

Abstract
A wide variety of RNA viruses have been shown to produce proteins that inhibit interferon (IFN) production and signaling. For human respiratory syncytial virus (RSV), the nonstructural NS1 and NS2 proteins have been shown to block IFN signaling by causing the proteasomal degradation of STAT2. In addition, recombinant RSVs lacking either NS1 or NS2 induce more IFN production than wild-type (wt) RSV in infected cells. However, the mechanisms by which the NS proteins perform this function are unknown. In this study, we focused on defining the mechanism by which NS2 inhibits the induction of IFN transcription. We find that NS2 is required for the early inhibition of IFN transcription since the infection of cells with NS2-deletion RSV resulted in a higher level of IRF3 activation at early time points postinfection compared with that of wt or NS1-deletion RSV infection. In addition, NS2 expression inhibits IFN transcription induced by both the RIG-I and TLR3 pathways. Furthermore, we show that NS2 inhibits RIG-I-mediated IFN promoter activation by binding to the N-terminal CARD of RIG-I and inhibiting its interaction with the downstream component MAVS (IPS-1, VISA, Cardif). Thus, the RSV NS2 protein is a multifunctional IFN antagonist that targets specific components of both the IFN induction and IFN signaling pathways.
Respiratory syncytial virus (RSV) is the most important etiologic agent of pediatric viral respiratory infection and remains a major cause of morbidity and mortality among infants as well as immunocompromised subjects and the elderly (9). RSV is the prototype member of the Pneumovirus genus in the family Paramyxoviridae. The nonsegmented, negative-sense RNA genome of RSV encodes 10 transcription units from which 11 proteins are translated. From this linear array, the two promoter-proximal genes encode the nonstructural NS1 and NS2 proteins which are dispensable for viral replication in vitro, although the deletion of either NS gene attenuates the replication of recombinant RSV (rRSV) significantly in vitro and markedly in vivo (7, 16, 32, 39, 42). Since the NS genes are the first two transcription units in the gene order, the transcription of NS1 and NS2 is thought to be abundant and to occur early in infection (9).
The RSV NS proteins are small (NS1, 139 amino acids [aa]; NS2, 124 aa) and have no significant sequence homology with each other or with any cellular protein in the database. NS1 and NS2 have been shown to encode multiple functions associated with viral pathogenesis. NS1 and NS2 appear to antagonize both the cellular antiviral response as well as the induction of interferon (IFN) transcription (5, 6, 32, 36). rRSVs lacking NS1 (ΔNS1) or NS2 (ΔNS2) induce significantly more IFN-β transcription in infected cells than wild-type (wt) rRSV; the deletion of both NS genes from rRSV results in a virus-induced IFN-β transcription to a greater extent than either single NS deletion rRSV (36). In addition, RSV infection results in the proteasome-mediated degradation of STAT2 by the formation of a ubiquitin ligase complex containing NS1 and NS2 (23, 29, 30). The IFN antagonism by NS1 and, particularly, NS2 has been implicated in altered immune responses in mice and human dendritic cells (19, 25). An analysis of immune responses to infection with NS-deletion rRSV in mice showed increased CD8+ T-cell responses in ΔNS2-infected mice compared with wt rRSV- and ΔNS1-infected mice (19). In addition, human dendritic cell maturation is enhanced by ΔNS1 and ΔNS1/2 infection, compared with that by wt rRSV and ΔNS2 infection (25). These studies indicate that NS1 and NS2 may play a critical role in altering immune responses. Recent studies also suggest that NS1 and NS2 are involved in apoptosis inhibition at early time points (<18 h) during infection (4). In addition, it has been reported that NS1 plays a role in regulating viral RNA synthesis (1). Since the NS proteins have multiple functions, the induction of IFN-β by ΔNS1 and ΔNS2 may be attributed to a complex interplay between the IFN antagonist and other functions of the NS proteins. However, it is difficult to segregate the contribution of these disparate effects of the NS proteins on replication, IFN antagonism, and apoptosis in the context of viral infection. Remarkably, few studies have been performed examining the functions of the NS proteins alone, in a virus-free environment. Thus, the ability of NS proteins to directly block IFN-β transcription has not been previously demonstrated.
RNA viruses induce IFN production mainly through two distinct signal transduction pathways. First, viral replication products are recognized by the retinoic acid induced gene (RIG)-like helicase family of cytosolic DEXD/H box RNA helicases, which includes RIG-I and melanoma differentiation antigen 5 (MDA-5) (17, 38). IFN induction by RIG-I and MDA-5 requires interaction with the mitochondrial protein MAVS (IPS-1/VISA/Cardif) through caspase recruitment domains (CARD), resulting in the phosphorylation of IRF3 by the IRF3 kinases IκB kinase ɛ (IKKɛ) and TANK binding kinase-1 (TBK-1) (11). Phosphorylated IRF3 then homodimerizes and translocates into the nucleus, where, together with AP-1 and NF-κB, it activates the transcription of IFN-β. Alternatively, Toll-like receptor (TLR) recognition of viral nucleic acids can induce IFN production after RNA virus infection. In RSV-infected epithelial cells, TLR3 has been shown to be important for IFN induction (22). TLR3 signal transduction results in the activation of the same IRF3 kinases (IKKɛ and TBK-1) via the adaptor protein TRIF (31).
Previous studies have shown that small interfering RNA knockdown of RIG-I, but not MDA-5, prevents the early induction of IFN after RSV infection (22). In contrast, the knockdown of TLR3 by small interfering RNA treatment blocked IFN induction at later times postinfection (p.i.) (22). In addition, RIG-I-mediated IFN production was responsible for the induction of TLR3 expression, underscoring the importance of RIG-I in IFN responses to RSV (22). We find that rRSV lacking NS2 induces increased IRF3 activation early in infection, suggesting that NS2 can inhibit RIG-I. In our study, we demonstrate that the RSV NS2 protein alone can inhibit both RIG-I- and TLR3-mediated IFN induction and investigate the mechanism of this inhibition.
MATERIALS AND METHODS
Plasmids and antibodies.
