

Abstract
APOBEC3 proteins are important cellular factors that restrict infection by a number of viruses, including human immunodeficiency virus type 1 (HIV-1). Previously, we found that the mouse APOBEC3 (mA3) restricts infection by mouse mammary tumor virus (MMTV) in its natural host. Dendritic cells (DCs) are the first in vivo targets of MMTV infection. In this study, we demonstrate that mA3 expressed in target cells restricts MMTV infection in DCs ex vivo and in vivo. By comparing infection of DCs from mA3+/+ and mA3−/− mice with one-hit viruses, we show that mA3 expression in target cells blocked MMTV infection at a postentry step and acted together with virion-packaged mA3 to inhibit infection. Similar results were obtained upon infection of mouse DCs with HIV-1 cores pseudotyped with vesicular stomatitis virus G protein. In addition, treatment of cells or mice with lipopolysaccharide (LPS) caused increased levels of mA3 expression and rendered them resistant to MMTV infection. Alpha interferon treatment had a similar effect. This LPS-induced resistance to infection was seen only in mA3+/+ mice and not in mA3−/− mice, arguing that mA3 is the major anti-MMTV restriction factor that is induced upon DC maturation. Thus, increasing the levels of this intrinsic antiretroviral factor in vivo can lead to increased levels of restriction because of higher levels of both cell-intrinsic as well as virion-packaged APOBEC3.
It is now well established that proteins belonging to the APOBEC3 (A3) family are potent restriction factors of viruses and retrotransposable elements (6, 16, 46). Several mechanisms have been proposed for the antiviral activity of APOBEC3. Most, if not all, family members have cytidine deaminase (CDA) activity. As for human immunodeficiency virus type 1 (HIV-1) lacking the viral infectivity factor (vif) gene, human APOBEC3G (hA3G) protein is incorporated into budding HIV-1 virions and deaminates cytidine to uridine residues during reverse transcription. This causes hypermutation in the nascent retroviral DNA and restricts infection through increased degradation of the DNA and the generation of nonfunctional proviral genomes (16, 26). This mechanism is counteracted by Vif, which binds and degrades hA3G via the ubiquitin/proteasome pathway in virus-producing cells, thereby preventing it from being packaged (27, 47, 49, 55). A3 proteins also inhibit replication by an undefined CDA-independent mechanism(s) (33, 48). A3 proteins packaged into virions have been shown to inhibit the accumulation of HIV-1 reverse transcription products, plus-strand DNA transfer, and provirus integration, at least in tissue culture cells (18, 24, 29).
In addition to A3 proteins inhibiting infection when packaged into virions, there is mounting evidence that these cellular proteins can restrict incoming virus particles. For example, hA3G expressed in recipient resting CD4+ T cells and mature dendritic cells (mDCs) functions as a postentry, Vif-resistant restriction for HIV infection (7, 39). Several A3 proteins are also expressed at higher levels in cells that restrict HIV-1 infection, such as monocytes and mDCs (36, 37, 39). Indeed, hA3G levels increase during dendritic cell (DC) maturation ex vivo, and treatment of several different cell types with alpha interferon (IFN-α) leads to higher levels of hA3G expression (5, 37, 39, 43, 50).
Unlike humans, whose genome carries seven A3 genes, mice have a single mA3 gene, and although the mouse protein restricts HIV-1 infection and is a functional CDA, the domain organization of this single gene is different from any of the human genes (8, 14, 23). Previously, we showed that mice with targeted deletion of the mA3 gene supported higher levels of infection by the betaretrovirus mouse mammary tumor virus (MMTV) than their wild-type counterparts (35). Moreover, we showed that this restriction was independent of A3 CDA activity. More recently, several groups have demonstrated that mA3 inhibits infection by murine leukemia virus and that this also occurs without cytidine deamination (3, 42). Thus, although both MMTV and murine leukemia virus are infectious in mice, it is likely that endogenous A3 plays a role in limiting infection and thereby pathogenesis.
In vivo, MMTV first infects DCs at the site of infection, which then carry the virus to lymph nodes (9). Infected DCs produce virus and, because they are professional antigen-presenting cells, present the MMTV-encoded superantigen (Sag) to T cells expressing Sag-specific T-cell receptor Vß chains (41). This results in the stimulation of Sag-reactive T cells, proliferation of B and T cells, and subsequent amplification of infection. MMTV's interactions with DCs also induce their maturation and migration, causing increased surface expression of costimulatory molecules and the MMTV entry receptor transferrin receptor 1 (TfR1) (4, 9, 28).
Although MMTV infection of DCs leads to maturation and increased TfR1 expression on the cell surface, MMTV nonetheless infects immature DCs (iDCs) more efficiently than it infects mDCs (51). In this study, we examined whether this preferential replication pattern was due to differential restriction of MMTV by mA3 in iDCs versus that in mDCs. We demonstrate that virion-packaged and DC-intrinsic mA3 function together to restrict MMTV infection ex vivo and in vivo and that this restriction is not due to cytidine deamination. We also show that DC activation and maturation increase mA3 levels in target cells and that this increase results in reduced viral infection ex vivo and in vivo. These studies point to the importance of examining the role of host antiviral mechanisms using natural pathogens and their hosts.
MATERIALS AND METHODS
Molecular constructs.
All the plasmids used were previously described, including V5-tagged mA3 (mA3.V5) (57); hemagglutinin (HA)-tagged mA3 (mA3.HA) (42); rat glucocorticoid receptor RSVGR (30); MMTV molecular clone HYB PRO (45); pENV (10); and lentiviral vector LVTHM, which expresses enhanced green fluorescent protein (EGFP) (53). MMTVΔEnv was created by site-directed mutagenesis of the receptor binding site encoded by the env gene of the HYB PRO clone, as previously described (56), and does not produce infectious virus in the absence of a functional Env-encoding plasmid provided in trans (not shown).
Cell culture.
Bone marrow-derived DCs (BMDCs) from mA3−/− and mA3+/+ mice were generated according to published procedures (25). The DCs were cultured for 8 days at 37°C with 5% CO2 in RPMI 1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.05 mM 2-mercaptoethanol, and 20 ng/ml recombinant murine GM-CSF (Peprotech Inc., Rocky Hill, NJ). DCs were matured by treatment with 100 ng/ml lipopolysaccharide (LPS) (Sigma, Inc., St. Louis, MO) for 24 h. Differentiation into iDCs or mDCs was assessed with flow cytometry by staining cell surface expression with anti-CD40 and -CD86 antibodies (BD Biosciences, Inc.). 293T, NIH 3T3, TRH3 (293T cells that stably express mouse TfR1 [56]), and CGRES6 (CrFK cells stably transfected with pGR102ES, a green fluorescent protein [GFP]-tagged molecular MMTV clone [20]) cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin; the TRH3 and CGRES medium was supplemented with Geneticin (100 μg/ml). 293T, TRH3, and CGRES cells were transfected with V5- or HA-tagged mA3 expression plasmid using Lipofectamine 2000, according to the manufacturer's instructions (Invitrogen, Inc., Carlsbad, CA).
