

Abstract
Endonuclease decay of nonsense-containing β-globin mRNA in erythroid cells generates 5′-truncated products that were reported previously to have a cap or caplike structure. We confirmed that this 5′ modification is indistinguishable from the cap on full-length mRNA, and Western blotting, immunoprecipitation, and active-site labeling identified a population of capping enzymes in the cytoplasm of erythroid and nonerythroid cells. Cytoplasmic capping enzyme sediments in a 140-kDa complex that contains a kinase which, together with capping enzyme, converts 5′-monophosphate RNA into 5′-GpppX RNA. Capping enzyme shows diffuse and punctate staining throughout the cytoplasm, and its staining does not overlap with P bodies or stress granules. Expression of inactive capping enzyme in a form that is restricted to the cytoplasm reduced the ability of cells to recover from oxidative stress, thus supporting a role for capping in the cytoplasm and suggesting that some mRNAs may be stored in an uncapped state.
The addition of the 5′ cap is the first posttranscriptional step in pre-mRNA processing (8, 25), and the cap plays a central role in subsequent steps of pre-mRNA processing, export, surveillance, translation, decay, and microRNA silencing through its binding by CBP80 (9) and eIF4E (22). The decay of most mammalian mRNAs begins with poly(A) shortening, after which the cap is removed and the body of the mRNA undergoes 3′-5′ decay by the cytoplasmic exosome or 5′-3′ decay by Xrn1 (7).
While the action of a cytoplasmic poly(A) polymerase can restore a shortened poly(A) tail to one capable of supporting efficient translation (13), there is no evidence for the reversibility of decapping (30). In Saccharomyces cerevisiae, decapping is the rate-limiting step in mRNA decay, and this is followed by rapid 5′-3′ degradation of the mRNA body by Xrn1 (7). Like in yeast, mammalian Dcp2 and Xrn1 are together recovered by immunoprecipitation and colocalize in P bodies (4), suggesting that unstable mRNAs decay similarly. However, until recently, no one had actually quantified the polarity of mammalian mRNA decay. Using a sensitive fluorescent resonance energy transfer-based assay to quantify the decay of each exon of a β-globin reporter mRNA, we found that the 5′ and 3′ ends decay simultaneously and showed that these processes are functionally linked (19). More surprisingly, we found that 5′ decay is slow and relatively inefficient.
mRNA containing a premature termination codon (PTC) is degraded by a process termed mRNA surveillance or nonsense-mediated mRNA decay. While it was previously thought that PTC-containing mRNAs are degraded while still associated with the nucleus (17), several recent studies point to P bodies as the major site of their decay (26, 31). An endonuclease activity in SMG6 also appears to be involved in this process, but Xrn1 must be knocked down to visualize downstream decay products (5, 10).
Mutations in the β-globin gene comprise one of the largest cohorts of inherited disorders, and a PTC in exon 1 or 2 activates nonsense-mediated mRNA decay for the mRNA from the affected allele. Previous work by Lim and coworkers (14, 16) showed that, in erythroid cells, the decay of this mRNA is associated with the appearance of 5′-truncated RNAs, and we showed that this results from endonuclease cleavage in the cytoplasm (28). Cleavage occurs primarily at UG dinucleotides, and subsequent work showed that the decay of this mRNA is catalyzed by the mammalian ortholog of the polysome-associated PMR1 endonuclease (2). The 5′-truncated forms of β-globin mRNA are remarkably stable, and a paper published in 1992 suggested that they are modified by a 5′ caplike structure (15). This finding was not followed up, and as part of our work characterizing the decay of this nonsense-containing mRNA, we asked whether these are indeed modified by a cap or caplike structure. Using several different criteria, we show that their 5′ ends are indistinguishable from the cap on the parent mRNA and identify a population of cytoplasmic capping enzymes. A capping enzyme contains two functional domains, an N-terminal triphosphatase domain and a C-terminal guanylyltransferase domain. In nuclear capping, the triphosphatase domain converts the 5′-triphosphate of the nascent transcript to a 5′-diphosphate, and the guanylyltransferase domain transfers covalently bound GMP onto this. Methyl groups are then added by cap methyltransferase. In cytoplasmic capping, 5′-monophosphate RNA is converted to a 5′-diphosphate capping substrate by a kinase that is present in a 140-kDa complex with capping enzyme. Capping enzyme then transfers covalently bound GMP onto this. Disrupting this process interferes with the ability of cells to recover from stress, suggesting that cells contain a population of uncapped mRNAs that can be reactivated by cytoplasmic capping.
MATERIALS AND METHODS
Cell culture and generation of stable cell lines.
The development of Thal10 murine erythroleukemia (MEL) cells stably transfected with the human β-globin gene bearing a PTC after codon 60 was described previously (28). Cells were cultured as described previously (2) in alpha-minimal essential medium plus 10% fetal bovine serum (FBS) and 100 μg/ml G418. The transcription of the transfected β-globin gene was induced by addition of 1.5% dimethyl sulfoxide to the medium for 48 h. COS-1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) plus 10% FBS and 2 mM l-glutamine; Huh7 cells in DMEM plus 10% FBS, 2 mM l-glutamine, 0.1 mM nonessential amino acids; and U2OS cells in McCoy 5A plus 10% FBS or in DMEM containing 10% FBS and 2 mM l-glutamine.
Tetracycline-inducible U2OS cells were created by stable transfection with the tetracycline repressor-expressing plasmid pcDNA6/TR, using FuGene 6 transfection reagent (Roche Molecular Biochemicals) according to the manufacturer's instructions. Individual clones were selected by growth in medium containing 6 μg/ml of blasticidin (Invitrogen), and these were tested for induction by tetracycline using the tetracycline-inducible pcDNA4/TO/LacZ reporter plasmid. The line showing the greatest range of regulation and lowest background was transfected with pcDNA4/TO/myc-mCEΔNLS+NES-Flag, pcDNA4/TO/myc-K294AΔNLS+NES-Flag, pcDNA4/TO/myc-mCE, or pcDNA4/TO/myc-GFP-Flag, and clones were selected in growth medium containing 400 μg/ml of Zeocin (Invitrogen) and 6 μg/ml of blasticidin. Selected clones were cultured and tested for tetracycline-inducible expression of target protein by Western blotting.
S1 nuclease protection.
For the S1 nuclease protection assay (24, 28), cytoplasmic RNA from Thal10 cells was dissolved in S1 hybridization buffer (80% formamide, 40 mM PIPES [piperazine-N,N-bis(2-ethanesulfonic acid)] [pH 6.4], 0.4 M NaCl, 1 mM EDTA) and incubated with the 5′-end-labeled antisense DNA probe overnight at 52°C. The antisense probe was obtained by asymmetric PCR using the human β-globin cDNA plasmid pSPkβC as a template. The 5′ end of the antisense primer 5′-TTTCTTGCCATGAGCCTTCACCTTA-3′ was labeled with [γ-32P]ATP (3,000 Ci/mmol) using T4 polynucleotide kinase. The first nucleotide of this corresponds to position 250. The unlabeled sense primer 5′-ATTTAGGTGACACTATA-3′ corresponded to the upstream SP6 promoter. The radiolabeled probes were purified by electrophoresis on denaturing polyacrylamide/urea gels. After hybridization, S1 nuclease solution (0.28 M NaCl, 0.05 M sodium acetate [pH 5.2], 4.5 mM ZnSO4, 20 μg/ml sheared salmon sperm DNA, 100 units S1 nuclease [Invitrogen]) was added to the hybridization mixture and incubated at 28°C for 2 h. Samples were precipitated with ethanol and electrophoresed on denaturing 6% polyacrylamide/urea gels, and protected fragments were visualized by a PhosphorImager.
Preparation of GST-eIF4E protein.
Escherichia coli BL21(DE3)pLysS (Promega) was transformed with the plasmid pGEX-meIF4E, expressing a glutathione S-transferase (GST) fusion protein with murine eIF4E cap-binding protein (GST-eIF4E). Cleared bacterial lysate was incubated with m7GTP-Sepharose (GE Biosciences) by end-over-end rotation at 4°C overnight. The beads were washed twice with washing buffer (20 mM HEPES [pH 7.5], 0.1 M KCl, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride [PMSF]) and once with washing buffer containing 50 μM GDP. GST-eIF4E was eluted with washing buffer containing 50 μM m7GDP at room temperature and dialyzed at 4°C twice against 20 mM HEPES (pH 7.5), 1 M KCl, 0.2 mM EDTA, and 0.5 mM PMSF for 2 h, followed by overnight dialysis with 20 mM HEPES (pH 7.5), 0.1 M KCl, 0.2 mM EDTA, and 0.5 mM PMSF to remove m7GDP.
