

Abstract
The Chagas’ disease parasite Trypanosoma cruzi commonly infects humans through skin abrasions or mucosa from reduviid bug excreta. Yet most studies on animal models start with subcutaneous or intraperitoneal injections, a distant approximation of the skin abrasion route. We show here that atraumatic placement of T. cruzi in the mouse nasal cavity produced low parasitemia, high survival rates, and preferential brain invasion compared to the case with subcutaneously injected parasites. Brain invasion was particularly prominent in the basal ganglia, peaked at a time when parasitemia was no longer detectable, and elicited a relatively large number of inflammatory foci. Yet, based on motor behavioral parameters and staining with Fluoro-Jade C, a dye that specifically recognizes apoptotic and necrotic neurons, brain invasion did not cause neurodegenerative events, in contrast to the neurodegeneration in the enteric nervous system. The results indicate that placement of T. cruzi on the mucosa in the mouse nasal cavity establishes a systemic infection with a robust yet harmless infection of the brain, seemingly analogous to disease progression in humans. The model may facilitate studies designed to understand mechanisms underlying T. cruzi infection of the central nervous system.
The protozoan parasite Trypanosoma cruzi causes Chagas' disease, a debilitating condition that afflicts millions of people in the Americas. Patients with chronic symptomatic disease typically present abnormalities of the gastrointestinal tract (megaesophagus and megacolon) and/or heart (cardiomegaly). Such megasyndromes are caused, in part, by damage of the peripheral nervous system, particularly the myenteric (Auerbach's) plexus, submucosal (Meissner's) plexus, and nerve fibers, which may be largely destroyed (1, 23, 30).
Prior to overt megasyndrome, patients exhibit normal electrocardiogram readings and digestive processes, despite parasitological and/or serological evidence of continued T. cruzi infection. This indeterminate phase of the disease may persist for decades while presenting minor peripheral neuropathy (sensory impairment and diminished tendon jerks) in a relatively small (∼10%) proportion of patients (17). Patients may show signs of neuroregeneration and/or neuroprotection, such as an age-dependent relative increase in the number of ganglion cells in the heart and enteric nervous system (23). Neuroregeneration in the enteric nervous system may occur even in chagasic megacolon (13).
Chronic indeterminate and symptomatic Chagas' disease is preceded by the acute phase of the disease, which commonly starts when T. cruzi gains access to the body through skin abrasions or undamaged mucosa, usually in the face, from the contaminated semiliquid excrement of hematophagous reduviid insects. Entry through the conjunctival mucosa is readily diagnosed by the swelling of the eyelids (Romaña's sign) (38). Another logical mucosal port of entry is the nasal cavity, which is directly accessible to moving trypanosomes deposited nearby within insect excreta. Notably, Triatoma infestans, a reduviid insect that frequently transmits Chagas' disease, is attracted to the human face by the carbon dioxide exhaled during respiration (44, 45), a behavior likely favoring T. cruzi intranasal transmission.
Most studies with animal models of Chagas' disease focus on the interaction of the parasite with the heart, gastrointestinal tract, and other organs. One notable exception is the central nervous system (CNS), even though T. cruzi infects the CNS in most patients with acute Chagas' disease (20). Paradoxically, T. cruzi infection of the CNS is symptomatically and pathologically silent in immunocompetent individuals. Infection of the brain by most other pathogenic microbes carries severe and enduring detrimental effects (40), including brain infection by the T. cruzi counterpart in Africa, Trypanosoma brucei, which causes sleeping sickness and significant brain abnormalities in humans and cattle (22). Curiously, while cardiac problems are the main cause of morbidity in chronic Chagas' disease, such problems are relatively benign in sleeping sickness, where conversely, CNS-dependent neurological problems dominate (5).
In the relatively few instances where T. cruzi invasion of the CNS was examined in animal models, parasites were inoculated into experimental suckling animals by the intraperitoneal route (12), or the brain infection was studied in the context of immunological responses, such as determining the prevailing inflammatory cell type (36), chemokine-dependent lymphocyte homing (37), or proinflammatory cytokine-dependent invasion (31).
As a first step toward understanding the molecular basis of T. cruzi interaction with cells in the CNS, we sought to develop an experimental mouse model that gives consistent invasion of the brain in immunocompetent adult animals. We found that atraumatic placement of T. cruzi in the nasal cavities of susceptible and resistant mice produced a systemic infection with preferential invasion of the brain, as assessed by quantitative PCR, parasitemia, histology, and immunohistochemistry. Intranasally inoculated susceptible animals survived acute infection which was otherwise lethal if the parasites were injected subcutaneously. Furthermore, brain invasion did not trigger detectable neurodegeneration.
MATERIALS AND METHODS
Parasites.
Trypanosoma cruzi (Tulahuén strain clone C2) was maintained in Vero cells at 37°C in 5% CO2 with 90% relative humidity in Dulbecco's minimal essential medium supplemented with 2.5% (vol/vol) fetal bovine serum, as previously described (33). Invasive and highly virulent T. cruzi trypomastigotes were washed serially with serum-free Dulbecco's minimal essential medium and sterile phosphate-buffered saline (PBS), counted in a hemacytometer, and resuspended in the appropriate volume of sterile PBS immediately prior to infection.