Virus production.
MMTV virions were produced by transient transfection of 293T cells. All transfections were performed using Lipofectamine, according to the manufacturer's instructions (Invitrogen, Inc., Carlsbad, CA). For production of one-hit, replication-incompetent MMTV virions with or without mA3 [ΔEnv(+mA3) or ΔEnv, respectively], 293T cells were cotransfected with the following four plasmids: MMTVΔEnv, pEnv, RSVGR, and mA3.V5 or pcDNA3.1. For GR102ES virions produced by CGRES cells, cells were transfected with RSVGR and mA3.V5 or pcDNA3.1. Twenty-four hours posttransfection, virion production for all cells was induced by the addition of 0.5 μM dexamethasone. Twenty-four hours postinduction, the virus-containing culture supernatants were harvested and DNase treated with 20 U/ml of DNase I (Roche, Inc., Nutley, NJ) for 30 min at 37°C to remove residual plasmid DNA. The supernatants were layered onto a 30% sucrose cushion in phosphate-buffered saline (PBS), and virions were pelleted by centrifugation at 105,000 × g for 1 h. Virus stocks were normalized for infection by Western blotting with anti-MMTV antisera, as previously described (35).
For the generation of LVTHM lentiviral vector stocks, 293T cells were transiently transfected with pLVTHM (EGFP-containing vector plasmid), pCMV-R8.91 and pMD2G-VSVG (helper plasmids), and mA3 expression or control pcDNA3.1 using the CalPhos mammalian transfection kit (BD Biosciences, Palo Alto, CA), according to the manufacturer's instructions. Supernatants containing the pseudovirions were harvested at 48 h posttransfection, filtered through a 0.45-μm filter, and then pooled and concentrated 500-fold using a Centricon Plus-70 100-kDa Ultracel-PL membrane (Millipore).
Purified MMTV and PLVTHM virions were used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis/Western blot analysis to examine mA3 packaging and the relative levels of virion production and to infect TRH3 cells, BMDCs, or mice. Purified lentivirus pseudovirion levels were quantified as follows. RNA was isolated from equal aliquots of virus using the RNeasy minikit (Qiagen, Inc.) according to the manufacturer's instructions. Isolated RNA was treated with DNase I (Qiagen, Inc.) and reverse transcribed with SuperScript III (Invitrogen, Inc.). The cDNA was amplified by the following primers to the GFP gene: 5′-ACGTAAACGGCCACAAGTTC-3′ and 5′-AAGTCGTGCTGCTTCATGTG-3′.
In vitro infection of BMDCs and TRH3 cells.
TRH3 cells and DCs were infected with ΔEnv, ΔEnv(+mA3), GR102ES, GR102ES(+mA3), or lentivirus virions for 24 h. Cells treated with 3 mg/ml of the reverse transcription inhibitor azidothymidine (AZT; Sigma, Inc.) at 37°C for 2 h prior to and during infection served as controls. Infectivity titrations of lentivirus vector stocks were performed on NIH 3T3 cells as follows. Cells were pretreated with 2 μg/ml Polybrene 1 h prior to infection, and the Polybrene was then aspirated. Then, 10-fold serial dilutions of diluted vector stock were applied, followed by incubation at 37°C for 1 h, after which medium was added to each plate. mA3−/− and mA3+/+ DCs were infected by the spinoculation method, as previously described (9). In some experiments, BMDCs from mA3+/+, mA3−/−, or C57BL/6 mice were matured with 100 ng/ml of LPS or incubated with the indicated amounts of IFN-α for 24 h prior to infection.
Infectivity assays.
For measurement of viral DNA in infected cells, DNA was isolated according to the manufacturer's protocol (DNeasy kit; Qiagen). DNA was used for real-time quantitative PCR (RT-qPCR) to detect integrated proviruses, as previously described, using primers specific to the MMTV long terminal repeat and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (9). Fluorescence-activated cell sorting (FACS) analysis was also used to detect infection by GR102ES and the lentivirus pseudovirions using FACSCalibur; data are presented as the percentage of GFP-positive (GFP+) cells or as relative infectivity levels normalized to viral RNA levels. For lentivirus infection of NIH 3T3 cells, infected plates of cells were observed under UV illumination 2 days postinfection, and green fluorescent cells (individuals or small groups) were counted. Infectivity titers were calculated from the number of fluorescent cells/colonies corrected for the dilution factors and then normalized to viral RNA levels. All infectivity assays were done in triplicate; each assay was performed two to four times with similar results.
Western blots.
Western blots of virus preparations were probed with anti-V5 or anti-HA (Invitrogen, Inc.) and anti-total MMTV or anti-p27 (capsid) (National Cancer Institute, Biochemical Carcinogenesis Branch Repository, Bethesda, MD). The species-appropriate horseradish peroxidase-conjugated secondary antibody was used, followed by detection with ECL reagents (Amersham Biosciences, Inc.). In some experiments (see Fig. 5), the Western blots were analyzed using anti-HA monoclonal antibody (EMSCO/VWR), followed by analysis using the Odyssey infrared imaging system (LI-COR Biosciences).
FIG. 5.

IFN-α treatment results in increased mA3 expression and decreased MMTV infection. (A and B) BMDCs from C57BL/6 (mA3+/+) and mA3−/− mice were grown in the presence of increasing amounts of 0, 10, or 100 U/ml IFN-α for 24 h, and then RNA was isolated and analyzed by RT-qPCR for mA3 expression (A) or the cells were infected with GR102ES virus for 24 h and DNA was isolated and analyzed for RT-qPCR for viral DNA (B). *, P ≤ 0.001; **, P ≤ 0.005; ***, P ≤ 0.01. P values are significant for comparisons of mA3+/+ and mA3−/− cells at each IFN-α concentration. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test.
In vivo infection of mice.
The mA3+/+ and mA3−/− mice were previously described (35). Toll-like receptor 4 (TLR4)−/− mice with a C57BL/6 background were developed by Hoshino et al. (19) and were a kind gift from Douglas Golenbock. Mice were housed according to the policies of the Institutional Animal Care and Use Committee of the University of Pennsylvania. In vivo footpad infectivity assays of mA3−/− and mA3+/+ mice were performed with ΔEnv or GR102ES virions, as indicated. Briefly, five mice of each genotype received subcutaneous footpad injections of virus. Injections also were performed in the presence of 3 mg of AZT (Sigma, Co., St. Louis, MO), which was administered intraperitoneally before injection, as described previously (17). Twenty-four hours after infection, the mice were sacrificed, and the draining lymph nodes from each group were harvested and pooled for DC purification. DCs were purified from the lymph nodes of infected mice using the CD11c isolation kit (Miltenyi Biotec Inc.). The purity of the populations was determined by FACS analysis using allophycocyanin-conjugated anti-CD11c (clone HL3) and was always >98%. Following DC purification, DNA was isolated and used for RT-qPCR to detect the integrated viral sequence.