RNA recovery with cap antibody or GST-eIF4E.
A total of 12.5 μg of purified GST-eIF4E was incubated with glutathione-Sepharose 4B (GE Biosciences), and 10 μl of trimethyl cap antibody (Synaptic Systems) was incubated with protein G-Sepharose (GE Biosciences) by end-over-end rotation for 2 h at 4°C and washed three times with phosphate-buffered saline (PBS). Glutathione-Sepharose coupled with GST-eIF4E or protein G-Sepharose coupled with anti-cap antibody were mixed with 40 μg of cytoplasmic RNA in 400 μl of binding buffer (10 mM KHPO4 [pH 8.0], 100 mM KCl, 2 mM EDTA, 5% glycerol, 6 mM dithiothreitol [DTT], 1.3% polyvinyl alcohol, 0.005% Triton X-100, 5 units RNaseOUT [Invitrogen]) supplemented with 15 μg/ml yeast tRNA. Before being mixed, RNA was denatured by incubation at 60°C for 10 min. Mixing was performed by end-over-end rotation at room temperature for 2 h. The resin was washed three times with 400 μl of binding buffer and twice with 400 μl of binding buffer containing 50 μM GDP. For Fig. 1A and B, capped RNA was specifically eluted by being mixed twice with 400 μl of binding buffer containing 50 μM, and for Fig. 1C, 20, 100, or 50 μM m7GDP was used to elute capped RNA. RNA that was recovered in each fraction was precipitated with isopropanol and analyzed by an S1 nuclease protection assay.
FIG. 1.

Recovery of decay intermediates with trimethyl cap antibody and eIF4E. (A) A total of 40 μg of cytoplasmic RNA from Thal10 cells was incubated with H20 cap antibody bound to protein G-Sepharose. The recovered beads were washed twice with binding buffer (lanes 5 and 6), twice with 50 μM GDP (lanes 7 and 8), and eluted twice with 50 μM m7GDP (lanes 9 and 10). RNA in each eluate was recovered by ethanol precipitation and analyzed by S1 nuclease protection assay using a 5′-labeled antisense probe (top) from nucleotide 250 past the 5′ end of β-globin mRNA. As a control, 8 μg of Thal10 cell cytoplasmic RNA was analyzed in lane 3 (input). A sample of the S1 probe plus 10 μg of yeast tRNA is in lane 1 (probe, −S1), and the same sample after S1 nuclease digestion is in lane 2 (probe, +S1). The positions of HinfI restriction fragments of ΦX174 DNA electrophoresed on the gel (molecular size markers) are indicated on the left, and the identified β-globin mRNA decay intermediates are indicated on the right. (B) Cytoplasmic RNA from Thal10 cells was analyzed as in panel A, with the exception that the RNA was recovered with GST-eIF4E bound to glutathione-Sepharose. (C) The relative amounts of full-length mRNA and each decay intermediate recovered in panels A and B were quantified by a PhosphorImager, and the results were normalized to full-length mRNA and each decay intermediate in the input RNA. (D) Cytoplasmic RNA from Thal10 cells was bound onto immobilized H20 cap antibody, washed as in panel A, and eluted with 50 μM GDP (lane 4) or the indicated concentrations of m7GDP (lanes 5, 6, and 7). (E) Cytoplasmic RNA from Thal10 cells was incubated with immobilized eIF4E in the presence of 0, 0.1, 1, 10, or 100 μM m7GpppG (lanes 5 to 8) or ApppG (lanes 9 to 12). Bound complexes were washed with binding buffer containing 50 μM GDP, eluted with 50 μM m7GDP, and analyzed by S1 nuclease protection assay. The positions of full-length mRNA and each of the decay intermediates are indicated on the right. As a control, 20% of Thal10 cell cytoplasmic RNA was applied to lane 3 (input).
In vitro decapping and 5′ exonuclease assay.
For the experiments shown in Fig. 2A, 10 μg of cytoplasmic RNA from dimethyl sulfoxide-treated Thal10 cells was incubated with 5 units of tobacco acid pyrophosphatase (TAP; Epicentre) for 2 h at 37°C in 50 mM sodium acetate (pH 6.0), 1 mM EDTA, 0.1% β-mercaptoethanol, and 0.01% Triton X-100. Decapping with recombinant human Dcp2 protein for Fig. 2B was performed as described by Piccirillo et al. (21). A total of 0.6 μg of His-hDcp2 (kindly provided by Mike Kiledjian) was incubated for 1 h at 37°C with 10 μg of cytoplasmic RNA from Thal10 cells in decapping buffer (10 mM Tris-HCl [pH 7.5], 100 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM MnCl2, 2 mM DTT, and 0.1 mM spermine). For the experiments shown in Fig. 2, the recovered RNA, after decapping with pyrophosphatase or Dcp2, was incubated for 1 h at 30°C with 1 unit of Terminator 5′-phosphate-dependent 5′ exonuclease (Epicentre) in 50 mM Tris-HCl (pH 8.0), 2 mM MgCl2, and 100 mM NaCl and analyzed by an S1 nuclease protection assay.
FIG. 2.
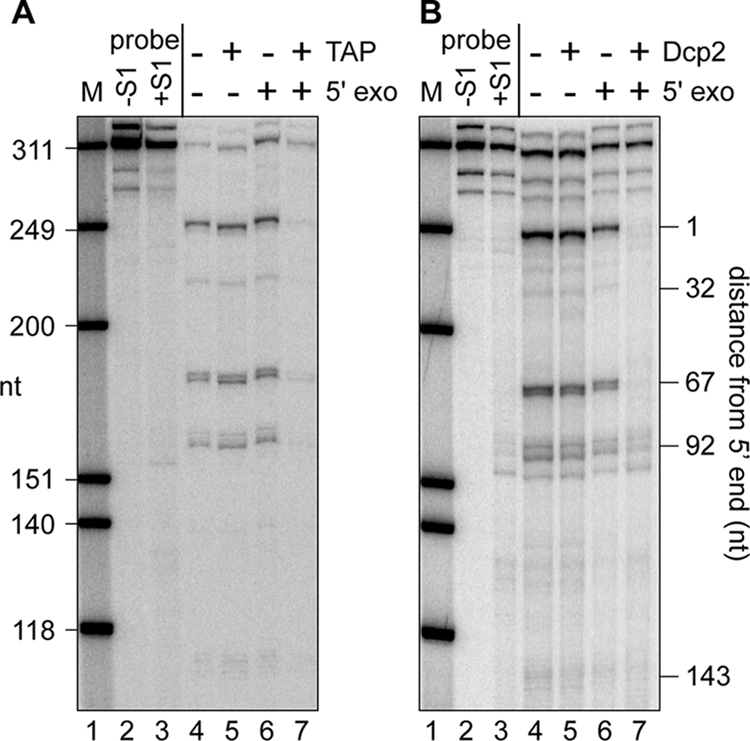
Susceptibility of full-length β-globin mRNA and decay intermediates to cap hydrolysis and cap analog competition for binding to eIF4E and cap antibody. Cytoplasmic RNA from Thal10 cells was treated without (−) (lanes 4 and 6) or with (+) (lanes 5 and 7) TAP (A) or Dcp2 (B). RNA recovered from each reaction was then incubated without (lanes 4 and 5) or with (lanes 6 and 7) a 5′-monophosphate-dependent 5′ exonuclease and analyzed by S1 nuclease protection assay.
In vitro guanylylation assay.
Guanylyltransferase activity was assayed as described by Yue et al. (34). Samples were incubated for 30 min at 37°C in 20 μl of capping buffer (25 mM Tris-HCl [pH 7.5], 5 mM MgCl2, and 0.5 mM DTT) containing [α-32P]GTP (3,000 Ci/mmol) and then denatured with sodium dodecyl sulfate (SDS) sample buffer and separated by electrophoresis on a 10% SDS-polyacrylamide gel electrophoresis (PAGE) gel. Guanylylated protein was identified by a PhosphorImager.