Expression constructs for human codon-optimized NS1 and NS2 (23) were kindly provided by Michael Holtzman (Washington University School of Medicine). Influenza hemagglutinin (HA) tags were added to the NS1 and NS2 open reading frames (ORFs) by annealing oligonucleotides bearing the HA tag and ligating the hybridized primers into the BamHI site of the corresponding vector. The correct insertion of the HA tag was determined by automated DNA sequencing. Plasmids for Flag-RIG-I, Flag-tagged N (residues 2 to 229)- or C (residues 218 to 925)-terminal fragments of RIG-I (N-RIG and C-RIG) were generous gifts from Takashi Fujita (Tokyo Metropolitan Institute of Medical Science) (45). Plasmids for Flag-MAVS and HA-VISA (MAVS) were provided by Zhijian Chen (University of Texas Southwestern Medical Center) (34) and Hong-Bing Shu (National Jewish Medical and Research Center; 43), respectively. Plasmids for Flag-TRIF, Flag-TBK-1, and Flag-IKKɛ were provided by Kathryn Fitzgerald (University of Massachusetts, Worcester, MA) (11) and Tom Maniatis (Harvard). The IRF3(5D), pIFNβ-luc, and pISG56-luc plasmids were from John Hiscott (McGill University) (20). Plasmid phRLTK, which bears a Renilla luciferase gene, was purchased from Promega. pCAGGS-P was generated by the insertion of the RSV P ORF into the SacI/XhoI window of pCAGGS-MCS.
Rabbit antisera against NS1 and NS2 were generated by immunizing rabbits with the recombinant proteins His6-NS1 and His6-NS2, respectively (21). Rabbit polyclonal antibodies against RIG-I and RSV P were from Tadaatsu Imaizumi (Hirosaki University) (45) and Paul Yeo (University Science Laboratories). Rabbit polyclonal antibody against IRF3 (SC-9082) was purchased from Santa Cruz Biotechnology. Anti-F monoclonal antibody (MAb1269) has been described previously (40). Monoclonal antibody against HA (12CA5) was a gift from Biao He (Penn State University). Anti-Flag antibody (M2) was purchased from Sigma.
Cells and viruses.
Human A549 pulmonary epithelial cells were grown in F-12 nutrient mixture (Ham) medium (Gibco) with 5% fetal bovine serum (FBS). African green monkey kidney Vero cells were cultured in Opti-MEM supplemented with 2% FBS and 1% l-glutamine. HEK293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 5% FBS. The rRSVs ΔNS1, ΔNS2, and ΔNS1/2 were described previously and are derivatives of the wt A2 strain (rA2) (35, 39, 41). To make the recombinant virus with HA-tagged NS2, BamHI and NotI restriction sites were engineered into pGEM-NS at the N and C termini of the NS2 ORF, respectively, by inverse PCR mutagenesis using Deep Vent DNA polymerase (NEB). The HA-tagged NS2 sequence was then subcloned from pcDNA5-HA-NS2 into pGEM-NS. The fragment for HA-NS2 was introduced back into the antigenome cDNA for RSV, resulting in D53-HANS2, which was then used to recover rRSV. The recovery of recombinant RSV from cloned DNA, multiple-step replication assays, and plaque assays were performed as described previously (10).
Immunofluorescence assay.
A549 cells were plated on coverslips, incubated overnight at 37°C, and then infected by the different rRSV at a multiplicity of infection (MOI) of 3 in triplicate. At 9, 12, and 16 h p.i., the cells were fixed in formalin and permeabilized by the addition of 0.1% Triton X-100. The subcellular localization of IRF3 was detected by staining with rabbit anti-human IRF3 (Santa Cruz), followed by Cy3-coupled goat anti-rabbit immunoglobulin (Ig) (Jackson Immunologicals). Virus-infected cells were detected by staining with anti-F monoclonal antibody (MAb1269 [40]) followed by Alexa488-coupled goat anti-mouse Ig (Jackson Immunologicals). The percentages of infected cells showing the nuclear localization of IRF3 were determined by counting approximately 300 infected cells in 10 fields per coverslip. Each experimental time point was performed in triplicate so that each percentage represents between 900 and 1,000 cells counted. Statistical analysis was performed using analysis of variance (ANOVA) to compare the NS deletion viruses to rA2. Micrographs were taken using a Zeiss Axioplan epifluorescence microscope with a 63× Apochromat oil immersion objective.
Luciferase reporter assay.
Transient transfection was carried out using GeneJuice (Novagen) or Lipofectamine (Invitrogen). For the luciferase reporter assays, 293T cells were seeded in 24-well plates at a cell density of 2.1 × 105 cells per well. On the second day, the cells were cotransfected with either 20 ng of pIFNβ-luc plasmid or 2.5 ng of pISG56-luc plasmid as a reporter gene, 0.5 ng phRLTK as an internal control for transfection efficiency, and the expression vectors indicated in the figure legends. The total DNA concentration was kept constant by supplementation with empty vector. Each sample was collected in triplicate, and each experiment was performed three times. Cells were harvested 24 h after transfection and lysed in 100 μl of passive lysis buffer (Promega). A total of 20 μl of cell lysates was used for the inspection of luciferase activity. For the activation of full-length RIG-I, the cells were first transfected with the Flag-RIG-I vector, luciferase constructs, and the expression vectors indicated in the figure legends. After 24 h, 0.1 μg of 5′-triphosphate short RNA, ssRNA-110 (provided by Craig Cameron of Penn State University [26]), was transfected into cells by using Lipofectamine (Invitrogen) under conditions specified by the manufacturer. The cells were harvested 16 h after the transfection of the RNA and lysed with passive lysis buffer (Promega). Luciferase activity was measured with dual luciferase reporter assay reagent (Promega) and a Veritas microplate luminometer. Statistical significance among the samples was determined by a one-way ANOVA test.
Immunoprecipitation.