LPS treatment of mice.
Three mice from each genotype (mA3−/−, mA3+/+, and TLR4−/− mice, all with a C57BL/6 background) were pretreated with 1 μg LPS. Twenty-four hours after LPS treatment, mice were inoculated subcutaneously with GR102ES MMTV virions produced by CGRES cells. Twenty-four hours after infection, the mice were sacrificed, and the lymph nodes from each group were harvested and pooled for DC purification. DCs were purified from the lymph nodes of mA3−/−-, mA3+/+-, and TLR4−/−-infected mice using the CD11c isolation kit (Miltenyi Biotec Inc.). Following DC purification, DNA was isolated and used for RT-qPCR to detect integrated viral sequence. In addition, RNA was isolated, and cDNA was synthesized and used to detect mA3 expression.
Viral sequence editing assay.
DNA was isolated from the BMDCs of mA3−/− and mA3+/+ mice infected with GR102ES and GR102ES(+mA3) virions using the DNeasy tissue kit (Qiagen, Inc.). Following DNA isolation, an ∼700-bp fragment was amplified by PCR using one primer specific to the GFP gene and the other to the MMTV long terminal repeat and cloned into pCR2.1-TOPO vector (Invitrogen, Inc.). At least 15 clones from each group of mice infected with the two different viruses were sequenced. Sequences were aligned using the ClustalW program (http://www.ebi.ac.uk/Tools/clustalw2/index.html).
Viral reverse transcript levels.
TRH3 cells were transfected with mA3.HA or the empty vector using Lipofectamine 2000, by following the manufacturer's recommendation (Invitrogen, Inc., Carlsbad, CA). Twenty-four hours after transfection, the cells were harvested and used either to assay for transfection efficiency by Western blotting and flow cytometry (FACS) or to assay for the formation of reverse transcription products. Transfection efficiency by FACS was measured by intracellular staining of permeabilized cells with anti-HA monoclonal antibody followed by anti-rabbit secondary antibody conjugated to allophycocyanin; mA3-positive (mA3+) cells were analyzed using FACSCalibur. For the analysis of viral reverse transcript formation, viral infectivity assays were performed by following a previously described protocol (18) and modified as follows. Transfected TRH3 cells were washed with ice-cold PBS, resuspended, and aliquoted into tubes for infection. MMTV virions were incubated with cells at 4°C for 2 h and shaken to allow synchronous virus binding. Following incubation, cells were washed three times with ice-cold PBS, resuspended in cold culture media, and incubated at 37°C with 5% CO2. Aliquots of cells (200 μl) were taken at different time points (0, 2, 4, 6, and 24 h), washed with PBS, and frozen until required for DNA extraction. Control cells were first treated with AZT 2 h prior to infection with MMTV virions. The DNAs were analyzed by RT-qPCR using the following primers: early reverse transcripts (ERT)/strong-stop DNA, 5′-CGTGTGTTTGTGTCTGTTCG-3′ and 5′-GACCCTCTGGAAAGTGAAGG-3′; late reverse transcripts (LRT), 5′-CCAACGAGAAGCGCGATCAC-3′ and 5′-GGACTGTTGCAAGTTTACAC-3′; and LRT/integrated DNA, 5′-CGTGAAAGACTCGCCAGAGCTA-3′ and 5′-TAATGTTCTATTAGTCCAGCCACTGT-3′. All results were normalized to those of GAPDH, as previously described (35). All assays were done in triplicate.
Reverse transcription-RT-qPCR to measure mA3 levels.
Total RNA was isolated from the BMDCs of mA3−/− and mA3+/+ mice using the RNeasy minikit (Qiagen, Inc.), according to the manufacturer's instructions. Isolated RNA was treated with DNase I (Qiagen, Inc.) and reverse transcribed with SuperScript III (Invitrogen, Inc.). The cDNA was amplified with mA3-exon 6/-exon 7-specific primers as follows: 5′-GAATGGACCCGCTAAGTGAA-3′ and 5′-ACAGGCGGGAGGTGTAGATA-3′; the amplification was normalized to that of GAPDH.
RESULTS
Both cell-intrinsic mA3 and virion-packaged mA3 restrict MMTV infection in tissue culture cells and BMDCs.
To determine if both virion-packaged and cell-intrinsic mA3 restrict MMTV infection, we first examined TRH3 cells, which are 293T cells that express the mouse TfR1. 293T cells do not express hA3G, and the MMTV produced in these cells is fully infectious (35). These cells were transfected with a mA3 expression vector (Fig. 1A) and then infected with a one-hit virus (ΔEnv) or an infection-competent, GFP-tagged virus, GR102ES, lacking or containing mA3 (Fig. 1B). Infection was analyzed by RT-qPCR (Fig. 1C) or FACS (Fig. 1D). With either virus, the lowest levels of infection were seen in TRH3 cells expressing mA3 and infected with MMTV containing mA3; conversely, TRH3 cells transfected with empty vector and infected with virus lacking mA3 showed the highest infection levels. Intermediate levels of infection were seen in mA3-expressing cells infected with virions lacking mA3 or in vector-transfected cells infected with virions containing mA3.
FIG. 1.

MMTV infection is restricted by cell-intrinsic and virion-packaged mA3. (A) Western blotting of cell extracts from mA3-transfected TRH3 cells probed with anti-HA or anti-β-actin antisera. The first two panels are the cells infected with the mA3− virions (ΔEnv), and the second two panels are the cells infected with the mA3+ virions [(ΔEnv(+mA3)]. (B) Western blotting of MMTV virions used in the study. The one-hit ΔEnv virus was made by transient transfection of 293T cells, with V5-tagged mA3 (+) or pcDNA3.1 (−). The pGR102ES virus was produced from CGRES6 cells transiently transfected with an expression vector containing HA-tagged mA3 (+) or pcDNA3.1 (−). Blots were probed with anti-V5, stripped, and reprobed with goat anti-MMTV antisera (top) or probed with anti-HA and reprobed with goat anti-MMTV Gag (p27) (bottom). (C) TRH3 cells were transfected with mA3 (mA3+) or pcDNA3.1 (mA3−) and then infected with ΔEnv or ΔEnv(+mA3) virions. Infection was analyzed by RT-qPCR; the relative infection levels are shown. Each bar represents the average of three wells. P values of ≤0.001 are significant for all comparisons [mA3+ for ΔEnv versus mA3− for ΔEnv, mA3+ for MMTVΔEnv(+mA3) versus mA3− for ΔEnv(+mA3), mA3+ for ΔEnv versus mA3+ for ΔEnv(+mA3), mA3− for ΔEnv versus mA3− for ΔEnv(+mA3)]. This experiment was performed three times with similar results. (D) TRH3 cells were transfected with mA3 (mA3+) or pcDNA3.1 (mA3−) and then infected with GR102ES virus with or without packaged mA3. FACS analysis was performed to determine the percentage of GFP+ cells. Each bar represents the average of three wells. *, P ≤ 0.01; **, P ≤ 0.001; ***, P ≤ 0.03. This experiment was performed twice with similar results. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test. α, anti-.