Recovery of cytoplasmic capping enzyme complex and use of RNA capping assay.
A total of 2 × 106 COS-1 cells were transiently transfected with 1.5 μg of pcDNA3-myc-GFP, pcDNA3-myc-mCE(WT), pcDNA3-myc-mCE(K294A), or pcDNA3-myc-mCE(211-597) plus 4.5 μg of vector (pcDNA3) using FuGene 6 (Roche Molecular Biochemicals). Cells were collected 24 h after transfection. Nuclear and cytoplasmic extracts were incubated with myc monoclonal antibody-coupled beads on a rocking platform for 5 h at 4°C. The beads were washed four times with IPP150 (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.1% Nonidet P-40) and then twice with capping buffer. The immunoprecipitates were suspended in capping buffer. For in vitro capping, the enzyme-GMP complex formation was first generated in capping buffer containing [α-32P]GTP (3,000 Ci/mmol), and the product was recovered by immunoprecipitation with myc antibody. This product was treated for 30 min at 37°C with 1 unit of shrimp alkaline phosphatase (SAP; Roche Molecular Biochemicals), followed by a second round of recovery with myc antibody-coupled beads. The beads were then incubated for 2 h at 30°C in 20 μl of capping buffer containing 5 μM high-pressure liquid chromatography (HPLC)-purified RNA oligonucleotides (5′-AAAGUUCCUUCAGCUGAAAAGAG) without a 5′-monophosphate (5′-OH-RNA) or with a 5′-monophosphate (5′-P-RNA) (obtained from IDT, Inc.), 1 μM ATP, and 4 units RNaseOUT. The reaction was stopped by chilling it on ice, and RNA was recovered by extraction with phenol-chloroform-isoamyl alcohol (25:24:1; Invitrogen) and two rounds of ethanol precipitation. The recovered RNA was dissolved in 20 μl of diethyl pyrocarbonate-treated water, and 5 μl of each was treated with or without 1 unit of TAP (Epicentre) for 2 h at 37°C prior to electrophoresis on a denaturing 15% polyacrylamide/6 M urea gel. The products were then visualized by a PhosphorImager. The 5′-end modification of these RNAs was determined by digesting 10 μl of each with 0.5 units of P1 nuclease (U.S. Biological) in 1 M sodium acetate, pH 5.2, and 1 mM ZnSO4 for 1 h at 37°C, followed by 30 min at 37°C with 1 unit of SAP. Half of each reaction was applied to an indicator dye-containing polyethylenimine (PEI)-cellulose plate (Sigma-Aldrich), together with standards of GTP, GDP, GMP, and GpppA, and developed with 0.45 M ammonium sulfate. The standards were visualized by UV shadowing, and radiolabeled products were visualized by a PhosphorImager.
Preparation of nuclear and cytoplasmic extracts, immunoprecipitation, and glycerol gradient analysis.
MEL, U2OS, Huh7, or COS-1 cells were washed twice with ice-cold PBS and then suspended in cell lysis buffer (10 mM HEPES-KOH [pH 7.5], 10 mM KCl, 10 mM MgCl2, 0.2% Nonidet P-40, 2 mM DTT, 0.5 mM PMSF, 25 μl/ml protease inhibitor mixture [Sigma-Aldrich], and 10 μl/ml RNaseOUT). After incubation for 10 min on ice, the cells were homogenized with 20 strokes of a Dounce homogenizer, and the homogenate was centrifuged for 10 min at 15,000 × g. The supernatant was kept as cytoplasmic extracts. The pellet was suspended in buffer A (10 mM HEPES-KOH [pH 7.5], 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 25 μl/ml protease inhibitor mixture, and 10 μl/ml RNaseOUT), incubated by end-over-end rotation at 4°C for 20 min, and then centrifuged for 5 min at 15,000 × g. The supernatant was kept as nuclear extracts. Both extracts were adjusted to equal volumes under the same buffer (10 mM HEPES-KOH [pH 7.5], 5 mM MgCl2, 1 mM DTT, 0.5 mM PMSF, 150 mM KCl, 10% glycerol, 0.2 mM EDTA, 0.15% Nonidet P-40, 25 μl/ml protease inhibitor mixture, and 10 μl/ml RNaseOUT). For immunoprecipitation with anti-capping enzyme antibody, nuclear or cytoplasmic extracts were incubated with the above-mentioned buffer or rabbit immunoglobulin G (IgG) (Santa Cruz) by end-over-end rotation for 4 h at 4°C, followed by addition of 20 μl of protein G-Sepharose (GE Biosciences) and incubation for 1.5 h. The beads were washed four times with IPP150 and twice with capping buffer. The immunoprecipitates were suspended in capping buffer for the guanylyltransferase activity assay. For glycerol gradient analysis, 3.5 × 106 U2OS cells that were stably transfected with plasmids expressing wild-type murine capping enzyme (mCE) with deletion of four amino acids from the putative C-terminal nuclear localization signal (NLS) and addition of the 15-amino-acid HIV Rev nuclear export signal (NES) (ΔNLS+NES) or green fluorescent protein (GFP) were plated into a 150-mm dish, and the next day were treated with 1 μg/ml of doxycycline. At 24 h, cells were washed once with PBS and then incubated with or without 0.25% formaldehyde in PBS for 10 min at 37°C (18). Cytoplasmic extracts were separated on 10 to 40% linear glycerol gradients by centrifugation for 20 h at 83,000 × g in a Sorvall TH-641 rotor. Molecular size markers containing a mixture of thyroglobulin (molecular weight [MW], 669,000), ferritin (MW, 440,000), catalase (MW, 232,000), lactate dehydrogenase (MW, 140,000), and bovine serum albumin (MW, 67,000) were fractionated on a parallel gradient.
Immunofluorescence microscopy.
U2OS cells stably transfected with tetracycline-inducible plasmids expressing myc-tagged mCE or mCE with the active-site K294A mutation (K294A) ΔNLS+NES form of the enzyme were grown in Dulbecco's minimum essential media containing 2 mM glutamine, 10% FBS, and 20 mM HEPES. Cells were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature and then permeabilized in absolute methanol (−20°C for 5 min). Samples were incubated for 1 h in PBS containing 5% horse serum (blocking buffer), followed by 1 h of incubation in a cocktail of primary antibodies. A 1/1,000 dilution was used for anti-myc monoclonal antibody or antibodies to YB1, DCP1a, or RCK, and a 1/200 dilution was used for antibodies against FXR1, TIA-1 and eIF4A as indicated in the figure legends.
Cells were washed twice in PBS (5 min per wash) and then incubated in a secondary antibody mixture for 1 h (1/200 donkey anti-mouse IgG-Cy2, 1/2,000 donkey anti-rabbit IgG-Cy3, and donkey anti-human IgG-Cy5; all were ML grade for multiple labeling). Cells were washed three times in PBS, mounted in a polyvinyl mounting medium, and viewed using a Nikon E800 upright microscope equipped for epifluorescence optics using a 100× objective (numerical aperture, 1.40). Images were obtained using a National Diagnostics CCD-SPOT RT digital camera and compiled using Adobe Photoshop CS.
Antibodies.
The H20 trimethyl cap monoclonal antibody was purchased from Synaptic Systems (Gottingen, Germany), and monoclonal antibody to the c-myc epitope tag (9E10), myc antibody (9E10)-coupled beads, and antibodies to FXR1 (sc-10544), TIA-1 (sc-1751), and eIF4A (sc-14211) were purchased from Santa Cruz. Horseradish peroxidase (HRP)-coupled goat anti-rabbit IgG and HRP-coupled goat anti-sheep IgG were also purchased from Santa Cruz, and HRP-coupled sheep anti-mouse IgG was purchased from GE Biosciences. Antibody against YB1 (rabbit polyclonal Ab12148) was purchased from Abcam (Cambridge, MA), and antibody to RCK (no. BL2139) was purchased from Bethyl Laboratories. The antibody to histone H4 was provided by Mark Parthun (The Ohio State University), capping enzyme antibodies were provided by Aaron Shatkin (Rutgers) and David Price (University of Iowa), the antibody against DCP1a was provided by Jens Lykke-Andersen (University of Colorado), and U2AF65 antibody was provided by Brent Graveley (University of Connecticut).