Cultures of HEK293T cells in 6-cm-diameter dishes were transfected with various combinations of plasmids. The total DNA concentration was kept constant by supplementation with empty vectors. Twenty-four hours after transfection, the cells were washed using phosphate-buffered saline and resuspended in 0.1 M phosphate buffer (pH 8). Dithiobis(succinimidyl propionate) (DSP; Pierce) was added into the suspension slowly at a final concentration of 1 mM, and this was incubated on ice for 30 min. The cross-linking reaction was stopped by the addition of 50 mM Tris buffer (pH 8) and further incubation on ice for 15 min. The cells were collected by centrifugation and lysed in 600 μl of lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 1% Triton X-100, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin). The lysates were subjected to centrifugation for 10 min at 13,000 × g, and the supernatant was collected. The supernatant of the cell lysate was precleared by incubation with 10 μl of protein A-Sepharose (Sigma-Aldrich) for 20 min at 4°C. For each immunoprecipitation, a 300-μl aliquot of lysate was incubated with 2 μg of antibody for 2 h at 4°C on a rotator. Antibody-antigen complexes were precipitated by adding 15 μl of a 1:1 slurry of protein A-Sepharose beads for 1 h at 4°C, followed by centrifugation for 1 min at 13,000 × g. The beads were washed three times with 1 ml of lysis buffer. The precipitates were subjected to Western blot (WB) analysis. For an analysis of the interaction of N-RIG and NS2, cells were lysed in lysis buffer and then first subjected to centrifugation for 3 min at 300 × g to remove nuclei. Sodium dodecyl sulfate (SDS) was added into the supernatant to a final concentration of 0.5% to lyse membranes and release membrane-associated proteins. Lysis buffer was then used to dilute the cell lysate to make a final concentration of 0.1% SDS. The cell lysates were then subjected to immunoprecipitation as described above. For the immunoprecipitation of the infected cells, A549 cells were infected as indicated with viruses at an MOI of 3 for 10 h and incubated with rHuIFN-α for 6 h to stimulate the expression of RIG-I. The cells were then harvested and subjected to immunoprecipitation as described above.
WB analysis.
For an analysis of the whole cell lysate, cells were harvested 24 h posttransfection and boiled in SDS sample buffer supplemented with 100 mM dithiothreitol for 10 min at 95°C. For an analysis of the immunoprecipitates, protein A-Sepharose beads were boiled in SDS sample buffer for 10 min at 95°C. Samples treated with the reversible cross-linker (DSP) were incubated at 37°C for 30 min for dithiothreitol to cleave the DSP prior to boiling. The protein samples were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (BioTrace NT; Pall). The membranes were probed with the appropriate primary antibodies. After being washed with phosphate-buffered saline containing 0.1% Tween 20, bound antibodies were probed by secondary incubation with horseradish peroxidase-coupled goat anti-rabbit IgG or goat anti-mouse IgG antibodies (KPL). The target bands were visualized by using Immobilon FL chemiluminescence reagent (Millipore) and exposed to BioMax X-ray film (Kodak).
RESULTS
Previous studies have found that rRSV lacking either NS1 or NS2 genes induces high levels of IFN-β transcription, suggesting that NS1 and NS2 play an important role in antagonizing IFN-β induction (36, 37). However, the mechanisms by which NS1 and NS2 accomplish this antagonism are unknown. As an initial indication of which step of IFN induction is affected by the NS proteins, we examined the time course of IRF3 activation infected by wt rRSV (rA2) and rRSVs lacking NS1 (ΔNS1), NS2 (ΔNS2), or both NS1 and NS2 (ΔNS1/2). The nuclear accumulation of IRF3 was used as a marker of activation in infected A549 cells. At 9, 12, and 16 h p.i., cells were fixed, permeabilized, and subjected to immunofluorescence for IRF3 and RSV F. The percentage of infected cells showing the nuclear localization of IRF3 was calculated by counting ∼30 infected cells in each of 10 fields for triplicate samples for each condition (∼900 to 1,000 infected cells/condition). We found substantially higher levels of IRF3 nuclear translocation in ΔNS2-infected cells by 9 h p.i. compared with those of the cells infected by rA2 or the NS1 deletion viruses (Fig. 1a). By 16 h p.i., cells infected by each of the deletion viruses displayed similar levels of IRF3 nuclear translocation at a level approximately twofold higher than the rA2-infected cells (Fig. 1c). The 12-h time point gave intermediate results, as would be expected (Fig. 1b). Thus, the deletion of either NS1 or NS2 resulted in a significant IRF3 translocation in infected cells, suggesting that both NS1 and NS2 can independently inhibit IFN activation.
FIG. 1.

IRF3 activation in rRSV-infected cells. A549 cells were mock infected or infected by wt rRSV (rA2) or rRSV lacking NS1 (ΔNS1), NS2 (ΔNS2), or both (ΔNS1/2). At 9 (top), 12 (middle), and 16 (bottom) h p.i., cells were fixed, permeabilized, and stained with antibodies to IRF3 (αIRF3; red) or RSV F (αF; green) for immunofluorescence. Infected (RSV F-positive) cells were scored for the presence of IRF3 in the nucleus. Approximately 30 cells for each of 10 fields were scored for triplicate samples of each condition (∼900 cells/virus/time p.i.). The photomicrographs show examples of individual fields that were counted for each sample. Shown in the bar graphs on the right are percentages (± standard errors of the means) of infected cells displaying nuclear IRF3. Asterisks denote a P value of <0.01 versus rA2-infected cells by ANOVA.