Infection.
Six- to 8-week-old female C57BL/6 and BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were anesthetized with tribromoethanol (Avertin) solution and infected subcutaneously in the hind-limb footpad (30 μl) or intranasally (2 μl/naris every 2 min for 15 min, for a 20-μl total volume equivalent to 0.5 × 103, 5 × 103, or 25 × 103 parasites/mouse).
To induce basal ganglion neurodegeneration, C57BL/6 mice (10 weeks of age) were injected intraperitoneally with increasing doses of 3-nitropropionic acid (3-NP) over 6 days (Sigma-Aldrich, St. Louis, MO) (20 mg/kg body weight every 12 h for 48 h, then 40 mg/kg body weight every 12 h for 48 h, and finally, 60 mg/kg body weight every 12 h for 48 h) (16). Semiquantitative behavioral assessment of motor disorders related to brain degeneration (general locomotor activity, truncal dystonia, and hind-limb dystonia) (16) was performed 1 day after the last injection of the neurotoxin. The same motor disorder assessment was performed at the peak of brain parasitism (25 days postinfection). For histological analysis, animals were sacrificed (on the indicated days) by CO2 asphyxiation and perfused intracardially with sterile PBS, and organs were removed, flushed with PBS, if necessary, and snap-frozen in liquid nitrogen or fixed in formalin solution. To determine parasitemia, tail vein blood was collected at the indicated days in a 1/10 volume of heparin (Sigma-Aldrich, St. Louis, MO), and parasites were counted by light microscopy (8). All procedures conducted were in accordance with the regulations set by the NIH Office of Laboratory Animal Welfare and were approved by the Institutional Animal Care and Use Committee at Tufts University.
Quantitative PCR.
Genomic DNAs were purified from uninfected and T. cruzi-infected brains by use of a DNeasy kit (Qiagen, Valencia, CA). T. cruzi was quantified from brain tissue by real-time PCR (11). A standard curve was generated from DNAs prepared from weighed, uninfected tissues spiked with known amounts of Vero cell-derived parasites to determine ΔCT values; these, in turn, were used to compute approximate numbers of T. cruzi/gram brain.
Histology/immunohistochemistry.
Formalin-fixed samples were embedded in paraffin wax, and sections (5 μm) were stained with hematoxylin and eosin (H&E) or Fluoro-Jade C, a fluorescent histological marker of degenerating neurons (35) (Invitrogen, Carlsbad, CA). Antibody specific for F4/80 antigen (Abcam) was used to immunochemically identify murine cells with macrophage-like properties in brain slices (presumably microglia) prepared with a Ventana 300 automatic immunohistochemical stainer (Ventana Medical Systems).
For quantification of inflammatory foci in T. cruzi-infected tissue, three noncontinuous H&E-stained brain sections were counted per mouse; for each section, 25 fields within the basal ganglia were counted at a magnification of ×200, and the average number of parasite nests per 25 fields was determined for each animal and used to determine the average number of foci for three animals.
Statistical analysis.
All experiments were carried out multiple times with more than three animals per experiment. Statistical analyses were conducted using GraphPad Prism software (version 4.0), and results are reported as means ± standard errors from one representative experiment. For analyses involving two treatments and comparison of multiple treatments, t tests and analysis of variance with post hoc Dunnett's multiple comparison tests were performed, respectively.
RESULTS
Intranasal inoculation of T. cruzi produced low parasitemia and high brain invasion levels.
Subcutaneous inoculation of T. cruzi into the footpad of resistant C57BL/6 mice infects the heart, gastrointestinal tract, and other organs, typically producing nonlethal disease in a broad dose range. To determine if such systemic infection is accompanied by brain invasion, we counted parasites in the tail vein blood (parasitemia) and in the brain by real-time PCR (11) after subcutaneous injection of 5 × 103 parasites into the mouse footpad. The results showed that parasitemia was maximal at 12 days postinoculation (PI) and undetectable a week thereafter (Fig. 1A). Brain parasitism was not detectable at the height of parasitemia, but it became measurable at the end of parasitemia, peaked at 25 days PI, and declined sharply thereafter (Fig. 1A).
FIG. 1.

Mice intranasally inoculated with T. cruzi develop more brain parasitism and less parasitemia than subcutaneously (s.c.) infected animals. C57BL/6 mice were inoculated subcutaneously (A and D) or intranasally (B and E) with 5 × 103 (A and B) or 25 × 103 (D and E) T. cruzi trypomastigotes. At the indicated days PI, mice were bled and sacrificed for measurements of parasitemia (black lines) and brain tissue parasitism (gray lines). (C and F) Bar graphs comparing brain and blood parasite loads, defined by the areas corresponding to black and gray curves, respectively, between subcutaneously (normalized to 1.0) and intranasally inoculated animals. Note that brain invasion increased 2.0- and 2.4-fold in intranasally infected mice at the two inoculating doses, respectively, in contrast to parasitemia, which decreased by one-third in both cases. **, P < 0.01. Experiments were repeated more than three times, using two to seven mice/time point, with similar results.