Although these experiments indicated that cell-intrinsic mA3 plays a role in restricting MMTV infection, they do not necessarily reflect the restriction that occurs when physiological levels of mA3 are expressed in natural target cells. We thus infected BMDCs derived from mA3−/− and mA3+/+ mice with ΔEnv or ΔEnv(+mA3) (Fig. 2A) or with GFP-tagged (Fig. 2B) viruses. Infection levels were analyzed by RT-qPCR or FACS, respectively, and AZT treatment served as the negative control. Results similar to those observed for transfected cells were seen, with the highest levels of infection detected in the complete absence of mA3 and the lowest levels detected when both cells and virus contained mA3 (Fig. 2). Interestingly, in transfected TRH3 cells and cultured BMDCs, cell-intrinsic mA3 was more effective at inhibiting MMTV infection than virion-packaged mA3 (mA3+ cells infected with virus lacking mA3 versus mA3-negative [mA3−] cells infected with virus containing mA3) (Fig. 1 and 2). Additionally, the difference in infection between mA3+ and mA3− cells and virions was more pronounced with the one-hit virus ΔEnv (Fig. 1A and 2A) than with the replication-competent GR102ES virus (Fig. 1B and 2B); this probably reflects the loss of mA3 in mA3+ virions that occurs upon infection of mA3− cells and, conversely, the acquisition of mA3 into mA3− virions after infection of mA3+ cells.
FIG. 2.

MMTV infection of BMDCs is restricted by cell-intrinsic and virus-packaged mA3 ex vivo. (A) BMDCs from mA3+/+ and mA3−/− mice were infected ex vivo with ΔEnv and ΔEnv(+mA3) viruses. Infection was analyzed by RT-qPCR; the relative infection levels are shown. Cells were also pretreated with AZT to block infection. Each bar represents the average of three independent BMDC cultures. P values of ≤0.001 are significant for all comparisons. (B) mA3+/+ and mA3−/− BMDCs infected with GR102ES virus. Infection was analyzed by FACS for GFP+ cells. Each bar represents the average of three independent cultures. P values of ≤0.001 are significant for all comparisons, except where marked with asterisks. *, P ≤ 0.02; **, P ≤ 0.003. This experiment was performed six times with similar results. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test.
To determine if intrinsic mA3 also inhibited HIV-1 infection, we used vesicular stomatitis virus G protein (VSV-G)-pseudotyped, GFP-tagged HIV virions to infect NIH 3T3 mouse cells and mA3−/− and mA3+/+ DCs. Infection levels were measured by counting GFP+ colonies (NIH 3T3 cells) or by using FACS analysis (DCs) to determine the percentage of GFP+ cells. As has been previously shown, HIV-1 efficiently packaged mA3 (Fig. 3A) and virions containing mA3-infected NIH 3T3 cells at much lower levels than did virions lacking this protein (Fig. 3B). As seen with MMTV, the highest levels of infection were seen in mA3−/− cells infected with pseudovirions produced in the absence of mA3, and the lowest levels were seen in mA3+/+ cells infected with mA3-packaged virions (Fig. 3C). Thus, the ability of DC-intrinsic mA3 to restrict incoming virus is not limited to MMTV but also occurs with HIV-1.
FIG. 3.

Cell-intrinsic mA3 restricts HIV-1 infection in murine DCs. (A) Virion-packaged mA3 was analyzed by Western blotting, using anti-HA antibody and the Odyssey infrared imaging system. Symbols: −, virions made with empty vector; +, virions made with mA3 expression vector. (B and C) NIH 3T3 cells (B) or DCs from mA3+/+ and mA3−/− mice (C) were infected with VSV-G-pseudotyped lentiviral constructs with or without packaged mA3 for 48 h (NIH 3T3 cells) or 24 h (DCs). The number of GFP+ colonies (B) or FACS analysis (C) to determine the percentage of GFP+ cells was used to measure infection levels. Bars represent relative infection levels normalized to viral RNA levels present in the infecting particles. For DC infection, filled bars represent mA3+/+ DCs, and open bars represent mA3−/− DCs. DCs pretreated with AZT showed no infection. P values of ≤0.001 are significant for all comparisons. This experiment was performed twice with similar results. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test.
Cell-intrinsic restriction of MMTV infection in mDCs is largely dependent on mA3 expression levels.
Work by others demonstrated that hA3G is expressed at higher levels in human mDCs than in iDCs (39). We therefore examined mA3 expression in iDCs versus that in mDCs; maturation was induced by LPS treatment for 24 h and was assessed by analyzing the expression of costimulatory molecules CD40 and CD86 (Fig. 4A). DC maturation caused increased mA3 RNA expression (Fig. 4B). We then tested whether the increased mA3 expression in mDCs resulted in lower levels of MMTV infection. BMDCs from both mA3+/+ and mA3−/− mice were treated with LPS, stained with anti-CD40 and -CD86 antibodies, and analyzed by FACS. Both mA3+/+ and mA3−/− BMDCs showed equivalent amounts of increased expression of these maturation markers (Fig. 4A and not shown). The mA3+/+ and mA3−/− iDCs and mDCs were then infected with ΔEnv and ΔEnv(+mA3) (Fig. 4C) or GR102ES and GR102ES(+mA3) (not shown). For the one-hit ΔEnv virions, mDCs from mA3+/+ mice showed significantly lower levels of infection than did iDCs, independent of whether the incoming virions lacked or contained mA3 (Fig. 4C). Similar results were seen with the replication-competent GR102ES and GR102ES(+mA3) virions, although with the GR102ES(+mA3) virions, where the infection levels in mA3+/+ iDCs and mDCs were near background levels, this difference was not statistically significant (not shown). For both sets of viruses, there was no difference in the level of infection of mDCs and iDCs from mA3−/− mice, independent of whether mA3 was packaged in the virion. However, virions containing mA3 were less infectious than those lacking mA3 in mA3−/− mDCs and iDCs.
FIG. 4.