Western blot analysis.
Proteins were separated on a 10% SDS-PAGE gel and electroblotted onto an Immobilon-P membrane (Millipore). The membrane was blocked for 1 h at room temperature with 5% nonfat dry milk in Tris-buffered saline containing Tween 20 (TBST) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.1% Tween 20), then incubated with the primary antibody overnight at 4°C, washed with TBST buffer, and incubated with HRP-conjugated secondary antibody for 1.5 h at room temperature. After being washed with TBST buffer, blots were developed with SuperSignal West Pico or Femto chemiluminescent substrate (Pierce) or ECL Plus Western blotting detection reagents (GE Healthcare).
Plasmid constructions.
The mCE cDNA clone was obtained from Invitrogen and PCR amplified using primers YO-85, 5′-GCTTATGGCCATGGAGGCCGCTTACAACAAGATCCCGCC, and YO-36, 5′-AAACGGGCCCTCAGGTTGGCCGATGCAGTCTTTTG. After digestion with SfiI and ApaI, it was cloned into pcDNA3-myc-TAP to generate pcDNA3-myc-mCE. The plasmid pcDNA3-myc-mCE(K294A) was generated by PCR amplification from pCR21-mCE(K294A) (provided by Aaron Shatkin, Rutgers) with the same primers as described above. The C-terminal guanylyltransferase domain of mCE in pGEX-4T3-mCE(211-597) (provided by Aaron Shatkin, Rutgers) was PCR amplified from the parent plasmid using the primers YO-26, 5′-TTATGGCCATGGAGGCCGAACCAGGGTCAAGTGCATC, and YO-27, 5′-TCTTCGCGGCCGCGGTTGGCCGATGCAGTCTTT, which contain restriction sites for SfiI and ApaI, respectively. The amplified products were digested with SfiI and ApaI and ligated into the corresponding sites in pcDNA3-myc-TAP to generate a plasmid, pcDNA3-myc-mCE(211-597). To generate the plasmid pcDNA3-myc+NES-mCE or pcDNA3-myc+NES-mCE(K294A), which has a NES (LQLPPLERLTLD) from a human immunodeficiency virus (HIV) Rev protein between the myc tag and mCE, the primers YO-86, 5′-GCTTATGGCCATGGAGGCCCTTCAGCTACCACCGCTTGAGAGACTTACTCTTGATATGGCCATGGAGGCCCAAGCT-3′, and YO-87, 5′-AGCTTGGGCCTCCATGGCCATATCAAGAGTAAGTCTCTCAAGCGGTGGTAGCTG AAGGGCCTCCATGGCCATAAGC-3′, which contain restriction sites for SfiI at both ends, were annealed, digested with SfiI, and ligated into the corresponding site in pcDNA3-myc-mCE or pcDNA3-myc-mCE(K294A). To delete a putative NLS (K573-Y576) from mCE or mCE(K294A), DNA fragments were PCR amplified from pcDNA3-myc-mCE or pcDNA3-myc-mCE(K294A) using the primers YO-128, 5′-TGTTTGAATTCATTGACAGATGTGCAGCAGCCGCCCAGGGACAGCCCCTGGACCCTGACACGGAG-3′, and YO-36, which contain restriction sites for EcoRI and ApaI, respectively. The amplified products were digested with EcoRI and ApaI and ligated into the corresponding sites in pcDNA3-myc-mCE, pcDNA3-myc+NES-mCE, or pcDNA3-myc+NES-mCE(K294A) to generate plasmids pcDNA3-myc-mCEΔNLS, pcDNA3-myc+NES-mCEΔNLS, or pcDNA3-myc+NES-mCE(K294A)ΔNLS, respectively.
To generate pcDNA4/TO-myc+NES-mCEΔNLS-Flag, pcDNA4/TO-myc+NES-mCE(K294A)ΔNLS-Flag, pcDNA4/TO-myc-mCE, or pcDNA4/TO-myc-GFP-Flag, DNA fragments were PCR amplified from pcDNA3-myc+NES-mCEΔNLS or pcDNA3-myc+NES-mCE(K294A)ΔNLS using T7 primer and YO-129, GAATAGGGCCCCTACTTATCGTCGTCATCCTTGTAATCGGTTGGCCGATGCAGTCTTTTGG, from pcDNA3-myc-mCE using T7 primer and YO-36 or from pcDNA3-myc-GFP using YO-130, AGCTTGGTACCATGGAACAAAAGCTGATTTCAGAAG, and YO-131, GAATAGGGCCCCTACTTATCGTCGTCATCCTTGTAATCATCCATGCCATGTGTAATCCCAGC, respectively. The amplified products were digested with KpnI and ApaI and ligated into the corresponding sites in pcDNA4/TO vector.
Cell viability assay.
A total of 0.8 × 104 to approximately 1 × 104 U2OS cells stably transfected with tetracycline-inducible plasmids expressing GFP, mCE ΔNLS+NES, or K294A ΔNLS+NES were plated into 96-well plates and treated the next day with or without 2 μg/ml of doxycycline for 24 h to induce each of these proteins. One-half of each set was exposed with 0.5 mM sodium arsenite for 40 min, then washed once with prewarmed growth medium, and cultured in growth medium for an additional 24 h. Cell viability was determined on triplicate samples of each treatment group using the CellTiter-Glo luminescent cell viability assay (Promega), a luminescent assay for ATP.
RESULTS
Full-length and 5′-truncated forms of PTC-containing β-globin mRNA are bound equally well by trimethyl cap antibody and eIF4E.
Stable, 5′-truncated forms of β-globin mRNA were originally observed by Lim and coworkers in erythroid cells of transgenic mice carrying the human β-globin gene with a nonsense codon in exon 2 (14). Subsequent work from that lab showed that the appearance of these shortened RNAs was dependent on the presence of a nonsense codon, their appearance did not change with the location of the nonsense codon, and they were restricted to the cytoplasm (16). Those authors also established a precursor-product relationship between these and parental β-globin mRNA. This erythroid-specific form of β-globin mRNA decay was replicated in stably transfected lines of MEL cells carrying the same nonsense-containing allele (Thal10 cells) (28). Importantly, the 5′ ends of the truncated RNAs were identified by both S1 nuclease protection and primer extension, and these matched the 5′ ends of RNAs recovered from erythroid cells of transgenic mice. Moreover, their appearance is linked to endonuclease cleavage (2), a process that generates products with a 5′-monophosphate end. Finally, the 5′-truncated RNAs were also reported to have a cap or caplike structure (15). This implies that a polynucleotide 5′-monophosphate kinase converts the 5′ ends of the proximal decay products to a diphosphate for GMP addition by capping enzyme, the only mammalian enzyme known to add a cap onto RNA.
We began by replicating the observations made by Lim and Maquat (15). For the experiment shown in Fig. 1, cytoplasmic RNA from Thal10 cells was bound to immobilized cap monoclonal antibody (Fig. 1A) or GST-eIF4E (Fig. 1B). The resins were washed twice with binding buffer, followed by two washes with 50 μM GDP and two washes with m7GDP. RNA recovered from each of these washes was analyzed by S1 nuclease protection using the probe shown above Fig. 1A and B. The positions of the 5′ ends of each of the decay intermediates that can be detected with this probe are shown in Fig. 1A and B. Unbound RNA was completely removed from both resins during the initial washes, and neither the full-length mRNA nor any of the decay intermediates was eluted with GDP. However, full-length mRNA and all of the decay intermediates were recovered equally when the resin was eluted with m7GDP (Fig. 1C).
Full-length β-globin mRNA and each of the 5′-truncated RNAs were eluted from immobilized trimethyl cap antibody with as little as 20 μM m7GDP but not with 50 μM GDP (Fig. 1D). Moreover, binding of full-length mRNA and each of the decay intermediates to GST-eIF4E was competed by m7GpppG but not by ApppG (Fig. 1E). Using the criteria of cap binding by these two proteins, there is no difference between the cap on the 5′ end of the parent mRNA and the modification present on each of the 5′-truncated RNAs. These results do not distinguish between cap 0 structure and the monomethylated (cap 1) or dimethylated (cap 2) caps present on most mRNAs.
Full-length and 5′-truncated RNAs are equally susceptible to cap hydrolysis.