Intranasally inoculated T. cruzi produced parasitemia and brain invasion analogous to those by subcutaneously injected parasites, except for the reversal in the extent of parasite load (Fig. 1B). Thus, parasitemia in intranasally infected mice was about one-third of that after subcutaneous injection, in contrast to the brain load, which increased about 2.0-fold (Fig. 1C). A similar progression and extent of parasite load were obtained when C57BL/6 mice were subcutaneously and intranasally infected with a five times greater parasite inoculum (25 × 103 parasites/mouse) (Fig. 1D to F).
Susceptible BALB/c mice intranasally inoculated with T. cruzi survived infection that would otherwise be lethal if they were injected subcutaneously.
Given that intranasally inoculated resistant C57BL/6 mice produced low parasitemia and given that resistance and susceptibility to T. cruzi infection are defined on the basis of parasitemia and survival produced by subcutaneous, intraperitoneal, or intravenous inoculation (21, 25, 41), we checked for the possibility of a paradigmatically susceptible mouse strain becoming relatively resistant if infected by the intranasal route. Intranasal administration of 500 T. cruzi trypomastigotes into susceptible BALB/c mice produced parasitemia following the kinetics and parasite load of resistant C57BL/6 mice (Fig. 2A). However, the intranasal inoculum, lethal if administered subcutaneously, did not kill infected BALB/c mice (Fig. 2B). A comparison of brain invasion in C57BL/6 and BALB/c mice was performed at the peak of infection, i.e., day 25 (Fig. 2C). All BALB/c mice had succumbed by 3 weeks after subcutaneous inoculation. Appropriately, C57BL/6 mice inoculated with 500 parasites exhibited less parasitism than those inoculated with 5,000 parasites, and BALB/c parasitism was similar to that found using the highest inoculum (25,000 parasites) in C57BL/6 mice. This finding could reflect a dampened immune response to T. cruzi infection noted previously in BALB/c mice. Regardless, intranasal doses provided a twofold increase compared to subcutaneous administration in both C57BL/6 and BALB/c mice at day 25 PI.
FIG. 2.

Susceptible BALB/c mice survive T. cruzi infection produced by an intranasal inoculum that would otherwise be lethal if they are injected subcutaneously. Parasitemia (A), survival (B), and brain parasite loads (C) of BALB/c mice inoculated with 0.5 × 103 T. cruzi trypomastigotes intranasally (i.n.) or subcutaneously (s.c.). Brain parasite loads of resistant C57BL/6 mice 25 days after subcutaneous or intranasal inoculation with the same number of parasites are shown in panel C for comparison with BALB/c mice. *, P < 0.05. Experiments were repeated three times, using two to four mice/time point.
T. cruzi targeted the basal ganglia whether the infection was initiated by subcutaneous or intranasal inoculation and triggered a strong inflammatory response.
To determine whether T. cruzi invades the brain randomly or targets a specific region, we determined parasite loads by real-time PCR in the cortex, basal ganglia, and cerebellum of C57BL/6 mice sacrificed at the peak of brain invasion (25 days PI). We found that T. cruzi invaded the basal ganglia preferentially, regardless of whether the parasites were inoculated by the subcutaneous or intranasal route. Furthermore, the parasite load in the basal ganglia following intranasal infection was greater (∼3.0-fold) than the basal ganglia parasitism resulting from subcutaneous inoculation (Fig. 3). Regardless of the inoculation route, T. cruzi invasion of the basal ganglia was more prominent than that of the brain cortex, which in turn was greater than that in the cerebellum (Fig. 3). Due to the nature of the assay to quantitate parasite load (real-time PCR), it was not practical to determine if T. cruzi targeted individual components of the basal ganglia, namely, the striatum (putamen and caudate nucleus), globus pallidus, subthalamic nucleus, and substantia nigra.
FIG. 3.
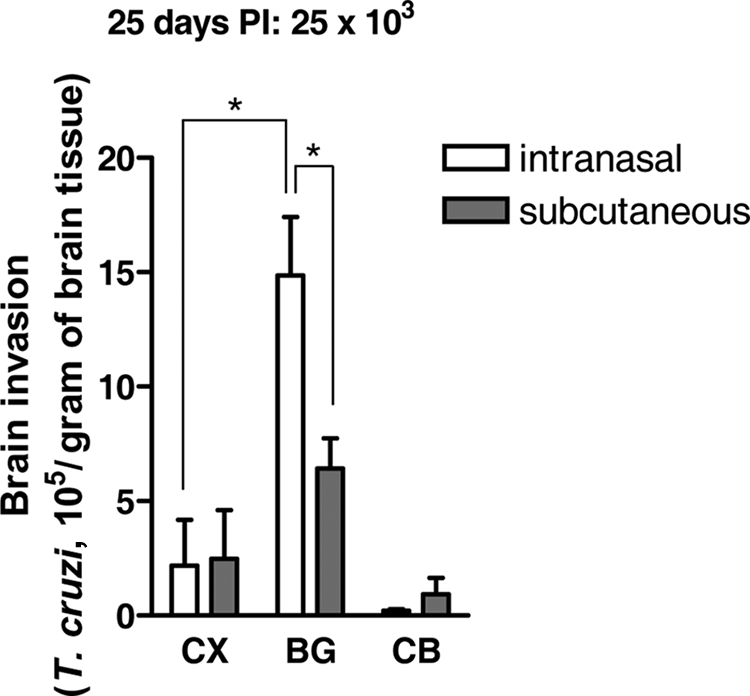
Location and abundance of parasites in the brain following intranasal and subcutaneous inoculation. C57BL/6 mice were infected intranasally and subcutaneously with 25 × 103 parasites, sacrificed at day 25 (peak brain parasitism), and assayed for T. cruzi by quantitative PCR in the brain cortex (CX), basal ganglia (BG), and cerebellum (CB). The experiment was repeated four times, using two or three mice per region tested. *, P < 0.001.