MMTV more highly infects iDCs and mDCs from mA3−/− mice than those from mA3+/+ mice. (A) BMDCs were treated with LPS for 24 h and then stained with anti-CD40 or anti-CD86 and anti-CD11c antibodies and analyzed by FACS. A representative plot for mA3+/+ BMDCs is shown. The table shows the percentage of CD40+ and CD86+ cells for triplicate samples of mA3−/− and mA3+/+ BMDCs. (B) RNA from the untreated or LPS-matured BMDCs was analyzed by reverse-transcribed RT-qPCR, using primers specific for mA3 exons 6 and 7. The relative levels normalized to those of GAPDH primers are shown. (C) BMDCs from mA3+/+ and mA3−/− mice were grown in the presence or absence of LPS for 24 h and then infected with ΔEnv or ΔEnv(+mA3). RT-qPCR analysis was carried out with DNA from immature (untreated) or matured (LPS-treated) BMDCs infected with ΔEnv mA3− and mA3+ virions; the relative infection levels are shown. Each bar represents the average of three independent BMDC cultures. *, P ≤ 0.001; **, P ≤ 0.1. Cells were also pretreated with AZT to block infection. This experiment was performed three times with similar results. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test.
LPS signals through the innate immune receptor TLR4 and causes both DC maturation and the release of cytokines (1), including IFN-α, which is known to increase hA3G expression. To determine whether IFN-α also caused increased mA3-mediated virus restriction, BMDCs from mA3−/− or C57BL/6 mice (the background strain of the mA3−/− mice) (21) were treated for 24 h with 10 or 100 units/ml IFN-α prior to infection with GR102ES virus. This treatment increased mA3 RNA levels in the C57BL/6 mice (Fig. 5A) and, at the same time, inhibited MMTV infection; at the highest concentration used (100 units), there was a twofold decrease in infection levels (Fig. 5B). In contrast, IFN-α had no effect on the ability of virus to infect mA3−/− DCs. These data suggest that LPS-mediated restriction of MMTV infection is due, at least in part, to the effects of IFN-α on mA3 expression.
mA3 caused no G-to-A transitions in murine DCs infected by MMTV but acts at an early stage of infection.
APOBEC3 proteins are CDAs, but whether this enzymatic activity is required for restriction of virus replication is unclear. Although hA3G hypermutates Vif-deficient HIV-1 virions on minus-strand DNA during reverse transcription in many cell types, in primary human CD4+ T cells, hA3G caused little or no deamination (7). Similarly, hA3G with a mutated CDA active site inhibits HIV-1 and MMTV infection (35), although this could be a function of protein overexpression in transfected cells (44). We therefore determined if mA3 induces G-to-A transitions in MMTV viral DNA in infected DCs expressing physiological levels of mA3. DNA from mA3−/− and mA3+/+ BMDCs infected with the GR102ES or GR102ES(+mA3) viruses was isolated at 24 h postinfection, PCR amplified, cloned, sequenced, and analyzed for mutations. No significant G-to-A hypermutation was observed in mA3+/+ or mA3−/− DCs infected with either virus (Table 1). This observation is in line with our previous report that mA3 does not cause deamination of MMTV virions in vivo (35).
TABLE 1.
Base changes detected in LTR sequences cloned from infected mA3−/− and mA3+/+ BMDCs
| Infected DC | Virusa | No. of clones | No. of base pairsb | No. of indicated base changes
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G→A | G→C | G→T | C→A | C→T | C→G | T→A | T→G | T→C | A→T | A→C | A→G | ||||
| mA3−/− | MMTV | 17 | 9,261 | 2 | |||||||||||
| MMTV(+mA3) | 17 | 10,580 | 3 | 1 | 1 | 3 | |||||||||
| mA3+/+ | MMTV | 19 | 10,198 | 1 | |||||||||||
| MMTV(+mA3) | 15 | 7,623 | 1 | 1 | 3 | ||||||||||
MMTV(+mA3), MMTV virions with mA3.
Indicates total number of base pairs sequenced.
To examine the step at which intrinsic mA3 acts, we transiently transfected TRH3 cells with an mA3 expression vector, infected them with GR102ES, and then used RT-qPCR to examine different reverse-transcribed products at different times after infection, as has previously been done for hA3 proteins and HIV-1 (18). Approximately 30% of the TRH3 cells expressed mA3, as determined by intracellular staining with anti-mA3 antibodies and FACS analysis (Fig. 6A). ERT (strong-stop DNA) were diminished in the mA3-expressing cultures as early as 2 h after infection (Fig. 6B); this diminution persisted through 24 h and was also seen in the LRT and integrated DNA (Fig. 6C and D). Similar results were obtained when we analyzed reverse transcripts in TRH3 cells infected with mA3− and mA3+ virus particles (C. M. Okeoma and S. R. Ross, unpublished data). Thus, it appears that cell-intrinsic mA3 as well as virion-packaged mA3 act at a step prior to or during early reverse transcription.
FIG. 6.

Cell-intrinsic mA3 inhibits ERT formation. TRH3 cells were transfected with mA3 (+A3) or with empty vector (−A3). (A) Twenty-four hours after transfection, aliquots of cells were taken, intracellularly stained for mA3, and analyzed by FACS, or extracts were made and analyzed by Western blotting (inset). (B to D) The mA3-transfected cells were infected with GR102ES, and at different time points (0, 2, 4, 6, and 24 h) after infection, aliquots of cells were taken for DNA isolation. RT-qPCR analyses were performed using primers that measure strong-stop DNA/ERT (B), LRT (C), or products from both LRT and integrated proviruses (D); all were normalized to those of GAPDH. The relative levels of PCR product are shown. Solid line, mA3+ cells; dashed line, mA3− cells. All error bars represent standard deviations.
Both cell-intrinsic and virion-packaged mA3 restrict MMTV infection in vivo.
To determine whether intrinsic mA3 expression in DCs also restricts MMTV infection in vivo, we infected mA3−/− and mA3+/+ mice by subcutaneous inoculation with ΔEnv virions containing or lacking mA3. Mice were sacrificed 24 h after the infection, and DCs purified from draining lymph nodes were analyzed for MMTV, using RT-qPCR as the readout. As was seen with the cultured BMDCs, we observed that cell-intrinsic and virion-packaged mA3 contributed to the inhibition of DC infection by MMTV in vivo (Fig. 7). Similar results were seen with the replication-competent GR102ES virus (not shown). As was seen ex vivo, cell-intrinsic expression of mA3 appeared to play a more dominant role in restricting infection than did virion-packaged mA3.
FIG. 7.

Cell-intrinsic and virion-packaged mA3 restricts MMTV infection in vivo. Five mA3+/+ and five mA3−/− mice were infected with ΔEnv and ΔEnv(+mA3), and DCs from their draining lymph nodes were pooled and analyzed for infection. Infection was analyzed by RT-qPCR; the relative infection levels are shown. Filled bars, mA3+/+ iDCs; open bars, mA3−/− iDCs. P values of ≤0.001 are significant for all comparisons. Mice pretreated with AZT showed no infection. This experiment was performed twice with similar results. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test.
Increasing mA3 expression in vivo reduces virus infection.