Under the appropriate conditions, Dcp2 is selective for the m7GpppX cap structure (32), whereas TAP nonspecifically hydrolyzes any pyrophosphate linkage. To further examine the cap or caplike modification on these RNAs, we compared the impact of pretreating Thal10 RNA with Dcp2 rather than with TAP on the susceptibility of parent mRNA and decay intermediates to degradation by a highly processive 5′-monophosphate-specific 5′-3′ exonuclease. We reasoned that the parent mRNA and decay intermediates should be degraded following nonspecific hydrolysis of the 5′-end modification by TAP. If the decay intermediates are not capped, only the parent mRNA should be degraded after Dcp2, whereas parent mRNA and decay intermediates will be degraded if they all are capped. The results shown in Fig. 2 indicate that without prior cap hydrolysis, the parent β-globin mRNA and all of the decay intermediates are resistant to the 5′ exonuclease (Fig. 2A and B, compare lanes 4 and 6) and, except for the 92-nucleotide (nt) product, become equally susceptible following prior treatment with TAP (Fig. 2A, lane 7) or Dcp2 (Fig. 2B, lane 7). It is not known why some of this remained while the others were lost, but it may be due to structural issues that impact decapping. In addition, binding to both GST-eIF4E and immobilized cap antibody is lost by prior treatment with TAP (see Fig. S1 in the supplemental material). Together with the results shown in Fig. 1, these data indicate that the decay intermediates have a cap or caplike structure that, by each of these criteria, is indistinguishable from that which is present on parent β-globin mRNA.
Capping enzyme is present in cytoplasmic extracts of erythroid and nonerythroid cells.
The presence of what appears to be an authentic cap on β-globin mRNA decay intermediates implied the existence of a novel cytoplasmic capping activity. Capping enzyme is the only mammalian enzyme known to add a cap. It is considered a nuclear protein, and while a novel viral capping activity has been described (20), the absence of any obvious mammalian ortholog of this type led us to examine the possibility that some capping enzyme might also be in the cytoplasm. Western blotting identified capping enzyme in cytoplasmic extracts of U2OS and Huh7 cells, although five times more cytoplasmic protein than nuclear protein had to be loaded to see a similar signal (Fig. 3A). For these experiments, cells were lysed with a Dounce homogenizer using a hypotonic buffer containing 10 mM KCl. Similar results were also obtained with MEL and COS-1 cells (not shown) and using other cytoplasmic isolation methods including NE-PER nuclear and cytoplasmic extraction reagents (Pierce), an isotonic buffer with detergent (without homogenization), or a hypotonic buffer and passage through a 25-gauge needle (not shown). To control nuclear leakage, the same samples were analyzed for U2AF65 and histone H4. Because histone H4 is tightly bound to chromatin, it should not be detected under any circumstances, whereas detection of U2AF65 in the cytoplasmic fraction would be indicative of nuclear leakage.
FIG. 3.

Biochemical evidence for cytoplasmic capping enzyme. (A) The indicated relative amounts of nuclear and cytoplasmic extracts of U2OS and Huh7 cells were analyzed by Western blotting (wb) with an affinity-purified sheep antibody against human capping enzyme (hCE). To control leakage of capping enzyme during cell lysis, the same samples were analyzed by Western blotting with antibodies to U2AF65 and histone H4 (lower panels). (B) The guanylylation activity of capping enzyme present in the indicated amounts of nuclear and cytoplasmic extracts from MEL cells, U2OS cells, and COS-1 cells was determined by incubating with [α-32P]GTP, followed by electrophoresis on a 10% SDS-PAGE gel and PhosphorImager analysis. In the lower panel, the same extracts were analyzed by Western blotting with antibody to U2AF65. (C) Cytoplasmic extract from COS-1 cells in panel B was immunoprecipitated with nonimmune IgG or antibody to human capping enzyme. A total of 25% of the immunoprecipitates and 2.5% of input protein were analyzed by guanylylation assay.
The Western blotting results were confirmed using a functional guanylylation assay that identifies capping enzyme as the active-site intermediate, with covalently bound [32P]GMP (34). For the experiment shown in Fig. 3B, nuclear and cytoplasmic extracts from MEL, U2OS, and COS-1 cells were incubated with [α-32P]GTP and separated on an SDS-PAGE gel. Both fractions from each of these cell lines yielded only a single 68-kDa band, the expected size for capping enzyme, and in spite of the large excess of cytoplasmic protein, U2AF65 was detected only in the nuclear fractions. In these experiments, six to eight times more cytoplasmic protein was added to the reaction, and the signal still did not match that of nuclear capping enzyme. It is unlikely that this is due to GTP in the extract, as inhibition is not relieved by dialysis but by immunoprecipitation (see below).
To confirm that the labeled protein is capping enzyme, the cytoplasmic extracts from COS-1 cells used for this experiment were immunoprecipitated using nonimmune IgG or anti-capping enzyme antibody, and the precipitated proteins were assayed for guanylylation activity by addition of [α-32P]GTP (Fig. 3C). Again, this labeled only the 68-kDa capping enzyme. More immunoprecipitated than input protein was used for this experiment, but the increased signal here compared to that shown in Fig. 3B indicates that immunoprecipitation separated cytoplasmic capping enzyme from the nondialyzable inhibitor noted above. Because MEL cells transfect with low efficiency, COS-1 and U2OS cells were used for subsequent experiments to characterize cytoplasmic capping.
Cytoplasmic capping enzyme can add a GMP onto 5′-monophosphate RNA.
Plasmids expressing wild-type myc-tagged capping enzyme, an inactive form of capping enzyme with the GMP-binding site changed to alanine (K294A), and capping enzyme lacking the N-terminal triphosphatase domain (211-597) were developed to study the biochemical properties of cytoplasmic capping enzyme. For Fig. 4A, each of these constructs or a plasmid expressing myc-GFP was transfected into COS-1 cells. Western blots of nuclear and cytoplasmic extracts show that a portion of each of these is recovered in the cytoplasmic fraction, much like the endogenous protein, and regardless of the time of exposure time, U2AF65 was not detectable in these fractions. Beads containing immobilized anti-myc monoclonal antibody were added to each of these extracts, and after being washed to remove unbound protein, a portion was analyzed by Western blotting to confirm the recovery of each of these proteins (Fig. 4B). We next examined GMP binding by each of the immunoprecipitated proteins. For the experiment shown in Fig. 4C, beads containing each of the immunoprecipitated proteins were incubated with [α-32P]GTP and washed to remove unbound isotope. Covalently bound GMP was identified by PhosphorImager analysis of a portion that was treated with SDS sample buffer and separated by SDS-PAGE (Fig. 4C). Regardless of whether they were recovered from nuclear or cytoplasmic extracts, immunoprecipitated capping enzyme and the 211 to 597 guanylylation domain were labeled following incubation with [α-32P]GTP (Fig. 4C, lanes 2, 4, 6, and 8). As anticipated, no labeling was observed for GFP (lanes 1 and 5) or the K294A active-site mutant (lanes 3 and 7).
FIG. 4.

In vitro capping of 5′-monophosphate RNA by immunoprecipitated cytoplasmic capping enzyme. (A) COS-1 cells were transfected with pcDNA3 expressing myc-tagged GFP (lanes 1 and 5), full-length mCE (lanes 2 and 6), mCE with the active-site K294A mutation (lanes 3 and 7), or the guanylylation domain (211-597) alone (lanes 4 and 8). Nuclear and cytoplasmic extracts were assayed by Western blotting with antibodies to the myc tag (top) or to U2AF65 (bottom). (B) Nuclear and cytoplasmic extracts from panel A were immunoprecipitated with immobilized anti-myc monoclonal antibody, and a portion was analyzed by Western blotting with an antibody to the myc tag. (C) Anti-myc antibody-containing beads with the remaining immunoprecipitated protein from panel B were incubated with [α-32P]GTP. A portion was removed after being washed to remove unincorporated GTP and separated on an SDS-PAGE gel, and the guanylylated proteins were visualized by a PhosphorImager. (D) The remaining beads from panel C were incubated with ATP and a 23-nt RNA with a 5′-monophosphate end (5′-P-RNA; top) or 5′-hydroxyl (5′-OH-RNA; bottom). The recovered RNA was treated with (+) or without (−) TAP and visualized by PhosphorImager analysis after electrophoresis on a 15% polyacrylamide/urea gel. (E) RNAs recovered from the reaction with 5′-P-RNA in panel D were digested with P1 nuclease and separated by PEI-cellulose TLC. The mobility of unlabeled nucleotide standards is indicated on the right side of the autoradiograph. (F) Anti-myc antibody-containing beads containing immunoprecipitated GFP, mCE, or K294A from panel B were incubated with 5′-P-RNA and [γ-32P]ATP. The recovered RNA was then electrophoresed on a 15% polyacrylamide/urea gel and visualized by a PhosphorImager.