Histological and histochemical analysis revealed T. cruzi nests in the cortex and basal ganglia (Fig. 4A and C) and T. cruzi inside F4/80-positive cells (Fig. 4C, inset). Quantitation of these nests or foci in the basal ganglia of C57BL/6 mice sacrificed 25 days after intranasal and subcutaneous inoculation with 25,000 parasites showed a preferential increase (about 9.0-fold) in the brains of mice infected by the intranasal route (Fig. 5). The strong inflammatory response to intranasal inoculation compared to that in uninfected tissue (Fig. 4B) reflects the preferential parasite load produced by this procedure (Fig. 1). Interestingly, abundance, location, and cellular constituents of foci in histological sections of brains from BALB/c mice inoculated intranasally with 500 parasites were virtually identical to those for mice infected with 25,000 parasites (data not shown).
FIG. 4.

Histology of the frontal cortex and basal ganglia of T. cruzi-infected C57BL/6 mouse. H&E stains of sagittal sections of the brain from a C57BL/6 mouse 18 days after intranasal inoculation are shown. (A) Three cellular foci in the frontal cortex (CX). Original magnification, ×200. Enhanced magnification (×400) of the indicated areas reveals microglia (open arrowhead) surrounding infected cells (closed arrow). (B) Uninfected basal ganglia. Magnification, ×200. (C) One cellular focus in the striatal tissue of the basal ganglia (BG) showing a microglial nodule (open arrowhead) surrounding infected cells (closed arrow). Original magnification, ×400. Additionally, infected F4/80-positive macrophage-like cells (presumably microglia) were found within nodules (inset). Bar, 50 μm.
FIG. 5.
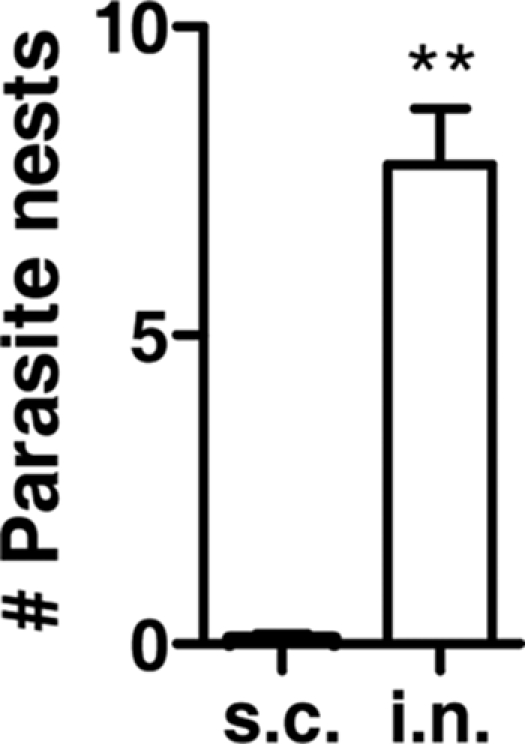
Increased numbers of inflammatory foci in the basal ganglia of intranasally infected compared to subcutaneously infected mice. C57BL/6 mice were infected intranasally (i.n.) with 25 × 103 or subcutaneously (s.c.) with 5 × 103 T. cruzi parasites, and at 25 days PI, their brains were fixed in formalin, embedded in paraffin, sectioned both coronally and sagittally, and stained with H&E. Three noncontinuous brain sections (basal ganglia) were counted per mouse. From each section, 25 fields within the basal ganglia (magnification, ×200) were counted. The average number of parasite nests per 25 fields for each mouse was determined. The numbers of T. cruzi nests (foci) were calculated from the averages for three mice. **, P < 0.001.