It has been suggested that increasing A3 expression in HIV-1-infected individuals could be used as an antiviral therapy (11, 22, 31, 34, 40). As we showed in Fig. 4, LPS treatment of DCs resulted in A3-dependent restriction of MMTV infection. To determine if this was also the case in vivo, mA3−/− and mA3+/+ mice were injected subcutaneously with LPS and, 24 h later, inoculated at the same site with GR102ES. Twenty-four hours after infection, the mice were sacrificed, and DNA and RNA isolated from DCs purified from the draining lymph nodes were analyzed by RT-qPCR for viral DNA and mA3 RNA levels, respectively. As a control for LPS stimulation, we also treated TLR4−/− mice, which are resistant to LPS treatment because they have targeted deletion of the cellular receptor for this bacterial ligand (19). LPS treatment increased A3 RNA levels in vivo (Fig. 8A), and correspondingly, DCs from mA3+/+ mice were more resistant to MMTV infection than those from untreated mA3+/+ mice (Fig. 8B). In contrast, DC infection of either mA3−/− or TLR4−/− mice was not affected by LPS treatment (Fig. 8B); infection of mA3−/− mice was highest in all cases. These data thus show that the treatment of mice with agents that increase cytokine production in vivo renders them more resistant to MMTV infection, primarily through increased A3 expression.
FIG. 8.

Increased mA3 expression in vivo results in reduced MMTV infection. Three mice of each genotype (mA3−/−, mA3+/+, and TLR4−/−), were pretreated with 1 μg LPS and, 24 h later, inoculated subcutaneously with GR102ES virions. Twenty-four hours after infection, the mice were sacrificed, and DNA and RNA were isolated from DCs isolated from pooled lymph nodes. (A) Reverse-transcribed RT-qPCR analysis to measure mA3 RNA levels. (B) RT-qPCR analysis to detect integrated viral sequences. Open bars, untreated mice; filled bars, LPS-treated mice. *, P ≤ 0.1; **, P ≤ 0.01. This experiment was performed twice with similar results. All error bars represent standard deviations. The statistical significance of differences between groups was tested using paired two-tailed Student's t test.
DISCUSSION
A3 proteins are potent retrovirus restriction factors which have been shown to inhibit infection by a number of viruses, most notably HIV-1. hA3G and human APOBEC3F (hA3F) inhibition of HIV-1 occurs through two mechanisms. In the absence of Vif, these hA3 proteins are packaged into viral particles and inhibit infection via CDA of the reverse-transcribed DNA and other more poorly defined means. In addition, cell-intrinsic hA3G and, to a lesser extent, hA3F can inhibit infection in DCs and resting T cells. However, little is known about which of these mechanisms is critical to the antiviral properties of A3 proteins in vivo.
Here, we showed that increasing A3 levels in vivo results in greater A3-mediated restriction of virus infection. We showed previously that MMTV infection of mA3−/− mice was dramatically increased compared to that of their wild-type littermates (35). Since MMTV depends on DCs to initiate infection in vivo (9) and little was known about mA3 expression in murine DCs, we first determined that mA3 expression levels increased upon treatment with LPS. LPS induces DC maturation and cytokine production, notably of type I IFNs, which are inducers of hA3G transcription, as well as of other retroviral restriction factors such as TRIM5α and tetherin (1, 32). The increase in mouse A3 levels upon LPS-induced DC activation and maturation or IFN-α treatment was similar to that observed by others for hA3G (5, 37, 39, 43, 50) and supports previous studies demonstrating that mDCs are less infected by MMTV than iDCs (51). Interestingly, most if not all of the antiviral effect induced by LPS was the result of mA3 restriction, since in mA3−/− DCs, LPS treatment had little or no effect on ex vivo (Fig. 4) or in vivo infection (Fig. 8). Thus, at least in murine DCs, mA3 and not other cytokine-induced factors such as tetherin seems to be the major anti-MMTV restriction factor induced upon their maturation.
We also examined the relative importance of virion-packaged and cell-intrinsic mA3 in restriction of MMTV infection using three different experimental systems. First, we demonstrated that mA3-transfected 293T cells, which do not express endogenous A3 proteins, restricted MMTV infection compared to their untransfected counterparts. More importantly, BMDCs from mA3+/+ mice expressing physiologically relevant levels of mA3 showed much lower levels of infection than those from mA3−/− mice. Finally and most importantly, we observed that infection of DCs was higher in mA3−/− mice than in mA3+/+ mice, demonstrating that cell-intrinsic expression of mA3 restricts virus infection in vivo. In all three cases, this restriction occurred whether the incoming particle contained or was deficient in packaged mA3. Interestingly, in all the experimental systems, cell-intrinsic mA3 inhibited MMTV infection to a greater extent than did packaged mA3 with either one-hit or replication-competent viruses.
Strikingly, packaged mA3 appeared to synergize with cell-intrinsic mA3 to decrease virus infection in most experiments. For example, in vivo infection of mA3+/+ mice was decreased by more than fourfold when virions contained mA3; in contrast, mA3 in virions decreased infection only ∼1.4-fold in mA3−/− mice (Fig. 8). This suggests that cell-intrinsic mA3 accesses the reverse transcription complex in incoming virions. Interestingly, packaged mA3 more potently restricted MMTV and HIV-1 infection in mA3+ cells than in mA3− cells in vitro, ex vivo, and in vivo; this direct comparison of the relative inhibitory effects of packaged versus cell-intrinsic A3 has not been previously described. CDAs like hA3G and mA3 are believed to function as dimers or tetramers, and one recent study by Wedekind and colleagues (52) suggested that hA3G self-associates into higher-order structures (13, 48, 52). Thus, mA3 that is already present in MMTV cores may “attract” cell-intrinsic mA3 through self-association, and if dimers, tetramers, or higher-order mA3 structures are more effective inhibitors of, for example, reverse transcription, this would explain the enhanced activity of virion-packaged mA3 in mA3+ cells. How cell-intrinsic mA3 accesses viral cores in infected cells and inhibits reverse transcription is currently under investigation.
We did not observe editing of proviral DNA when mA3 was packaged into virions or when intrinsically expressed in DCs (Table 1) (35). Instead, we found that intrinsic mA3 exerts its effects at an early step of virus infection, since even strong-stop, ERT levels were reduced (Fig. 6). This supports previous observations that both T cell- and DC-intrinsic expression of hA3G and hA3F inhibited HIV-1 infection but resulted in only low-level hypermutation of LRT (7, 39, 39). A number of in vitro studies have suggested that A3 proteins in particles can inhibit binding of the 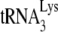 primer to the viral RNA or accumulation of HIV-1 reverse transcription products (2, 12, 18, 21, 24, 29). Whether mA3 inhibition of MMTV infection, either in the intrinsic or virion-packaged form, is preventing reverse transcriptase processivity or functions at an earlier step is currently under investigation.
primer to the viral RNA or accumulation of HIV-1 reverse transcription products (2, 12, 18, 21, 24, 29). Whether mA3 inhibition of MMTV infection, either in the intrinsic or virion-packaged form, is preventing reverse transcriptase processivity or functions at an earlier step is currently under investigation.
