallel, monolayers were tested for MXF transport (Table 1). In the absorptive direction, 10 and 25 μM rifampin treatment decreased the permeability by 17% and 22%, respectively. In the secretory direction, increases were observed with 10 and 25 μM of rifampin (12% and 18%, respectively). Rifampin treatment increased the polarized transport of MXF, as indicated by the ERs (Table 1).
FIG. 3.

Rifampin upregulates expression of P-gp in Calu-3 cells. Cells were exposed to rifampin for 15 days at 10 and 25 μM concentrations. P-gp expression was analyzed by immunoblotting with specific antibodies to MDR1 and actin. (A) Representative immunoblot. (B) Intensity of C219 band relative to actin band intensity and normalized to control (0 μM). Data are expressed as means ± SEM (n = 4 or 5). One-way ANOVA and Bonferroni's post hoc test were used to compare the data against the control. *, P < 0.05; **, P < 0.01.
DISCUSSION
The current results clearly indicate that MXF is in part actively secreted through Calu-3 lung epithelial cells. Indeed, the Papp values for the secretory and absorptive directions are dissymmetric, and the secretory permeability is concentration dependent. The model used to describe the data for MXF transport through the cell monolayer establishes that the Papp is the addition of passive (Papp Min) and active permeabilities. As a consequence, at the highest concentrations (over 350 μg/ml), active transport is saturated and the Papp is due essentially to passive permeability (Papp Min = 7.9 × 10−6 cm·s−1). This relatively high passive permeability is consistent with the MXF apparent log P of −0.28, which is characteristic of a lipophilic compound (13). On the other hand, the Papp Max obtained for the lowest concentrations (below 10 μg/ml) was 13.6 × 10−6 cm·s−1, representing the active plus passive transport of MXF. When the Papp Min is subtracted, the resulting value reflects the maximum active transport (Papp Max − Papp Min = 5.7 × 10−6 cm·s−1), which then accounts for 42% of the maximum permeability, at most. This can be compared with the situation in vivo. Following a single dose of 400 mg in healthy volunteers, the maximum concentration of MXF in plasma (Cmax) was close to 5 μg/ml (31). In the clinical range of concentrations, the active secretion of MXF in the lung is expected to be fully effective, since the Cmax is about 10 times lower than the apparent Km (65 μg/ml). This P-gp-dependent secretion should lead to higher MXF epithelial lining fluid concentrations than those in serum and could therefore possibly explain the severalfold higher MXF concentrations in bronchoalveolar lavage fluid than in serum observed for patients 24 h after a single 400-mg dose (28). However, extrapolation of these in vitro data to the in vivo situation should be done with caution. Calu-3 cells are derived from a tumor cell line and may express a higher level of P-gp than pneumocytes I and II of the alveolar epithelium. Few studies have been performed to compare the performances of the different in vitro models for pulmonary drug disposition in terms of P-gp-dependent transport. For instance, primary ATI cells or ATI-like human alveolar epithelial cells have been shown to express P-gp (8), and rhodamine 123, used as a P-gp substrate, was dissymmetrically transported through human alveolar epithelial cells, with an ER of 3.1, whereas this ratio was reported to be 11 with the Calu-3 cell model (12). This difference in ERs might be explained by an overexpression of P-gp in the tumor cell line Calu-3 leading to an overestimation of the P-gp-dependent transport in the lung (9). In line with this, the apparent Km for MXF permeability of Calu-3 monolayers was lower than that obtained in studying the uptake of MXF in oral carcinoma epithelial cells (240 μg/ml) (6). The discrepancy between these results could be explained by the differences in study design and levels of P-gp expression in different cell lines. One should note that the apparent Km reflects the cell monolayer transport kinetics of MXF in Calu-3 cells and does not reflect the Michaelis P-gp-MXF Km constant, since diffusion rates of MXF through the lipid membrane and cytosol are not taken into account and vary with cell type (4). However, the apparent Km is a useful value to characterize active mediated transport across a cell monolayer. A concentration of MXF equal to 50 μM (i.e., 21.9 μg/ml) was used for the inhibition studies. This concentration is three times lower than the apparent Km for membrane transport, so at this