The remaining beads shown in Fig. 4C were used to determine the ability of immunoprecipitated protein to transfer covalently bound [32P]GMP onto RNA. For the experiment shown in Fig. 4D, the beads containing [32P]GMP-labeled cytoplasmic proteins were incubated in buffer containing ATP and a 23-nt HPLC-purified RNA that was synthesized with a 5′-monophosphate (5′-P-RNA) or a 5′-hydroxyl (5′-OH-RNA) terminus. RNA recovered from each of these reactions was then separated on a denaturing 15% polyacrylamide/urea gel and visualized by PhosphorImager analysis. No labeling was observed when immunoprecipitated proteins were incubated with RNA containing a 5′-hydroxyl end (5′-OH-RNA) (Fig. 4D, bottom). However, 5′-monophosphorylated RNA (5′-P-RNA) was labeled by immunoprecipitated capping enzyme (Fig. 4D, top, lane 3) and the 211 to 597 guanylylation domain (lane 7). The label was removed by digestion with TAP, indicating that the reaction generated a product with a pyrophosphate bond.
Adenosine is the first nucleotide of the substrate RNA, and if the [32P]GMP labeling shown in Fig. 4D is generated by capping, 5′-P-RNA would have to be converted to a diphosphate, which upon GMP addition would become Gp*ppA. This was shown first in Fig. 4E, where the reaction products from Fig. 4D were digested with P1 nuclease and separated by PEI-cellulose thin-layer chromatography (TLC). In keeping with the results shown in Fig. 4D, full-length capping enzyme and the 211 to 597 guanylylation domain generated RNA with GpppA at the 5′ end, and no product was generated by GFP or the inactive K294A mutant. We suspect that the strong signal that remained at the origin was due to insufficient digestion with P1 (see below). These data also suggest that a kinase capable of converting 5′-P-RNA into a diphosphate capping substrate was recovered with immunoprecipitated cytoplasmic capping enzyme. Evidence for such an enzyme was described more than 30 years ago (23, 27), but there are no contemporary reports. This was examined by incubation of the beads containing immunoprecipitated GFP, mCE, or K294A used for Fig. 4B with 5′-P-RNA and [γ-32P]ATP, followed by electrophoresis on a denaturing 15% polyacrylamide gel (Fig. 4F). Kinase activity was recovered with mCE and with K294A, even though the latter lacks the ability to bind and transfer GMP. For reasons that are not known, significantly less radioactivity was incorporated in this reaction than in reactions using [α-32P]GTP to label the cap, thus limiting our ability to characterize the products of this reaction by TLC.
Further evidence of the requirement for a 5′-monophosphate end was obtained by comparing products generated from 5′-P-RNA or 5′-OH-RNA that was converted to 5′-P-RNA with T4 polynucleotide kinase. For the experiment shown in Fig. 5A, immunoprecipitated K294A or the 211 to 597 forms of capping enzyme were incubated with [α-32P]GTP, ATP, and each of the 5′-monophosphate substrates, and the products that remained after P1 nuclease digestion were analyzed by PEI-cellulose TLC. The immunoprecipitated 211 to 597 guanylylation domain generated GpppA on each of these RNAs, and no labeling was observed with catalytically inactive enzyme. Digestion of the P1 product of kinased 5′-OH-RNA confirmed that the label came from GMP (Fig. 5B).
FIG. 5.
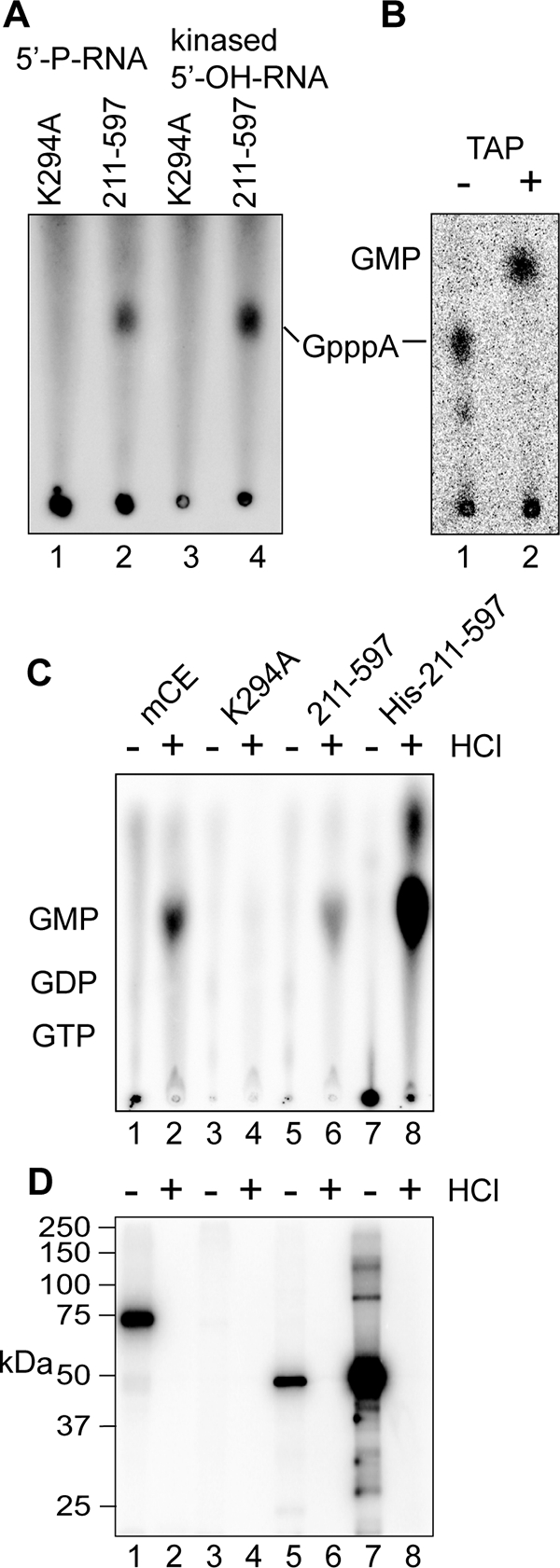
Identification of GMP as the nucleoside that is transferred from capping enzyme onto 5′-monophosphate RNA. (A) The 5′-OH-RNA used in Fig. 4D was phosphorylated by incubation with T4 polynucleotide kinase and ATP (kinased 5′-OH-RNA). This and the HPLC-purified 5′-P-RNA used in Fig. 4D were incubated as in that experiment with immunoprecipitated K294A or the 211-597 guanylylation domain plus [α-32P]GTP and ATP. The products recovered by ethanol precipitation were digested with P1 nuclease and separated by PEI-cellulose TLC, as in Fig. 4E. (B) Kinased 5′-OH-RNA was incubated as in panel A except with immunoprecipitated full-length mCE. The RNA recovered by ethanol precipitation was digested with P1 nuclease and analyzed without further treatment on PEI-cellulose (lane 1, −), or treated with TAP prior to TLC (lane 2, +). (C and D) Full-length mCE (lanes 1 and 2), K294A (lanes 3 and 4), the 211 to 597 guanylylation domain (lanes 5 and 6), or a His-tagged recombinant form of the 211 to 597 guanylylation domain expressed in E. coli was immunoprecipitated from U2OS cells expressing each form of the enzyme (lanes 1 to 6) using beads containing anti-myc monoclonal antibody or bound onto Ni-nitrilotriacetic acid agarose (lanes 7 and 8). The immobilized proteins were incubated with [α-32P]GTP, as in Fig. 4C. After being washed to remove unbound GTP, the odd-numbered samples were adjusted to 0.1 N HCl, the even-numbered samples received water, and both were heated for 15 min at 70°C. (C) The acid-treated samples were then neutralized and the supernatants were applied directly to PEI-cellulose and separated as in panel A. (D) SDS sample buffer was added to the beads, and the guanylylated proteins recovered after being heated were separated by SDS-PAGE and visualized by a PhosphorImager.