T. cruzi invasion of the brain did not trigger neurodegeneration.
Alterations in the structure and physiology of the basal ganglia give rise to motor disorders such as parkinsonism and Huntington's disease. One widely used model of basal ganglion neurodegeneration results from systemic intoxication with the fungal toxin 3-NP, which irreversibly inhibits mitochondrial respiration and selectively damages the striatum in most species, including humans, after accidental ingestion of contaminated mildewed sugar cane (4, 6, 29). Motor and behavioral disorders of adult C57BL/6 mice treated with 3-NP (16) served as a positive control for possible basal ganglion dysfunction of mice intranasally inoculated with T. cruzi. The 10 mice intoxicated with 3-NP displayed severe motor slowing, akinesia, hind-limb dystonia, and kyphosis. These disorders were not observed in the 12 mice intranasally infected with T. cruzi at the peak of brain invasion (25 days PI), similar to the behavior of uninfected mice (Table 1). This result suggests that functioning of basal ganglia is normal in T. cruzi-infected mice, despite the high parasite load and inflammatory response (Fig. 4 and 5). The inflammatory response, as demonstrated by increased focal cellularity around parasitized cells, is primarily microglial in origin, as shown previously in rodent and human studies (Fig. 4B) (7, 12).
TABLE 1.
Absence of motor behavioral disorders in mice infected with T. cruzi
| Group | Neurotoxina | No. of animals with disorder/total no. of animals
|
||
|---|---|---|---|---|
| Hind-limb dystoniab | Truncal dystoniac | General locomotor activityd | ||
| Control group | − | 0/12 | 0/12 | 0/12 |
| Infected mice | − | 0/12 | 0/12 | 0/12 |
| Uninfected mice | + | 10/10 | 10/10 | 10/10 |
Animals were treated with the neurotoxin 3-NP (10 mice) or intranasally infected with 25 × 103 T. cruzi organisms (12 mice) (see Materials and Methods). Symptoms related to basal ganglion degeneration were assessed 1 day after the last injection of the neurotoxin or at the peak of brain parasitism (25 days postinfection).
Scores for hind-limb dystonia were as follows: 0, absent; 1, intermittent or increased hind-limb space, abnormal crouching, and poor hind-limb movement and coordination.
Scores for truncal dystonia were as follows: 0, absent; 1, slightly or markedly abnormal flexed posture with visible kyphosis.
Scores for general locomotor activity were as follows: 0, normal; 1, slight or marked (<50%) reduction in general activity: the mouse stayed in place and showed reduced displacement velocity, rearing, or grooming.
The lack of basal ganglion-dependent motor symptoms in T. cruzi-infected mice was consistent with the absence of neurodegeneration in the infected basal ganglia (Fig. 6A), including areas heavily infected with T. cruzi (Fig. 6A, inset). Neurodegeneration was unambiguously verified with the dye Fluoro-Jade C (Fig. 6A), which selectively stains degenerating (apoptotic and/or necrotic) neurons (35). Areas devoid of neurodegeneration, including infected and uninfected basal ganglia as well as uninfected intestine, did not fluoresce with Fluoro-Jade C staining.
FIG. 6.

Absence of neurodegeneration in the brains of mice infected with T. cruzi. C57BL/6 mice were infected intranasally with 25,000 T. cruzi parasites for 25 days. Animals were sacrificed and perfused with PBS, and their brains were fixed with formalin. Slides were stained with H&E to detect parasites (arrows) or with Fluoro-Jade C to visualize degenerating neurons. (A) Basal ganglia. Erythrocytes (arrowheads) reacted with the Fluoro-Jade C dye. (Inset) Enlarged view of infected tissues to indicate infected cells and the absence of detectable neurodegeneration. Bar = 50 μm. (B) Large intestine. Magnification, ×600. Note the extensive neurodegeneration in colon myenteric neurons.
In contrast, Fluoro-Jade C readily identified degenerating neurons in the enteric nervous systems (myenteric plexus) (Fig. 6B) of infected animals, in agreement with findings by others (3, 19).
DISCUSSION
Our results show that nasal colonization with T. cruzi results in robust infection of the CNS that maxes out after the rise and fall of a modest parasitemia. This pattern is similar to but quantitatively the reverse of the parasite load produced by injecting T. cruzi into soft tissues (footpad). Because of the relatively subdued systemic infection following intranasal administration, we could quantitate the rise and fall of brain invasion in susceptible BALB/c mice, which would otherwise be impractical if the parasites were injected subcutaneously, normally a lethal route of infection in this mouse strain. This suggests that it may be possible to study brain invasion over a reasonable time course in immunocompromised animals intranasally infected with T. cruzi, which would be a useful model of T. cruzi reactivation, a phenomenon that occurs in chagasic patients coinfected with human immunodeficiency virus or subjected to immunosuppressant drugs. Reactivation leads to robust parasite growth in the heart and severe myocarditis, while in the CNS tumor-like parasite masses form in the brain, resulting in an inexorably fatal disease progression (14, 24, 27, 34, 42) Because the brain is an area that is impenetrable for most therapeutics, this method could also be useful for targeted delivery of antiparasitic agents, such as in chagasic patients coinfected with human immunodeficiency virus.