concentration 80% of the maximum secretory permeability should be reached, allowing characterization of this active transport. P-gp and MRP1 have been identified in healthy human lung cells and Calu-3 cells (11, 12, 14), and effective inhibitors were used in this study to identify the MXF transporters. The results indicate for the first time that P-gp is mainly involved in the active transport of MXF in this model. The PSC-833 inhibitor, which is highly effective and relatively more specific for P-gp, affected permeability in both the absorptive and secretory directions. In the presence of 3 μM of PSC-833, both permeabilities were identical and equivalent to the passive permeability, or Papp Min. The corresponding ER of MXF was close to 1, indicating that P-gp was the main efflux transporter involved in the transport of MXF in the Calu-3 model. This was also supported by the results obtained with verapamil and probenecid. Although verapamil inhibits P-gp (18) and MRPs (5), MRP-mediated transport can be excluded since probenecid, a highly effective inhibitor of MRPs (26), did not significantly affect the transport of MXF. Statistical analysis of MXF permeability values in the presence of PSC-833 showed no difference in the secretory and absorptive directions. However, the slight difference observed in the secretory and absorptive directions in Fig. 2 and Table 1 could have been abolished by a higher concentration of PSC-833, although higher concentrations are not recommended since they could induce cell toxicity. Since no statistical differences between the two permeabilities were found, further experiments were not conducted to test this hypothesis. P-gp has been found predominantly at the luminal membrane in Calu-3 cells (12) as well as in human epithelial cells in vivo (8). According to the results of this study and considering that P-gp works as a membrane efflux pump, MXF transport through the cell is assumed to be mostly passive at the BL side and in part active at the luminal side. MXF affinities for efflux transporters may be different for different cell types. For instance, MRP2 has been reported to mediate the biliary elimination of MXF and its metabolites (1). However, in a study with J774 macrophages, MXF uptake was shown to be slightly affected by a ciprofloxacin transporter and not by MRPs (19). As a P-gp substrate, MXF may interact with another P-gp substrate or modulator drugs in terms of tissue distribution. In cases of rifampin-resistant tuberculosis, MXF is often used as a second-line therapy after rifampin treatment, and a pharmacokinetic interaction between the two drugs, leading to lower MXF serum concentrations, has already been observed in patients (30). Since rifampin is a known inducer of P-gp, rifampin-MXF interaction may also lead to a modification of MXF lung disposition. An increase in P-gp expression following rifampin treatment has been described for LS180 human colonic cells (7) and LCC-PK1 cells (15). The current Western blot results also showed an increase in the expression of P-gp after rifampin treatment in Calu-3 cells. When tested for affects on permeability, rifampin treatment induced a dose-dependent effect on active transport of MXF: the secretory permeability was increased, and the absorptive permeability was decreased. It can be concluded that rifampin treatment increases P-gp expression and P-gp-dependent MXF transport in Calu-3 cells. Although the rifampin concentration may not be high enough to produce this interaction in the clinical situation, these results show the in vitro mechanistic effect of rifampin-MXF interaction on MXF transport, and in the case of a combination of rifampin and MXF treatments, the possibility exists that a drug-drug interaction could occur at the lung level and affect the secretion of MXF into the lung. These effects on pulmonary distribution have not been investigated to date, and the current results call for further in vivo experiments.
In conclusion, this study clearly indicates that MXF is subject to active transport in the Calu-3 model and that this transport is mainly P-gp dependent. Moreover, a drug-drug interaction could occur at the lung level and affect the MXF distribution when it is combined with other P-gp substrates or modulators, such as rifampin. Further studies will be conducted to compare the MXF permeability with that of other members of the FQ family and to characterize their passive and active transport through lung epithelial cells.
Footnotes
Published ahead of print on 2 February 2009.