Lastly, we confirmed that GMP was the nucleoside that was bound covalently to immunoprecipitated capping enzyme. For the experiment shown in Fig. 5C and D, the indicated forms of capping enzyme were immunoprecipitated as shown in Fig. 4. We also expressed the recombinant guanylylation domain (211 to 597) as a His-tagged protein in bacteria. Each protein bound to anti-myc monoclonal antibody or Ni-nitrilotriacetic acid beads was incubated with [α-32P]GTP to generate a covalent intermediate. After being washed and treated with SAP to remove unincorporated GTP, the samples were divided in half. One portion received HCl to 0.1 N, and water was added to the other, followed by 15 min of incubation at 70°C. The acid-hydrolyzed samples were neutralized, and the beads were recovered by centrifugation. The supernatants were analyzed by PEI-cellulose TLC to identify nucleosides that were released by acid hydrolysis (Fig. 5C), and SDS sample buffer was added to the beads to elute bound protein for analysis by SDS-PAGE (Fig. 5D). GMP was detected in the supernatant after acid hydrolysis of immunoprecipitated mCE, 211 to 597 and His-tagged 211 to 597 (Fig. 5C, lanes 2, 6, and 8), but not K294A (lane 4). The converse was seen by the guanylylation assay of the unhydrolyzed proteins shown in Fig. 5D. These data confirm that one or more proteins recovered with immunoprecipitated capping enzyme convert 5′-P-RNA into a substrate for the addition of GMP bound covalently to capping enzyme.
Development of cytoplasmic forms of capping enzyme.
The absence of U2AF6s and histone H4 in cytoplasmic extracts made it is unlikely that cytoplasmic capping enzyme leaked from the nucleus during cell lysis. However, to remove this potential complication from our focus on cytoplasmic capping, we developed forms of capping enzyme that are restricted to the cytoplasm. The functional domains of capping enzyme are shown in Fig. 6A. To create forms of the enzyme that are restricted to the cytoplasm, we deleted four amino acids from the putative C-terminal NLS (ΔNLS), added the 15-amino-acid HIV Rev NES (+NES), or engineered forms of the enzyme with both modifications. The effects of these on the distribution of capping enzyme determined by Western blotting are shown in Fig. 6B. Individually, each of the changes increased the relative amount of capping enzyme in the cytoplasm, but both modifications together (ΔNLS+NES) were needed to create forms of capping enzyme that are completely cytoplasmic (Fig 6B, lanes 8 and 10).
FIG. 6.
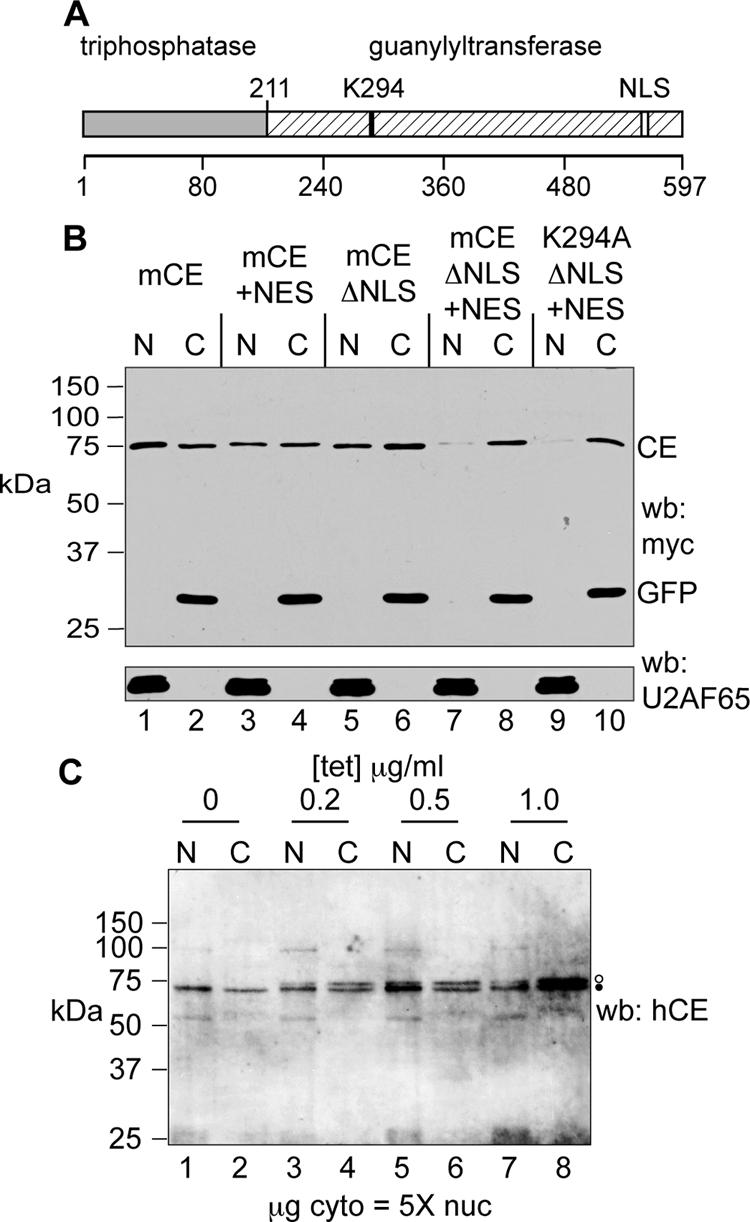
Development of forms of capping enzyme that are restricted to the cytoplasm. (A) The organization of mammalian capping enzyme is shown with the location of the guanylylation site (K294) and the NLS highlighted. (B) U2OS cells were transfected with plasmids expressing wild-type mCE (lanes 1 and 2), mCE with addition of the HIV Rev NES (mCE +NES; lanes 3 and 4), mCE with four amino acids deleted from the NLS (mCE ΔNLS; lanes 5 and 6), mCE ΔNLS+NES (lanes 7 and 8), and K294A ΔNLS+NES (lanes 9 and 10). Each of these proteins has an N-terminal myc tag and was cotransfected with a plasmid expressing myc-tagged GFP. Nuclear and cytoplasmic extracts were analyzed as in Fig. 3 by Western blotting with antibodies to the myc tag (top) and U2AF65 (bottom). (C) A stable line of tetracycline-inducible U2OS cells was generated that expresses mCE ΔNLS+NES with an N-terminal myc tag and a C-terminal FLAG tag. These were cultured overnight with the indicated concentrations of tetracycline, and nuclear (N) and cytoplasmic (C) extracts were analyzed by Western blotting (wb) with an affinity-purified rabbit anti-human capping enzyme antibody (hCE). Five times more cytoplasmic extract (on a μg of protein basis) than nuclear extract was applied to the gel. Endogenous capping enzyme is identified with a filled circle, and the slightly larger epitope-tagged protein is identified with an open circle.
Tetracycline-inducible forms of each of these proteins with an N-terminal myc tag and a C-terminal Flag tag were stably transfected into tetracycline-inducible U2OS cells, and individual clones were selected based on the absence of expression without added tetracycline and the overall range of induction. We also developed lines of these cells expressing myc-tagged wild-type mCE and GFP as a control. Before using these to further characterize cytoplasmic capping enzyme, we examined the inducible expression of the wild-type protein compared to that of the endogenous capping enzyme. For the experiment shown in Fig. 6C, mCE-expressing cells were cultured overnight in medium containing the indicated concentrations of tetracycline, and nuclear or five times more cytoplasmic protein was analyzed by Western blotting with an affinity-purified sheep anti-human capping enzyme antibody. Only the endogenous protein was detected in nuclear and cytoplasmic fractions of cells cultured in antibiotic-free medium (Fig. 6C, lanes 1 and 2). Expression of the slightly larger epitope-tagged protein became evident at 0.2 μg/ml tetracycline, and at 0.5 μg/ml, it approximated that of endogenous capping enzyme. Expression was only incrementally greater at 1 μg/ml.
Immunofluorescence identification of cytoplasmic capping enzyme.