Why does intranasally inoculated T. cruzi preferentially invade the brain? The olfactory neuroepithelium is the only site in the body that directly links the environment to the CNS, and thus it could serve as a vehicle for proteins and small particles to reach the CNS, thereby bypassing the blood-brain barrier. Such a possibility was proven experimentally about 70 years ago with the demonstration that intranasally administered Prussian blue migrated to the brains of mice and rabbits via the olfactory route (32). Growth factors and other proteins can, likewise, access the brain via the olfactory and trigeminal nerve pathways independently of the bloodstream (39). Olfactory nerve-dependent, circulation-independent brain entry is not restricted to inert particles or proteins. The microbial pathogen Streptococcus pneumoniae, which typically causes meningitis through the hematogenous route, invades the CNS via olfactory tissues, without detectable bacteremia, if inoculated into the nasal cavity (43).
Therefore, intranasally administered T. cruzi likely gains access to the brain via olfactory nerve tissues.
Additionally, it may also reach the CNS after invading cells in the nasal cavity, amplifying the infection locally, and subsequently migrating to the brain via the olfactory tissues. It is also possible that a smaller subset of intranasally administered parasites will invade distant sites and subsequently the brain via the circulation. Regardless, it is clear that T. cruzi administered intranasally on the mucosa in the nasal cavity preferential invades the CNS. Interestingly, a recent study demonstrated that T. cruzi atraumatically deposited in the mouse conjunctiva migrated through the nasolacrimal duct to reach the nasal cavity, from where it invaded ductal and respiratory epithelia and then local lymph nodes and distant tissues via the bloodstream (18). Although it was not determined if the conjunctiva/nasal cavity circuit led to brain invasion, we predict, based on our findings and the data in the literature about brain entry by proteins and bacteria administered into the nasal cavity, that conjunctival infection with T. cruzi results in robust brain invasion.
In the brain, T. cruzi targeted the basal ganglia regardless of whether the infection was initiated via the intranasal mucosa or subcutaneous tissues. In the basal ganglia, T. cruzi grew abundantly and triggered a strong inflammatory response. The strong inflammatory response is in accordance with reports by others (31, 36, 37). One would expect T. cruzi infection and the ensuing inflammatory response in the basal ganglia to cause abnormal control of movement and posture and changes in muscular tone, which normally accompany alterations in the striatum and substantia nigra, as in parkinsonism, Huntington's disease, and African sleeping sickness (10, 22, 26). Puzzlingly, we did not find evidence of motor disorders in any of the T. cruzi-infected mice at the peak of brain invasion and inflammation, in contrast to the obvious signs of basal ganglion disorders in the mouse model of Huntington's disease (16) (Table 1). The absence of motor disorders agrees with the lack of detectable neurodegeneration in the brains of T. cruzi-infected mice (Fig. 6). A similar paradox is common to acute chagasic patients, whose CNS are infected with T. cruzi and who nevertheless do not present symptoms normally related to brain infection (20).
The intranasal inoculation mouse model described here should be useful for studying the molecular basis of T. cruzi invasion of the brain, such as the binding of parasite-derived neurotrophic factor/trans-sialidase to the nerve growth factor receptor TrkA (2, 9, 15) and of a T. cruzi mimic of glial cell-derived neurotrophic family ligands to glial family ligand receptors (28). It may also facilitate studies aimed to understand the recruitment of inflammatory cells in resistant and susceptible immunocompetent mice and immunocompromised animals.
Acknowledgments
We thank Joseph Alroy and Rolf Pfannl for their analyses of H&E sections and Alejandro Luquetti for useful comments on the manuscript.
This work was sponsored by NIH grants NS40574 and NS429660S.
Editor: W. A. Petri, Jr.
Footnotes
Published ahead of print on 21 January 2009.