REFERENCES
- 1.Ahmed, S., N. T. Vo, T. Thalhammer, F. Thalhammer, K. B. Gattringer, and W. Jager. 2008. Involvement of Mrp2 (Abcc2) in biliary excretion of moxifloxacin and its metabolites in the isolated perfused rat liver. J. Pharm. Pharmacol. 60:55-62. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez, A. I., M. Perez, J. G. Prieto, A. J. Molina, R. Real, and G. Merino. 2008. Fluoroquinolone efflux mediated by ABC transporters. J. Pharm. Sci. 97:3483-3493. [DOI] [PubMed] [Google Scholar]
- 3.Ballow, C., J. Lettieri, V. Agarwal, P. Liu, H. Stass, and J. T. Sullivan. 1999. Absolute bioavailability of moxifloxacin. Clin. Ther. 21:513-522. [DOI] [PubMed] [Google Scholar]
- 4.Bentz, J., T. T. Tran, J. W. Polli, A. Ayrton, and H. Ellens. 2005. The steady-state Michaelis-Menten analysis of P-glycoprotein mediated transport through a confluent cell monolayer cannot predict the correct Michaelis constant Km. Pharm. Res. 22:1667-1677. [DOI] [PubMed] [Google Scholar]
- 5.Binaschi, M., R. Supino, R. A. Gambetta, G. Giaccone, E. Prosperi, G. Capranico, I. Cataldo, and F. Zunino. 1995. MRP gene overexpression in a human doxorubicin-resistant SCLC cell line: alterations in cellular pharmacokinetics and in pattern of cross-resistance. Int. J. Cancer 62:84-89. [DOI] [PubMed] [Google Scholar]
- 6.Brayton, J. J., Q. Yang, R. J. Nakkula, and J. D. Walters. 2002. An in vitro model of ciprofloxacin and minocycline transport by oral epithelial cells. J. Periodontol. 73:1267-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collett, A., J. Tanianis-Hughes, and G. Warhurst. 2004. Rapid induction of P-glycoprotein expression by high permeability compounds in colonic cells in vitro: a possible source of transporter mediated drug interactions? Biochem. Pharmacol. 68:783-790. [DOI] [PubMed] [Google Scholar]
- 8.Endter, S., U. Becker, N. Daum, H. Huwer, C. M. Lehr, M. Gumbleton, and C. Ehrhardt. 2007. P-glycoprotein (MDR1) functional activity in human alveolar epithelial cell monolayers. Cell Tissue Res. 328:77-84. [DOI] [PubMed] [Google Scholar]
- 9.Forbes, B., and C. Ehrhardt. 2005. Human respiratory epithelial cell culture for drug delivery applications. Eur. J. Pharm. Biopharm. 60:193-205. [DOI] [PubMed] [Google Scholar]
- 10.Geys, J., L. Coenegrachts, J. Vercammen, Y. Engelborghs, A. Nemmar, B. Nemery, and P. H. Hoet. 2006. In vitro study of the pulmonary translocation of nanoparticles: a preliminary study. Toxicol. Lett. 160:218-226. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton, K. O., G. Backstrom, M. A. Yazdanian, and K. L. Audus. 2001. P-glycoprotein efflux pump expression and activity in Calu-3 cells. J. Pharm. Sci. 90:647-658. [DOI] [PubMed] [Google Scholar]
- 12.Hamilton, K. O., E. Topp, I. Makagiansar, T. Siahaan, M. Yazdanian, and K. L. Audus. 2001. Multidrug resistance-associated protein-1 functional activity in Calu-3 cells. J. Pharmacol. Exp. Ther. 298:1199-1205. [PubMed] [Google Scholar]
- 13.Langlois, M.-H., M. Montagut, J.-P. Dubost, J. Grellet, and M.-C. Saux. 2005. Protonation equilibrium and lipophilicity of moxifloxacin. J. Pharm. Biomed. Anal. 37:389-393. [DOI] [PubMed] [Google Scholar]
- 14.Lehmann, T., C. Kohler, E. Weidauer, C. Taege, and H. Foth. 2001. Expression of MRP1 and related transporters in human lung cells in culture. Toxicology 167:59-72. [DOI] [PubMed] [Google Scholar]
- 15.Magnarin, M., M. Morelli, A. Rosati, F. Bartoli, L. Candussio, T. Giraldi, and G. Decorti. 2004. Induction of proteins involved in multidrug resistance (P-glycoprotein, MRP1, MRP2, LRP) and of CYP 3A4 by rifampicin in LLC-PK1 cells. Eur. J. Pharmacol. 483:19-28. [DOI] [PubMed] [Google Scholar]