As is believed to be the case for HIV-1, DCs are critical early targets for MMTV infection (9, 54). Expression of A3 in DCs may play a significant role in restriction of infection because they are the first cells to be infected by retroviruses like HIV and MMTV in vivo. Increased levels of cell-intrinsic A3 would render the host cell nonpermissive to initial infection and also increase the level of virion-packaged A3 protein. Thus, treatments that increase A3 expression would have a dual effect on suppression of virus infection in DCs. Whether this would also be the case for other lymphoid targets of MMTV infection such as B and T cells in mice is currently under investigation. In addition, we can test whether long-term administration of agents that increase A3 expression, such as IFN-α, will suppress viral loads in chronically infected mice.
While mice have a single A3 gene in contrast to humans, who have seven genes, these model studies suggest that increasing A3 expression in vivo has potential as an antiretroviral therapy, particularly because hA3G and hA3F levels are induced by IFN-α and other factors in many of the cell types that are targets for HIV-1 infection (5, 37, 39, 43, 50). Because A3 proteins are CDAs, it has been speculated that their misexpression could lead to increased genomic instability or even cancer (15, 38), although there is, at present, no direct evidence that this occurs in mammalian cells. The mouse thus also provides a model system for testing whether increasing A3 levels as a means of antiretroviral therapy does indeed lead to genomic changes in vivo.
Acknowledgments
We thank Stacy Hultine for technical assistance. The TLR4−/− mice were a kind gift from Doug Golenbock.
This research was supported by a grant from the Penn Center for AIDS Research (CFAR), an NIH-funded program (grant P30 AI 045008), and an NIH Center grant (grant P50 GMO82250-01). C.M.O. was supported by PHS training grant T32-CA-009140.
We have no conflicting financial interests.
Footnotes
Published ahead of print on 19 January 2009.
REFERENCES
- 1.Akira, S. 2006. TLR signaling. Curr. Top. Microbiol. Immunol. 341-16. [DOI] [PubMed] [Google Scholar]
- 1a.Asaoka, K., K. Ikeda, T. Hishinuma, K. Horie-Inoue, S. Takeda, and S. Inoue. 2005. A retrovirus restriction factor TRIM5alpha is transcriptionally regulated by interferons. Biochem. Biophys. Res. Commun. 3381950-1956. [DOI] [PubMed] [Google Scholar]
- 2.Bishop, K. N., R. K. Holmes, and M. H. Malim. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 808450-8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Browne, E. P., and D. R. Littman. 2008. Species specific restriction of Apobec3 mediated hypermutation. J. Virol. 821305-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burzyn, D., J. C. Rassa, D. Kim, I. Nepomnaschy, S. R. Ross, and I. Piazzon. 2004. Toll-like receptor 4-dependent activation of dendritic cells by a retrovirus. J. Virol. 78576-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, K., J. Huang, C. Zhang, S. Huang, G. Nunnari, F. X. Wang, X. Tong, L. Gao, K. Nikisher, and H. Zhang. 2006. Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T cells. J. Virol. 807645-7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu, Y. L., and W. C. Greene. 2006. Multifaceted antiviral actions of APOBEC3 cytidine deaminases. Trends Immunol. 27291-297. [DOI] [PubMed] [Google Scholar]
- 7.Chiu, Y. L., V. B. Soros, J. F. Kreisberg, K. Stopak, W. Yonemoto, and W. C. Greene. 2005. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435108-114. [DOI] [PubMed] [Google Scholar]
- 8.Conticello, S. G., C. J. Thomas, S. K. Petersen-Mahrt, and M. S. Neuberger. 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 22367-377. [DOI] [PubMed] [Google Scholar]
- 9.Courreges, M. C., D. Burzyn, I. Nepomnaschy, I. Piazzon, and S. R. Ross. 2007. Critical role of dendritic cells in mouse mammary tumor virus in vivo infection. J. Virol. 813769-3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golovkina, T. V., J. L. Dzuris, B. van den Hoogen, A. B. Jaffe, P. C. Wright, S. M. Cofer, and S. R. Ross. 1998. A novel membrane protein is a mouse mammary tumor virus receptor. J. Virol. 723066-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greene, W. C., Z. Debyser, Y. Ikeda, E. O. Freed, E. Stephens, W. Yonemoto, R. W. Buckheit, J. A. Este, and T. Cihlar. 2008. Novel targets for HIV therapy. Antivir. Res. 80251-265. [DOI] [PubMed] [Google Scholar]
-
12.Guo, F., S. Cen, M. Niu, Y. Yang, R. J. Gorelick, and L. Kleiman. 2007. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits
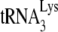 annealing to viral RNA. J. Virol. 8111322-11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
annealing to viral RNA. J. Virol. 8111322-11331. [DOI] [PMC free article] [PubMed] [Google Scholar] - 13.Hakata, Y., and N. R. Landau. 2006. Reversed functional organization of mouse and human APOBEC3 cytidine deaminase domains. J. Biol. Chem. 28136624-36631. [DOI] [PubMed] [Google Scholar]
- 14.Harris, R. S., and M. T. Liddament. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4868-877. [DOI] [PubMed] [Google Scholar]
- 15.Harris, R. S., S. K. Petersen-Mahrt, and M. S. Neuberger. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 101247-1253. [DOI] [PubMed] [Google Scholar]
- 16.Harris, R. S., A. M. Sheehy, H. M. Craig, M. H. Malim, and M. S. Neuberger. 2003. DNA deamination: not just a trigger for antibody diversification but also a mechanism for defense against retroviruses. Nat. Immunol. 4641-643. [DOI] [PubMed] [Google Scholar]
- 17.Held, W., G. A. Waanders, H. Acha-Orbea, and H. R. MacDonald. 1994. Reverse transcriptase-dependent and -independent phases of infection with mouse mammary tumor virus: implications for superantigen function. J. Exp. Med. 180:2347-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmes, R. K., F. A. Koning, K. N. Bishop, and M. H. Malim. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: comparisons with APOBEC3G. J. Biol. Chem. 2822587-2595. [DOI] [PubMed] [Google Scholar]
- 19.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 1623749-3752. [PubMed] [Google Scholar]
- 20.Indik, S., W. H. Gunzburg, B. Salmons, and F. Rouault. 2005. Mouse mammary tumor virus infects human cells. Cancer Res. 656651-6659. [DOI] [PubMed] [Google Scholar]
- 21.Iwatani, Y., D. S. Chan, F. Wang, K. S. Maynard, W. Sugiura, A. M. Gronenborn, I. Rouzina, M. C. Williams, K. Musier-Forsyth, and J. G. Levin. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 357096-7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izumi, T., K. Shirakawa, and A. Takaori-Kondo. 2008. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini Rev. Med. Chem. 8231-238. [DOI] [PubMed] [Google Scholar]