Previous studies have implicated both the RIG-I and TLR3 pathways as being utilized for IFN activation in RSV-infected cells, with RIG-I activation occurring early in infection and TLR3 activation occurring later (22). Since NS2 appears to affect IRF3 activation at earlier times p.i. (Fig. 1), we wanted to determine whether NS2 has differential effects in inhibiting these two pathways. To examine the effect of NS2 in isolation, we performed promoter assays in transfected 293T cells using constructs encoding firefly luciferase under the control of the IFN-β or ISG56 promoter (Fig. 2). The RIG-I pathway was activated by the cotransfection of an expression construct encoding the constitutively active N-terminal 229 aa of RIG-I containing the CARD. We found that the expression of NS2 inhibited the induction of IFN-β transcription induced by N-RIG in a dose-dependent fashion (Fig. 2a). Moreover, this effect was due to the inhibition of IRF3 activation since transcription from the IRF3-responsive ISG56 promoter was also decreased (Fig. 2b). This inhibition of IRF3 activation was not due to the NS2-mediated degradation of N-RIG, as the expression levels of the IFN activators were constant regardless of NS2 expression (Fig. 2c). In contrast, the expression of P had no effect on either ISG56 promoter activity or N-RIG expression (Fig. 2d and e). Similarly, the TLR3 pathway was activated by the cotransfection of an expression construct encoding the downstream adaptor TRIF. Increasing amounts of expression plasmid encoding NS2 were added to the transfection mixture to determine whether NS2 can inhibit the TLR3 pathway. Transcription from both IFN-β and ISG56 promoters induced by TRIF expression was inhibited by NS2 in a dose-dependent fashion (Fig. 3a and b). As with N-RIG, NS2 did not affect TRIF expression (Fig. 3c). In addition, P did not affect TRIF expression or TRIF-induced IRF3 activation (Fig. 3d and e). Thus, NS2 expression alone is sufficient to inhibit IFN-β transcription induced via either the RIG-I or TLR3 pathway.
FIG. 2.
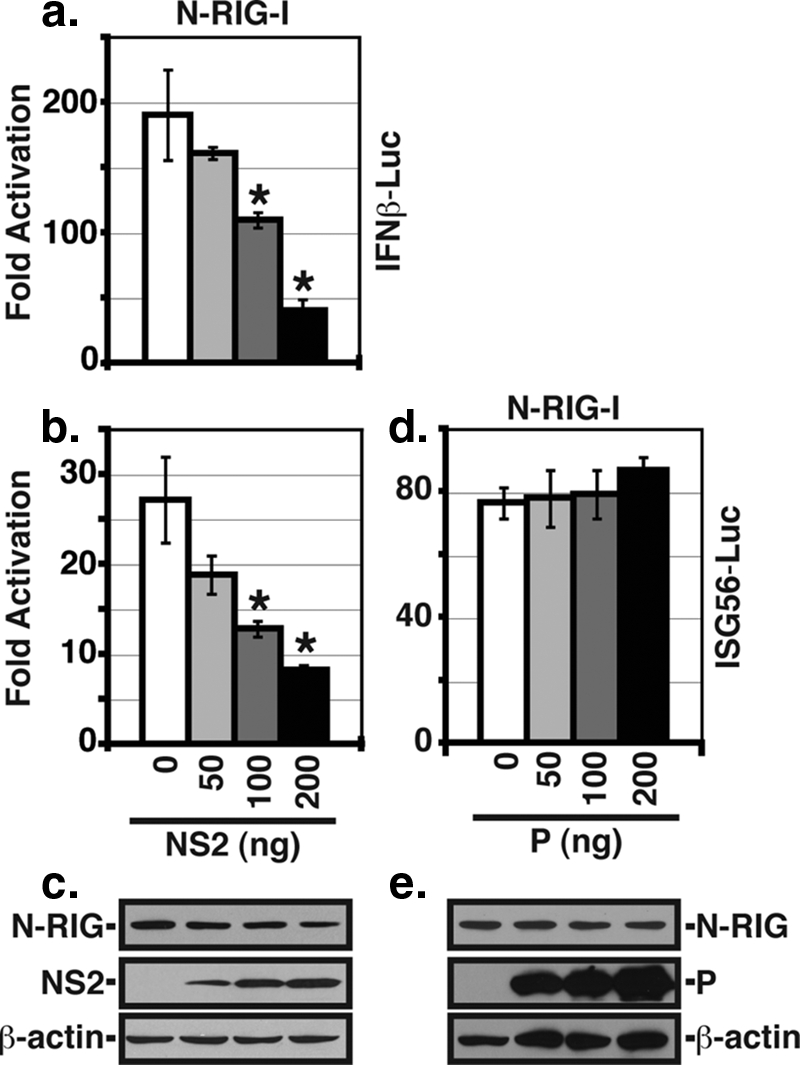
Inhibition of RIG-I-induced IFN promoter activity by NS2. 293T cells were transfected by an expression construct encoding N-RIG and either pIFNβ-luc (a) or pISG56-luc (b). Increasing amounts of the NS2 expression plasmid were added to the transfection. phRL-TK was used as a transfection control and empty vector was used to standardize the amount of plasmid transfected. Luciferase activities were determined at 24 h posttransfection. Shown are the means (± standard errors of the means) of triplicate samples. One representative experiment (of three) is shown. Asterisks denote a P value of <0.01 versus that of the control by one-way ANOVA. (c) Expression levels of N-RIG, NS2, and β-actin (as a control) were determined by WB analysis. (d) The ISG56 reporter assay was performed as described above using N-RIG to induce promoter activity and increasing amounts of an expression plasmid encoding P. (e) WB analysis of protein expression from the experiment described for panel d.
FIG. 3.