REFERENCES
- 1.Adad, S. J., C. G. Cancado, R. M. Etchebehere, V. P. Teixeira, U. A. Gomes, E. Chapadeiro, and E. R. Lopes. 2001. Neuron count reevaluation in the myenteric plexus of chagasic megacolon after morphometric neuron analysis. Virchows Arch. 438254-258. [DOI] [PubMed] [Google Scholar]
- 2.Akpan, N., K. Caradonna, M. V. Chuenkova, and M. PereiraPerrin. 2008. Chagas' disease parasite-derived neurotrophic factor activates cholinergic gene expression in neuronal PC12 cells. Brain Res. 1217195-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arantes, R. M., H. H. Marche, M. T. Bahia, F. Q. Cunha, M. A. Rossi, and J. S. Silva. 2004. Interferon-gamma-induced nitric oxide causes intrinsic intestinal denervation in Trypanosoma cruzi-infected mice. Am. J. Pathol. 1641361-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal, M. F., E. Brouillet, B. G. Jenkins, R. J. Ferrante, N. W. Kowall, J. M. Miller, E. Storey, R. Srivastava, B. R. Rosen, and B. T. Hyman. 1993. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 134181-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blum, J. A., M. J. Zellweger, C. Burri, and C. Hatz. 2008. Cardiac involvement in African and American trypanosomiasis. Lancet Infect. Dis. 8631-641. [DOI] [PubMed] [Google Scholar]
- 6.Brouillet, E., F. Conde, M. F. Beal, and P. Hantraye. 1999. Replicating Huntington's disease phenotype in experimental animals. Prog. Neurobiol. 59427-468. [DOI] [PubMed] [Google Scholar]
- 7.Chimelli, L., and F. Scaravilli. 1997. Trypanosomiasis. Brain Pathol. 7599-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chuenkova, M., and M. E. Pereira. 1995. Trypanosoma cruzi trans-sialidase: enhancement of virulence in a murine model of Chagas' disease. J. Exp. Med. 1811693-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuenkova, M. V., and M. PereiraPerrin. 2004. Chagas' disease parasite promotes neuron survival and differentiation through TrkA nerve growth factor receptor. J. Neurochem. 91385-394. [DOI] [PubMed] [Google Scholar]
- 10.Cowan, C. M., and L. A. Raymond. 2006. Selective neuronal degeneration in Huntington's disease. Curr. Top. Dev. Biol. 7525-71. [DOI] [PubMed] [Google Scholar]
- 11.Cummings, K. L., and R. L. Tarleton. 2003. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Mol. Biochem. Parasitol. 12953-59. [DOI] [PubMed] [Google Scholar]
- 12.Da Mata, J. R., M. R. Camargos, E. Chiari, and C. R. Machado. 2000. Trypanosoma cruzi infection and the rat central nervous system: proliferation of parasites in astrocytes and the brain reaction to parasitism. Brain Res. Bull. 53153-162. [DOI] [PubMed] [Google Scholar]
- 13.da Silveira, A. B., M. A. Freitas, E. C. de Oliveira, S. G. Neto, A. O. Luquetti, J. B. Furness, R. Correa-Oliveira, and D. d'Avila Reis. 2008. Neuronal plasticity of the enteric nervous system is correlated with chagasic megacolon development. Parasitology 1351337-1342. [DOI] [PubMed] [Google Scholar]
- 14.Del Castillo, M., G. Mendoza, J. Oviedo, R. P. Perez Bianco, A. E. Anselmo, and M. Silva. 1990. AIDS and Chagas' disease with central nervous system tumor-like lesion. Am. J. Med. 88693-694. [DOI] [PubMed] [Google Scholar]
- 15.de Melo-Jorge, M., and M. PereiraPerrin. 2007. The Chagas' disease parasite Trypanosoma cruzi exploits nerve growth factor receptor TrkA to infect mammalian hosts. Cell Host Microbe 1251-261. [DOI] [PubMed] [Google Scholar]
- 16.Fernagut, P. O., E. Diguet, N. Stefanova, M. Biran, G. K. Wenning, P. Canioni, B. Bioulac, and F. Tison. 2002. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57BL/6 mice: behavioural and histopathological characterisation. Neuroscience 1141005-1017. [DOI] [PubMed] [Google Scholar]
- 17.Genovese, O., C. Ballario, R. Storino, E. Segura, and R. E. Sica. 1996. Clinical manifestations of peripheral nervous system involvement in human chronic Chagas disease. Arq. Neuropsiquiatr. 54190-196. [DOI] [PubMed] [Google Scholar]
- 18.Giddings, O. K., C. S. Eickhoff, T. J. Smith, L. A. Bryant, and D. F. Hoft. 2006. Anatomical route of invasion and protective mucosal immunity in Trypanosoma cruzi conjunctival infection. Infect. Immun. 745549-5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez Cappa, S. M., O. P. Sanz, L. A. Muller, H. A. Molina, J. Fernandez, M. T. Rimoldi, and R. E. Sica. 1987. Peripheral nervous system damage in experimental chronic Chagas' disease. Am. J. Trop. Med. Hyg. 3641-45. [DOI] [PubMed] [Google Scholar]
- 20.Hoff, R., R. S. Teixeira, J. S. Carvalho, and K. E. Mott. 1978. Trypanosoma cruzi in the cerebrospinal fluid during the acute stage of Chagas' disease. N. Engl. J. Med. 298604-606. [DOI] [PubMed] [Google Scholar]
- 21.Hunter, C. A., T. Slifer, and F. Araujo. 1996. Interleukin-12-mediated resistance to Trypanosoma cruzi is dependent on tumor necrosis factor alpha and gamma interferon. Infect. Immun. 642381-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy, P. G. 2008. The continuing problem of human African trypanosomiasis (sleeping sickness). Ann. Neurol. 64116-126. [DOI] [PubMed] [Google Scholar]