- 23.LaRue, R. S., V. Andresdottir, Y. Blanchard, S. G. Conticello, D. Derse, M. Emerman, W. C. Greene, S. R. Jonsson, N. R. Landau, M. Lochelt, H. S. Malik, M. H. Malim, C. Munk, S. J. O'Brien, V. K. Pathak, K. Strebel, S. Wain-Hobson, X. F. Yu, N. Yuhki, and R. S. Harris. 2009. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 83494-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo, K., T. Wang, B. Liu, C. Tian, Z. Xiao, J. Kappes, and X. F. Yu. 2007. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 817238-7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lutz, M. B., N. Kukutsch, A. L. Ogilvie, S. Rossner, F. Koch, N. Romani, and G. Schuler. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 22377-92. [DOI] [PubMed] [Google Scholar]
- 26.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defense by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 42499-103. [DOI] [PubMed] [Google Scholar]
- 27.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 91398-1403. [DOI] [PubMed] [Google Scholar]
- 28.Martin, P., S. R. Ruiz, G. Martinez del Hoyo, F. Anjuere, H. H. Vargas, M. Lopez-Bravo, and C. Ardavin. 2002. Dramatic increase in lymph node dendritic cell numbers during infection by the mouse mammary tumor virus occurs by a CD62L-dependent blood-borne DC recruitment. Blood 991282-1288. [DOI] [PubMed] [Google Scholar]
- 29.Mbisa, J. L., R. Barr, J. A. Thomas, N. Vandegraaff, I. J. Dorweiler, E. S. Svarovskaia, W. L. Brown, L. M. Mansky, R. J. Gorelick, R. S. Harris, A. Engelman, and V. K. Pathak. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 817099-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miesfeld, R., S. Rusconi, P. J. Godowski, B. A. Maler, S. Okret, A. C. Wikstrom, J. A. Gustafsson, and K. R. Yamamoto. 1986. Genetic complementation of a glucocorticoid receptor deficiency by expression of cloned receptor cDNA. Cell 46389-399. [DOI] [PubMed] [Google Scholar]
- 31.Nathans, R., H. Cao, N. Sharova, A. Ali, M. Sharkey, R. Stranska, M. Stevenson, and T. M. Rana. 2008. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 261187-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neil, S. J., T. Zang, and P. D. Bieniasz. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451425-430. [DOI] [PubMed] [Google Scholar]
- 33.Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa, M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15166-170. [DOI] [PubMed] [Google Scholar]
- 34.Ohsugi, T., and A. Koito. 2008. Current topics in prevention of human T-cell leukemia virus type I infection: NF-κB inhibitors and APOBEC3. Int. Rev. Immunol. 27225-253. [DOI] [PubMed] [Google Scholar]
- 35.Okeoma, C. M., N. Lovsin, B. M. Peterlin, and S. R. Ross. 2007. APOBEC3 inhibits mouse mammary tumor virus replication in vivo. Nature 445927-930. [DOI] [PubMed] [Google Scholar]
- 36.Peng, G., T. Greenwell-Wild, S. Nares, W. Jin, K. J. Lei, Z. G. Rangel, P. J. Munson, and S. M. Wahl. 2007. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110393-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng, G., K. J. Lei, W. Jin, T. Greenwell-Wild, and S. M. Wahl. 2006. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 20341-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pham, P., R. Bransteitter, and M. F. Goodman. 2005. Reward versus risk: DNA cytidine deaminases triggering immunity and disease. Biochemistry 442703-2715. [DOI] [PubMed] [Google Scholar]
- 39.Pion, M., A. Granelli-Piperno, B. Mangeat, R. Stalder, R. Correa, R. M. Steinman, and V. Piguet. 2006. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J. Exp. Med. 2032887-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reeves, J. D., and A. J. Piefer. 2005. Emerging drug targets for antiretroviral therapy. Drugs 651747-1766. [DOI] [PubMed] [Google Scholar]
- 41.Ross, S. R. 1997. MMTV and the immune system. Adv. Pharmacol. 3921-46. [DOI] [PubMed] [Google Scholar]
- 42.Rulli, S. J., J. Mirro, S. A. Hill, P. Lloyd, R. J. Gorelick, J. M. Coffin, D. Derse, and A. Rein. 2008. Interactions of murine APOBEC3 and human APOBEC3G with murine leukemia viruses. J. Virol. 826566-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarkis, P. T., S. Ying, R. Xu, and X. F. Yu. 2006. STAT1-independent cell type-specific regulation of antiviral APOBEC3G by IFN-alpha. J. Immunol. 1774530-4540. [DOI] [PubMed] [Google Scholar]
- 44.Schumacher, A. J., G. Haché, D. A. MacDuff, W. L. Brown, and R. S. Harris. 2008. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 822652-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shackleford, G. M., and H. E. Varmus. 1988. Construction of a clonable, infectious, and tumorigenic mouse mammary tumor virus provirus and a derivative genetic vector. Proc. Natl. Acad. Sci. USA 859655-9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418646-650. [DOI] [PubMed] [Google Scholar]
- 47.Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 91404-1407. [DOI] [PubMed] [Google Scholar]
- 48.Shindo, K., A. Takaori-Kondo, M. Kobayashi, A. Abudu, K. Fukunaga, and T. Uchiyama. 2003. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J. Biol. Chem. 27844412-44416. [DOI] [PubMed] [Google Scholar]
- 49.Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12591-601. [DOI] [PubMed] [Google Scholar]
- 50.Stopak, K. S., Y. L. Chiu, J. Kropp, R. M. Grant, and W. C. Greene. 2007. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 2823539-3546. [DOI] [PubMed] [Google Scholar]
- 51.Vacheron, S., S. J. Luther, and H. Acha-Orbea. 2002. Preferential infection of immature dendritic cells and B cells by mouse mammary tumor virus. J. Immunol. 1683470-3476. [DOI] [PubMed] [Google Scholar]
- 52.Wedekind, J. E., R. Gillilan, A. Janda, J. Krucinska, J. D. Salter, R. P. Bennett, J. Raina, and H. C. Smith. 2006. Nanostructures of APOBEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J. Biol. Chem. 28138122-38126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiznerowicz, M., and D. Trono. 2003. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 778957-8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu, L., and V. N. KewalRamani. 2006. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat. Immunol. 6859-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu, X., Y. Yu, B. Liu, K. Luo, W. Kong, P. Mao, and X. F. Yu. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 3021056-1060. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, Y., J. C. Rassa, M. E. deObaldia, L. M. Albritton, and S. R. Ross. 2003. Identification of the receptor binding domain of the mouse mammary tumor virus envelope protein. J. Virol. 7710468-10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng, Y. H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 786073-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]