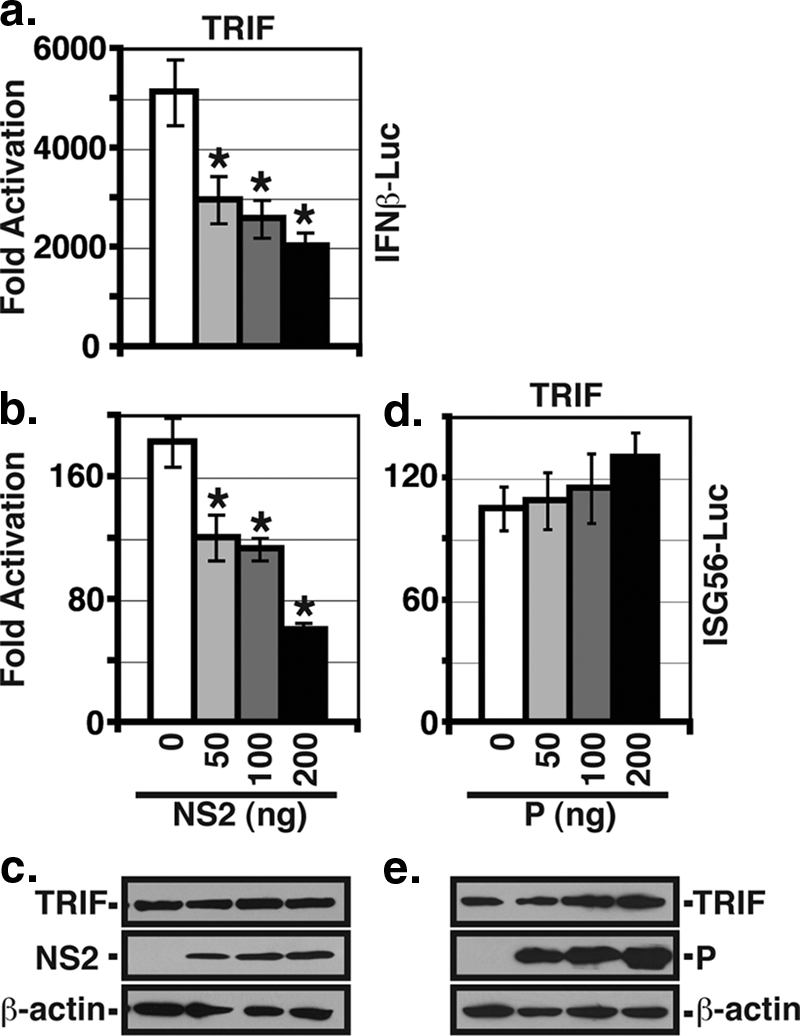
NS2 inhibits TRIF-induced IFN promoter activity. 293T cells were transfected by an expression construct encoding TRIF and the pIFNβ-luc (a) or pISG56-luc (b) reporter constructs with increasing amounts of NS2 plasmids as in Fig. 2. Luciferase activities were determined at 24 h posttransfection. Shown are the means (± standard errors of the means) of triplicate samples. One representative experiment (of three) is shown. Asterisks denote a P value of <0.01 versus that of the control by one-way ANOVA. (c) The expression levels of TRIF, NS2, and β-actin were determined by WB analysis. (d) The ISG56 reporter assay was performed as described above using TRIF to induce promoter activity and increasing amounts of an expression plasmid encoding P. (e) WB analysis of protein expression from the experiment described for panel d.
Since N-RIG is not a physiologic stimulator of IFN transcription, we wanted to determine whether NS2 could block RIG-I signaling activated by a more relevant stimulus. Binding to 5′-triphosphorylated RNA activates RIG-I (15, 28). Therefore, we cotransfected an expression construct encoding full-length RIG-I with 5′-triphosphorylated RNA to activate IRF3 and then asked if the expression of NS2 could block this activation (Fig. 4). Indeed, NS2 inhibited ISG56 promoter activity induced by RNA-activated RIG-I in a dose-dependent manner, suggesting that NS2 inhibits activated RIG-I and N-RIG by similar mechanisms.
FIG. 4.
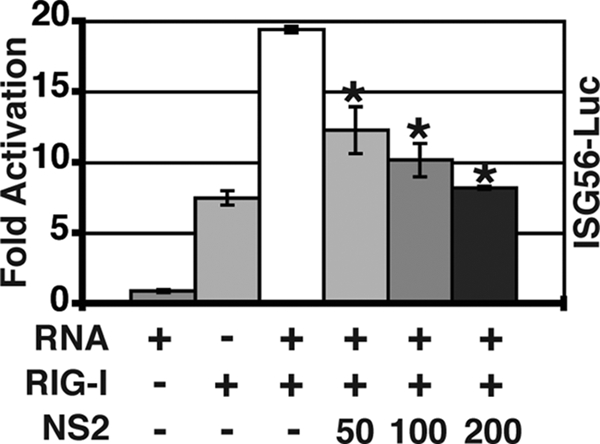
NS2 inhibits IRF3 activation by RNA-activated RIG-I. 293T cells were transfected with pISG56-luc and expression constructs encoding full-length RIG-I and the indicated amounts of NS2. phRL-TK was used as a transfection control, and empty vector was used to standardize the amount of plasmid transfected. Twenty-four hours posttransfection, RIG-I was activated by the transfection of 5′-triphosphorylated RNA, and luciferase activities were determined 16 h later. Shown are the means (± standard errors of the means) of triplicate samples. One representative experiment (of two) is shown. Asterisks denote a P value of <0.01 versus that of the control by one-way ANOVA.
We next wanted to determine at what level NS2 was acting on the RIG-I and TLR3 pathways. The two pathways are known to have common and unique intermediates. Since NS2 could inhibit both pathways, we first examined whether NS2 could inhibit a common intermediate of RIG-I- and TLR3-mediated IRF3 activation. RIG-I and TLR3 both ultimately activate the IRF3 kinases (IKKɛ and TBK-1); therefore, we determined whether NS2 could inhibit transcription from the ISG56 promoter activated by the IRF3 kinases. We found that the expression of NS2 did not inhibit ISG56 promoter activity induced by either TBK-1 or IKKɛ (Fig. 5a and c, respectively). In fact, there was a slight increase of ISG56 promoter activity induced by TBK1 at higher levels of NS2, which was likely due to the enhanced expression of TBK1 in the presence of NS2 (Fig. 5b), although the same does not appear to hold true for IKKɛ (Fig. 5d). We next examined whether NS2 could inhibit ISG56 promoter activity induced by MAVS (IPS-1, VISA, Cardif), which is directly downstream of RIG-I and upstream of TBK-1 and IKKɛ. However, the cotransfection of NS2 did not inhibit MAVS-induced ISG56 promoter activity or protein expression (Fig. 5e and f). Thus, it appears that NS2 inhibits IRF3, and thus IFN, activation at the level of RIG-I.
FIG. 5.

NS2 does not inhibit IRF3 activation by the IRF3 kinases or MAVS. The ISG56 promoter assay was performed as described in the legend to Fig. 2 using expression constructs encoding TBK-1 (a), IKKɛ (c), or MAVS (e) to induce luciferase activity and increasing amounts of NS2 expression plasmid. Shown are the means (± standard errors of the means) of triplicate samples for one representative experiment (of three) for each condition. Protein expression levels for the inducing protein, NS2, and β-actin, as determined by WB analysis, are shown in the bottom panels.