- 23.Koberle, F. 1968. Chagas' disease and Chagas' syndromes: the pathology of American trypanosomiasis. Adv. Parasitol. 663-116. [DOI] [PubMed] [Google Scholar]
- 24.Kohl, S., L. K. Pickering, L. S. Frankel, and R. G. Yaeger. 1982. Reactivation of Chagas' disease during therapy of acute lymphocytic leukemia. Cancer 50827-828. [DOI] [PubMed] [Google Scholar]
- 25.Kress, Y., B. R. Bloom, M. Wittner, A. Rowen, and H. Tanowitz. 1975. Resistance of Trypanosoma cruzi to killing by macrophages. Nature 257394-396. [DOI] [PubMed] [Google Scholar]
- 26.Le, W., S. Chen, and J. Jankovic. 2009. Etiopathogenesis of Parkinson disease: a new beginning? Neuroscientist 1528-35. [DOI] [PubMed] [Google Scholar]
- 27.Leiguarda, R., A. Roncoroni, A. L. Taratuto, L. Jost, M. Berthier, M. Nogues, and H. Freilij. 1990. Acute CNS infection by Trypanosoma cruzi (Chagas' disease) in immunosuppressed patients. Neurology 40850-851. [DOI] [PubMed] [Google Scholar]
- 28.Lu, B., and M. PereiraPerrin. 2008. A novel immunoprecipitation strategy identifies a unique functional mimic of the glial cell line-derived neurotrophic factor family ligands in the pathogen Trypanosoma cruzi. Infect. Immun. 763530-3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ludolph, A. C., F. He, P. S. Spencer, J. Hammerstad, and M. Sabri. 1991. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 18492-498. [DOI] [PubMed] [Google Scholar]
- 30.Meneghelli, U. G. 1985. Chagas' disease: a model of denervation in the study of digestive tract motility. Braz. J. Med. Biol. Res. 18255-264. [PubMed] [Google Scholar]
- 31.Michailowsky, V., N. M. Silva, C. D. Rocha, L. Q. Vieira, J. Lannes-Vieira, and R. T. Gazzinelli. 2001. Pivotal role of interleukin-12 and interferon-gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am. J. Pathol. 1591723-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olitsky, P. K., and H. R. Cox. 1934. Temporary prevention by chemical means of intranasal infection of mice with equine encephalomyelitis virus. Science 80566-567. [DOI] [PubMed] [Google Scholar]
- 33.Prioli, R. P., J. S. Mejia, and M. E. Pereira. 1990. Monoclonal antibodies against Trypanosoma cruzi neuraminidase reveal enzyme polymorphism, recognize a subset of trypomastigotes, and enhance infection in vitro. J. Immunol. 1444384-4391. [PubMed] [Google Scholar]
- 34.Rosemberg, S., C. J. Chaves, M. L. Higuchi, M. B. Lopes, L. H. Castro, and L. R. Machado. 1992. Fatal meningoencephalitis caused by reactivation of Trypanosoma cruzi infection in a patient with AIDS. Neurology 42640-642. [DOI] [PubMed] [Google Scholar]
- 35.Schmued, L. C., C. C. Stowers, A. C. Scallet, and L. Xu. 2005. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 103524-31. [DOI] [PubMed] [Google Scholar]
- 36.Silva, A. A., E. Roffe, A. P. Marino, P. V. dos Santos, T. Quirico-Santos, C. N. Paiva, and J. Lannes-Vieira. 1999. Chagas' disease encephalitis: intense CD8+ lymphocytic infiltrate is restricted to the acute phase, but is not related to the presence of Trypanosoma cruzi antigens. Clin. Immunol. 9256-66. [DOI] [PubMed] [Google Scholar]
- 37.Silva, A. A., E. Roffe, H. Santiago, A. P. Marino, K. Kroll-Palhares, M. M. Teixeira, R. T. Gazzinelli, and J. Lannes-Vieira. 2007. Trypanosoma cruzi-triggered meningoencephalitis is a CCR1/CCR5-independent inflammatory process. J. Neuroimmunol. 184156-163. [DOI] [PubMed] [Google Scholar]
- 38.Tanowitz, H. B., L. V. Kirchhoff, D. Simon, S. A. Morris, L. M. Weiss, and M. Wittner. 1992. Chagas' disease. Clin. Microbiol. Rev. 5400-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorne, R. G., G. J. Pronk, V. Padmanabhan, and W. H. Frey II. 2004. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127481-496. [DOI] [PubMed] [Google Scholar]
- 40.Townsend, G. C., and W. M. Scheld. 1998. Infections of the central nervous system. Adv. Intern. Med. 43403-447. [PubMed] [Google Scholar]
- 41.Trischmann, T., H. Tanowitz, M. Wittner, and B. Bloom. 1978. Trypanosoma cruzi: role of the immune response in the natural resistance of inbred strains of mice. Exp. Parasitol. 45160-168. [DOI] [PubMed] [Google Scholar]
- 42.Vaidian, A. K., L. M. Weiss, and H. B. Tanowitz. 2004. Chagas' disease and AIDS. Kinetoplastid Biol. Dis. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Ginkel, F. W., J. R. McGhee, J. M. Watt, A. Campos-Torres, L. A. Parish, and D. E. Briles. 2003. Pneumococcal carriage results in ganglioside-mediated olfactory tissue infection. Proc. Natl. Acad. Sci. USA 10014363-14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiesinger, D. 1956. Importance of environmental factors for the blood sucking activity of Triatoma infestans. Acta Trop. 1397-141. [PubMed] [Google Scholar]
- 45.Zeledón, R. 1975. Effects of triatomine behavior on trypanosoma transmission. PAHO Sci. Publ. 318326-329. [Google Scholar]