- 16.Marchand, S., M. Chenel, I. Lamarche, C. Pariat, and W. Couet. 2003. Dose ranging pharmacokinetics and brain distribution of norfloxacin using microdialysis in rats. J. Pharm. Sci. 92:2458-2465. [DOI] [PubMed] [Google Scholar]
- 17.Marchand, S., D. Frasca, C. Dahyot-Fizelier, C. Breheret, O. Mimoz, and W. Couet. 2008. Lung microdialysis study of levofloxacin in rats following intravenous infusion at steady state. Antimicrob. Agents Chemother. 52:3074-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marier, J. F., J. L. Deschenes, A. Hage, E. Seliniotakis, A. Gritsas, T. Flarakos, F. Beaudry, and P. Vachon. 2005. Enhancing the uptake of dextromethorphan in the CNS of rats by concomitant administration of the P-gp inhibitor verapamil. Life Sci. 77:2911-2926. [DOI] [PubMed] [Google Scholar]
- 19.Michot, J. M., C. Seral, F. Van Bambeke, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2005. Influence of efflux transporters on the accumulation and efflux of four quinolones (ciprofloxacin, levofloxacin, garenoxacin, and moxifloxacin) in J774 macrophages. Antimicrob. Agents Chemother. 49:2429-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mills, J. B., K. A. Rose, N. Sadagopan, J. Sahi, and S. M. de Morais. 2004. Induction of drug metabolism enzymes and MDR1 using a novel human hepatocyte cell line. J. Pharmacol. Exp. Ther. 309:303-309. [DOI] [PubMed] [Google Scholar]
- 21.Miravitlles, M., and A. Anzueto. 2008. Moxifloxacin: a respiratory fluoroquinolone. Expert Opin. Pharmacother. 9:1755-1772. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee, J. S., M. L. Rich, A. R. Socci, J. K. Joseph, F. A. Viru, S. S. Shin, J. J. Furin, M. C. Becerra, D. J. Barry, J. Y. Kim, J. Bayona, P. Farmer, M. C. Smith Fawzi, and K. J. Seung. 2004. Programmes and principles in treatment of multidrug-resistant tuberculosis. Lancet 363:474-481. [DOI] [PubMed] [Google Scholar]
- 23.Nijland, H. M., R. Ruslami, A. J. Suroto, D. M. Burger, B. Alisjahbana, R. van Crevel, and R. E. Aarnoutse. 2007. Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin. Infect. Dis. 45:1001-1007. [DOI] [PubMed] [Google Scholar]
- 24.Oliphant, C. M., and G. M. Green. 2002. Quinolones: a comprehensive review. Am. Fam. Physician 65:455-464. [PubMed] [Google Scholar]
- 25.Schentag, J. J. 2000. Clinical pharmacology of the fluoroquinolones: studies in human dynamic/kinetic models. Clin. Infect. Dis. 31(Suppl. 2):S40-S44. [DOI] [PubMed] [Google Scholar]
- 26.Schinkel, A. H., and J. W. Jonker. 2003. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 55:3-29. [DOI] [PubMed] [Google Scholar]
- 27.Shen, B. Q., W. E. Finkbeiner, J. J. Wine, R. J. Mrsny, and J. H. Widdicombe. 1994. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl− secretion. Am. J. Physiol. 266:L493-L501. [DOI] [PubMed] [Google Scholar]
- 28.Soman, A., D. Honeybourne, J. Andrews, G. Jevons, and R. Wise. 1999. Concentrations of moxifloxacin in serum and pulmonary compartments following a single 400 mg oral dose in patients undergoing fibre-optic bronchoscopy. J. Antimicrob. Chemother. 44:835-838. [DOI] [PubMed] [Google Scholar]
- 29.Van Bambeke, F., J. M. Michot, J. Van Eldere, and P. M. Tulkens. 2005. Quinolones in 2005: an update. Clin. Microbiol. Infect. 11:256-280. [DOI] [PubMed] [Google Scholar]
- 30.Weiner, M., W. Burman, C. C. Luo, C. A. Peloquin, M. Engle, S. Goldberg, V. Agarwal, and A. Vernon. 2007. Effects of rifampin and multidrug resistance gene polymorphism on concentrations of moxifloxacin. Antimicrob. Agents Chemother. 51:2861-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wise, R., J. M. Andrews, G. Marshall, and G. Hartman. 1999. Pharmacokinetics and inflammatory-fluid penetration of moxifloxacin following oral or intravenous administration. Antimicrob. Agents Chemother. 43:1508-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]