We next wanted to determine the mechanism by which NS2 inhibits RIG-I-stimulated IFN transcription. We first wanted to determine whether NS2 could directly interact with RIG-I. Therefore, we cotransfected the RIG-I and NS2 expression vectors into 293T cells and performed coimmunoprecipitation (Co-IP)/WB analysis using antibodies specific to the Flag (RIG-I) or influenza HA (NS2) tag. We found that NS2 was coprecipitated with RIG-I by anti-Flag antibody (Fig. 6a); conversely, RIG-I coprecipitated with NS2 using anti-HA antibody (Fig. 6b). By contrast, an interaction of RIG-I with NS1 was not detected by co-IP/WB (Fig. 6a and b). To confirm that NS2 interacts with RIG-I during viral infection, we infected A549 cells with wt (rA2) or HA-NS2-expressing rRSV (rHA-NS2) and then subjected the infected cells to co-IP/WB analysis using anti-HA antibody for IP and an anti-RIG-I antiserum for WB. We found that endogenous RIG-I coprecipitated with HA-NS2 (Fig. 6d).
FIG. 6.

NS2 interacts with RIG-I. 293T cells were transfected by expression constructs encoding full-length RIG-I and either HA-NS1 or HA-NS2. Cytoplasmic extracts were subjected to immunoprecipitation with anti-Flag (αFlag) or anti-HA (αHA) antibodies. The presence of RIG-I or NS protein was detected by WB analysis of the precipitates using anti-Flag (a) or anti-HA (b) antibodies, respectively. (c) WB analysis of unprecipitated cytoplasmic extracts. (d and e) A549 cells were mock infected or infected by rA2 or rHA-NS2 at an MOI of 3. RIG-I expression was induced by treatment with IFN-α at 10 h p.i. for 6 h. Cytoplasmic extracts were then generated and subjected to immunoprecipitation using anti-HA antibody. Precipitates (top) and unprecipitated cytoplasmic extracts (Input, bottom) were analyzed by WB using anti-RIG-I antiserum (d) or anti-HA antibody (e).
To determine which region of RIG-I bound to NS2, we performed cotransfection experiments as described for Fig. 6, using expression constructs encoding the N terminus (229 aa), the C terminus (218 to 925 aa), or the full-length version of RIG-I. Co-IP/WB analysis using anti-Flag antibody to precipitate RIG-I showed that NS2 interacts with full-length RIG-I and N-RIG but not C-RIG (Fig. 7a). Precipitation with anti-HA antibody confirmed the interaction between NS2 and full-length RIG-I and N-RIG (Fig. 7b). Interestingly, the interaction of N-RIG and NS2 appears to be more robust than that of full-length RIG-I and NS2 (compare the intensity of the bands in the 3rd and 5th lanes in Fig. 7).
FIG. 7.

NS2 interacts with the N-terminal CARD of RIG-I. 293T cells were transfected by expression constructs encoding NS2 and either full-length (FL) RIG-I or the N- or C-terminal fragments of RIG-I (N-RIG [N] or C-RIG [C], respectively). Immunoprecipitation with anti-Flag (αFlag; a) or anti-HA (αHA; b) antibodies and WB analysis for the transfected proteins were performed as described in the legend to Fig. 4. (c) WB analysis of unprecipitated cytoplasmic extracts. HC, immunoglobulin heavy chain.
We wanted to determine if the N-RIG-NS2 interaction had functional consequences. RIG-I, upon activation, is known to bind to MAVS through the N-terminal CARD, and this interaction is essential for signal transduction resulting in IFN activation (33). Therefore, we investigated whether NS2 binding of N-RIG could block its downstream interaction with MAVS. Co-IP/WB analysis of 293T cells transfected with expression vectors encoding Flag-tagged N-RIG and HA-tagged MAVS showed that the precipitation of N-RIG with anti-Flag antibody efficiently coprecipitates MAVS (Fig. 8a). However, the coexpression of NS2 with N-RIG and MAVS showed that the interaction between NS2 and N-RIG completely abrogates the N-RIG-MAVS interaction (Fig. 8a). Co-IP/WB analysis using anti-HA antibody to precipitate MAVS confirms the interaction with N-RIG and the abrogation of that interaction in the presence of NS2 (Fig. 8b).
FIG. 8.

NS2 disrupts N-RIG interaction with MAVS. 293T cells were transfected with the indicated combinations of expression vectors encoding Flag-N-RIG, HA-MAVS, and NS2. Cytoplasmic extracts were harvested 24 h posttransfection and subjected to immunoprecipitation using anti-Flag (αFlag; a) or anti-HA (αHA; b) antibodies. The presence of N-RIG, MAVS, and NS2 were detected by WB analysis using anti-Flag (top), anti-HA (middle), or anti-NS2 (bottom) antibodies, respectively. (c) WB analysis of unprecipitated cytoplasmic extracts.
DISCUSSION
While other studies suggested that the RSV NS proteins are involved in IFN inhibition, a direct role of the NS1 or NS2 protein in antagonizing IFN transcription has not previously been shown. In addition, the mechanisms by which this IFN antagonism occurs have not previously been elucidated. Our studies show NS2 blocks the activation of IRF3 early during infection by RSV. Furthermore, we have found that NS2 inhibits IFN activation at the level of RIG-I via a specific interaction requiring the N-terminal 229 aa of RIG-I. The functional consequence of this binding is the inhibition of RIG-I-MAVS interaction and thus signal transduction leading to IRF3 activation. Thus, NS2 encodes activities responsible for antagonizing both IFN induction and IFN signaling, in addition to its functions in viral replication and apoptosis inhibition.
We have tried to examine the effect of NS2 on the interaction between full-length RIG-I and MAVS. However, we have not been able to detect RIG-I-MAVS interaction, either in the presence or absence of RSV infection (data not shown). Other groups have also had difficulty detecting RIG-I-MAVS interaction, though they could detect N-RIG-MAVS interaction (18, 34). NS2 also appears to inhibit TLR3 signaling at or downstream of the TRIF adaptor molecule. We are currently examining the mechanism by which this inhibition occurs. Our preliminary evidence indicates that NS2 does not bind specifically to TRIF itself (data not shown), suggesting that NS2 may act on a downstream mediator (e.g., TRAF3).
Our immunofluorescence results differ significantly from a previous study in that we find that IRF3 nuclear translocation is sustained in rA2-infected cells for at least 16 h p.i., whereas Spann et al. found a precipitous decrease in nuclear IRF3 at 14 h p.i. (37). In addition, our overall percentages of infected cells displaying nuclear IRF3 are considerably higher than those previously reported (∼80% for ΔNS1/2 in our study versus ∼30% as reported in reference 37). This discrepancy is likely due to a difference in methodology, since we determined the number of infected cells displaying nuclear IRF3 (double-staining with anti-F and anti-IRF3) rather than the total percentages of cells in the culture with either nuclear IRF3 or NS protein expression (single staining [37]). Thus, our data likely better reflect the actual events in infected cells. An intriguing finding is that IRF3 nuclear translocation appears to occur earlier in ΔNS2-infected cells compared with those infected by ΔNS1/2 (Fig. 1). While the reasons for this difference are not known, ΔNS2 replicates more rapidly and to higher final titers than ΔNS1/2 (36), suggesting that the synthesis of viral replication products recognized by RIG-I (5′-triphosphorylated RNA, double-stranded RNA) may be delayed in the absence of NS1.
That viruses have developed mechanisms for inhibiting antiviral activities induced by IFNs has been well documented. Paramyxoviruses, in particular, have developed mechanisms for inhibiting both IFN production and signal transduction. In large part, paramyxoviruses induce IRF3 activation and IFN transcription via the MDA-5 pathway and thus express V proteins that interact with the C terminus of MDA-5 in order to block IFN induction (8). A number of other viral interferon antagonists block IFN production on downstream effectors such as MAVS (e.g., hepatitis C virus NS3/4A [12]) or even IRF3 itself (rotavirus NSP1 [3]). However, there are examples of viral interferon antagonists that specifically inhibit RIG-I-mediated signal transduction. First, several groups have recently shown that influenza A virus NS1 can block RIG-I-mediated IFN induction (14, 24, 27, 28). Furthermore, influenza NS1 binds to RIG-I, likely in an RNA-dependent manner, although this interaction may not encompass the totality of the RIG-I antagonist activity of this protein (28). More recently, HMPV G was shown to bind RIG-I and inhibit IFN (2). Our data indicate that NS2 binds to the N-terminal 229 aa of RIG-I and that the C terminus of RIG-I is dispensable for this interaction. This region includes the two CARD that are essential for interaction with and signal transduction via MAVS and does not bind RNA (44). It has been hypothesized that the recognition of RNA by the helicase domain in the C terminus of RIG-I results in a conformational exchange in the protein, thereby exposing the CARD so that they are accessible for downstream interactions. In addition, the ubiquitylation of the RIG-I CARD by tripartite motif protein 25 (TRIM25) is required for IFN induction (13). Although we do not know whether NS2 interacts directly with the N terminus of RIG-I, we did not detect additional cellular proteins in equimolar amounts nor were we able to detect the presence of RNA bound in NS2-N-RIG immunoprecipitates (data not shown). We are currently dissecting the mechanism by which NS2 inhibits RIG-I-MAVS interaction and the structural determinants necessary for this function. We hypothesize that NS2 directly interacts with the N terminus of RIG-I and inhibits signal transduction through the downstream MAVS activator.
In addition to NS2, the RSV NS1 protein also has IFN antagonist activity (36, 37) (Fig. 1). The mechanism by which NS1 accomplishes this effect is not known, but our data suggest that NS1 inhibits IFN production in a different manner than NS2 since NS1 does not bind RIG-I (Fig. 4) and acts downstream of MAVS (Z. Ling, K. C. Tran, and M. N. Teng, unpublished data). Thus, RSV encodes two distinct IFN antagonist proteins that likely act by different mechanisms, underscoring the importance of inhibiting IFN response in viral infection.
Acknowledgments
We thank Todd Borland for technical assistance and Craig Cameron and Biao He for their support and help with the luciferase assays. We also thank Biao He for critical reading of the manuscript. The gifts of plasmids and reagents from M. Holtzman, T. Fujita, Z. Chen, H.-B. Shu, K. Fitzgerald, T. Maniatis, J. Hiscott, T. Imaizumi, and P. Yeo are gratefully acknowledged. We thank P. Collins for use of the RSV reverse genetics system and the NS deletion viruses.
Footnotes
Published ahead of print on 4 February 2009.
REFERENCES
- 1.Atreya, P. L., M. E. Peeples, and P. L. Collins. 1998. The NS1 protein of human respiratory syncytial virus is a potent inhibitor of minigenome transcription and RNA replication. J. Virol. 721452-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bao, X., T. Liu, Y. Shan, K. Li, R. P. Garofalo, and A. Casola. 2008. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS Pathog. 4e1000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barro, M., and J. T. Patton. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. USA 1024114-4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bitko, V., O. Shulyayeva, B. Mazumder, A. Musiyenko, M. Ramaswamy, D. C. Look, and S. Barik. 2007. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-κB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 811786-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bossert, B., and K. K. Conzelmann. 2002. Respiratory syncytial virus (RSV) nonstructural (NS) proteins as host range determinants: a chimeric bovine RSV with NS genes from human RSV is attenuated in interferon-competent bovine cells. J. Virol. 764287-4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bossert, B., S. Marozin, and K. K. Conzelmann. 2003. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J. Virol. 778661-8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchholz, U. J., S. Finke, and K. K. Conzelmann. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Childs, K., N. Stock, C. Ross, J. Andrejeva, L. Hilton, M. Skinner, R. Randall, and S. Goodbourn. 2006. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed]
- 9.Collins, P. L., R. M. Chanock, and B. R. Murphy. 2001. Respiratory syncytial virus, p. 1443-1485. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott, Williams and Wilkins, Philadelphia, PA.
- 10.Collins, P. L., M. G. Hill, E. Camargo, H. Grosfeld, R. M. Chanock, and B. R. Murphy. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. USA 9211563-11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4491-496. [DOI] [PubMed] [Google Scholar]
- 12.Foy, E., K. Li, R. Sumpter, Jr., Y. M. Loo, C. L. Johnson, C. Wang, P. M. Fish, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA 1022986-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gack, M. U., Y. C. Shin, C. H. Joo, T. Urano, C. Liang, L. Sun, O. Takeuchi, S. Akira, Z. Chen, S. Inoue, and J. U. Jung. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446916-920. [DOI] [PubMed] [Google Scholar]
- 14.Guo, Z., L. M. Chen, H. Zeng, J. A. Gomez, J. Plowden, T. Fujita, J. M. Katz, R. O. Donis, and S. Sambhara. 2007. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 36263-269. [DOI] [PubMed] [Google Scholar]
- 15.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314994-997. [DOI] [PubMed] [Google Scholar]
- 16.Jin, H., H. Zhou, X. Cheng, R. Tang, M. Munoz, and N. Nguyen. 2000. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology 273210-218. [DOI] [PubMed] [Google Scholar]
- 17.Kawai, T., and S. Akira. 2007. Antiviral signaling through pattern recognition receptors. J. Biochem. (Tokyo) 141137-145. [DOI] [PubMed] [Google Scholar]
- 18.Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii, O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6981-988. [DOI] [PubMed] [Google Scholar]
- 19.Kotelkin, A., I. M. Belyakov, L. Yang, J. A. Berzofsky, P. L. Collins, and A. Bukreyev. 2006. The NS2 protein of human respiratory syncytial virus suppresses the cytotoxic T-cell response as a consequence of suppressing the type I interferon response. J. Virol. 805958-5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 182986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ling, Z., K. C. Tran, J. J. Arnold, and M. N. Teng. 2008. Purification and characterization of recombinant human respiratory syncytial virus nonstructural protein NS1. Protein Expr. Purif. 57261-270. [DOI] [PubMed] [Google Scholar]
- 22.Liu, P., M. Jamaluddin, K. Li, R. P. Garofalo, A. Casola, and A. R. Brasier. 2007. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 811401-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo, M. S., R. M. Brazas, and M. J. Holtzman. 2005. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J. Virol. 799315-9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mibayashi, M., L. Martinez-Sobrido, Y. M. Loo, W. B. Cardenas, M. Gale, Jr., and A. Garcia-Sastre. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81514-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munir, S., C. Le Nouen, C. Luongo, U. J. Buchholz, P. L. Collins, and A. Bukreyev. 2008. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J. Virol. 828780-8796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nallagatla, S. R., J. Hwang, R. Toroney, X. Zheng, C. E. Cameron, and P. C. Bevilacqua. 2007. 5′-Triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 3181455-1458. [DOI] [PubMed] [Google Scholar]
- 27.Opitz, B., A. Rejaibi, B. Dauber, J. Eckhard, M. Vinzing, B. Schmeck, S. Hippenstiel, N. Suttorp, and T. Wolff. 2007. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 9930-938. [DOI] [PubMed] [Google Scholar]
- 28.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314997-1001. [DOI] [PubMed] [Google Scholar]
- 29.Ramaswamy, M., L. Shi, M. M. Monick, G. W. Hunninghake, and D. C. Look. 2004. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 30893-900. [DOI] [PubMed] [Google Scholar]
- 30.Ramaswamy, M., L. Shi, S. M. Varga, S. Barik, M. A. Behlke, and D. C. Look. 2005. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed]
- 31.Sato, S., M. Sugiyama, M. Yamamoto, Y. Watanabe, T. Kawai, K. Takeda, and S. Akira. 2003. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappaB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 1714304-4310. [DOI] [PubMed] [Google Scholar]
- 32.Schlender, J., B. Bossert, U. Buchholz, and K. K. Conzelmann. 2000. Bovine respiratory syncytial virus nonstructural proteins NS1 and NS2 cooperatively antagonize alpha/beta interferon-induced antiviral response. J. Virol. 748234-8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seth, R. B., L. Sun, and Z. J. Chen. 2006. Antiviral innate immunity pathways. Cell Res. 16141-147. [DOI] [PubMed] [Google Scholar]
- 34.Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122669-682. [DOI] [PubMed] [Google Scholar]
- 35.Spann, K. M., P. L. Collins, and M. N. Teng. 2003. Genetic recombination during coinfection of two mutants of human respiratory syncytial virus. J. Virol. 7711201-11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spann, K. M., K. C. Tran, B. Chi, R. L. Rabin, and P. L. Collins. 2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J. Virol. 784363-4369. (Erratum, 78:6705, 2005.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spann, K. M., K. C. Tran, and P. L. Collins. 2005. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J. Virol. 795353-5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeuchi, O., and S. Akira. 2008. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2017-22. [DOI] [PubMed] [Google Scholar]
- 39.Teng, M. N., and P. L. Collins. 1999. Altered growth characteristics of recombinant respiratory syncytial viruses which do not produce NS2 protein. J. Virol. 73466-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teng, M. N., and P. L. Collins. 2002. The central conserved cystine noose of the attachment G protein of human respiratory syncytial virus is not required for efficient viral infection in vitro or in vivo. J. Virol. 766164-6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng, M. N., S. S. Whitehead, A. Bermingham, M. St. Claire, W. R. Elkins, B. R. Murphy, and P. L. Collins. 2000. Recombinant respiratory syncytial virus that does not express the NS1 or M2-2 protein is highly attenuated and immunogenic in chimpanzees. J. Virol. 749317-9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitehead, S. S., A. Bukreyev, M. N. Teng, C. Y. Firestone, M. St. Claire, W. R. Elkins, P. L. Collins, and B. R. Murphy. 1999. Recombinant respiratory syncytial virus bearing a deletion of either the NS2 or SH gene is attenuated in chimpanzees. J. Virol. 733438-3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu, L. G., Y. Y. Wang, K. J. Han, L. Y. Li, Z. Zhai, and H. B. Shu. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19727-740. [DOI] [PubMed] [Google Scholar]
- 44.Yoneyama, M., and T. Fujita. 2007. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 18545-551. [DOI] [PubMed] [Google Scholar]
- 45.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5730-737. [DOI] [PubMed] [Google Scholar]