Several of the inducible cell lines were used to examine the subcellular distribution of capping enzyme because the available antibodies are limited in their ability to detect low levels of capping enzyme by immunofluorescence. For the experiment shown in Fig. 7, cells expressing mCE or the K294A ΔNLS+NES protein were cultured with increasing concentrations of tetracycline, and fixed cells were examined by immunofluorescence using antibodies to the myc tag, YB1 (to visualize P bodies), and TIA-1. Staining for all three proteins is shown in the top panels of Fig. 7, and capping enzyme staining alone is shown in the lower panels. In the absence of tetracycline, there is no detectable myc staining even at 3× magnification of the field. As shown in Fig. 6C, recombinant capping enzyme appeared at 0.2 μg/ml tetracycline and increased with increasing concentrations in both cell lines. In agreement with Western blotting data, the K294A ΔNLS+NES protein was restricted to the cytoplasm, and wild-type capping enzyme showed predominantly nuclear staining at all concentrations examined. However, a 3× magnification of the indicated area showed diffuse staining punctate in the cytoplasm. In addition, the relative amount of cytoplasmic versus nuclear signal appeared to be similar to that determined by Western blotting shown in Fig. 3A and 5C.
FIG. 7.

Tetracycline-inducible lines of U2OS cells expressing wild-type mCE or the K294A ΔNLS+NES were cultured for 8 h in medium containing 0, 0.05, 0.2, 0.5, 1, and 2 μg/ml of tetracycline. Fixed cells were stained with antibodies to the myc tag on each form of capping enzyme (CE) (red), YB1 (green), and TIA-1 (blue). The merged micrographs are shown in the upper two panels, and the lower two panels show only the distribution of capping enzyme. Enlargements (3×) of the indicated areas of cells cultured without tetracycline or with 0.5 μg/ml tetracycline are shown beneath.
Neither form of capping enzyme shown in Fig. 7 appeared associated with YB1-positive P bodies. The absence of cytoplasmic capping enzyme at P bodies or at stress granules is shown using immunofluorescence in Fig. S2 in the supplemental material, where the localization of the various forms of capping enzyme in normal and arsenite-stressed cells was compared with that of YB1, Dcp1a, RCK (P bodies) and TIA-1, FXR1, and eIF4A (stress granules).
Identification of a cytoplasmic capping enzyme complex.
The recovery of a kinase activity with an immunoprecipitated capping enzyme suggested that cytoplasmic capping may be catalyzed by a complex that generates a capping substrate from 5′-P-RNA and transfers GMP onto this. This was first examined by glycerol gradient sedimentation. In the first experiment, the cytoplasmically restricted mCE ΔNLS+NES form of capping enzyme was induced to a level similar to that of endogenous cytoplasmic capping enzyme (Fig. 8A). Postmitochondrial extract was separated on a 10 to 40% glycerol gradient, and individual fractions were assayed by Western blotting with antibody to the myc tag on mCE ΔNLS+NES (Fig. 8B, top) and by guanylylation assay (Fig. 8B, middle). These proteins could be distinguished by guanylylation assay because the epitope-tagged recombinant protein is slightly larger than the endogenous capping enzyme. Both endogenous protein and mCE ΔNLS+NES sediment at 140 kDa, whereas GFP expressed in a matching tetracycline-inducible cell line is restricted to the top of the gradient.
FIG. 8.
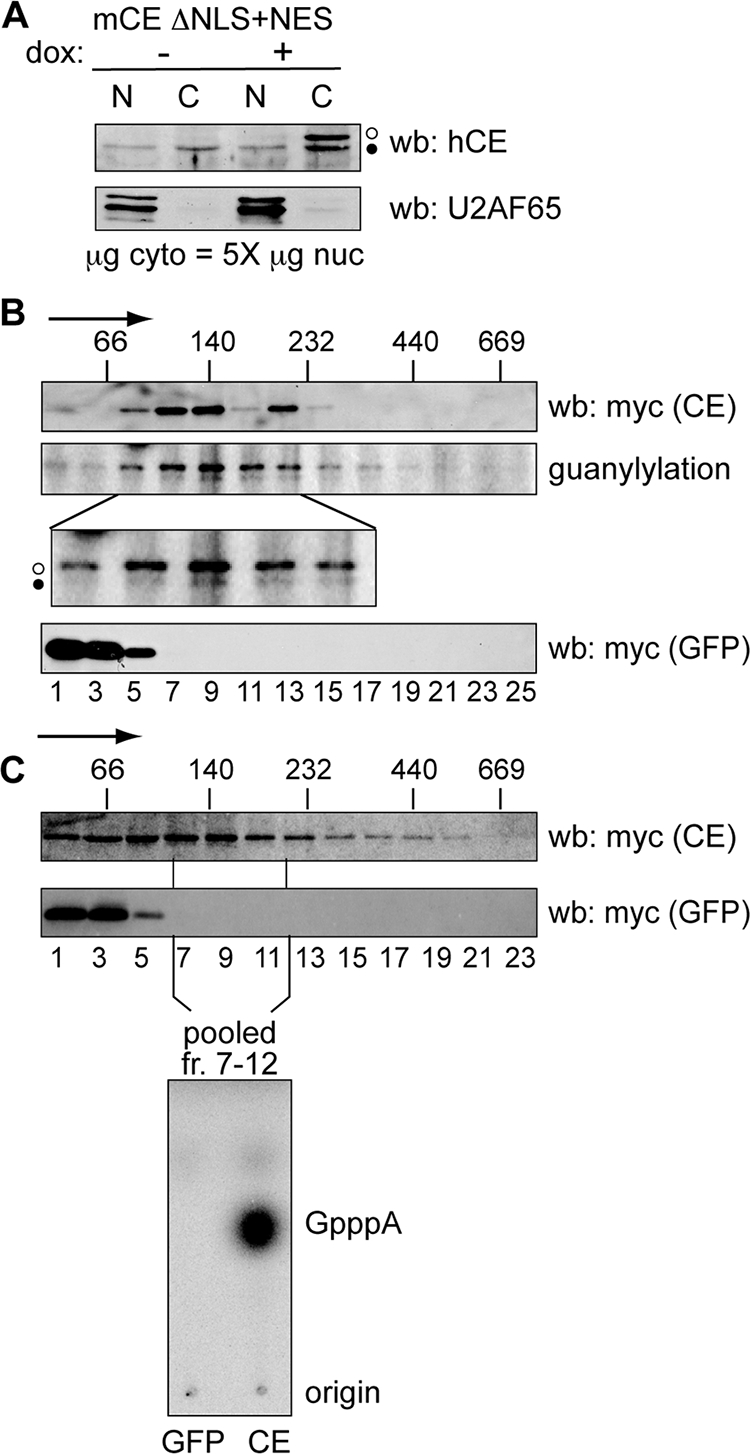
Identification of a cytoplasmic capping enzyme complex. (A) U2OS cells stably expressing mCE ΔNLS+NES were cultured for 24 h in the absence (−) or presence (+) of doxycycline (dox), and nuclear (N) and a fivefold-greater amount of cytoplasmic (C) protein was analyzed by Western blotting (wb) with affinity-purified capping enzyme antibody (top) or antibody to U2AF65 (bottom). The open circles correspond to recombinant protein and the filled circles correspond to endogenous capping enzyme. (B) Doxycycline was added to induce recombinant protein expression in cell lines stably transfected with tetracycline-inducible forms of mCE ΔNLS+NES or GFP. Cells were treated with 0.25% formaldehyde for 15 min prior to lysis, and cytoplasmic extracts were separated on linear 10 to 40% glycerol gradients. The sedimentation of mCE ΔNLS+NES or GFP was determined by Western blotting of individual fractions with antibody to the myc tag on each of these proteins, and the cosedimentation of mCE ΔNLS+NES (middle, open circle) with endogenous capping enzyme (CE) (filled circle) was determined by guanylylation assay. The lower panel is an enlargement of that portion of the gradient containing the 140-kDa capping enzyme complex. The positions of MW standards that were run in parallel gradients are indicated on the top panel, with the arrow indicating the direction of sedimentation. (C) mCE ΔNLS+NES and GFP were induced as in panel B by addition of tetracycline to lines of U2OS cells expressing each of these proteins. Cytoplasmic extracts were separated on 10 to 40% glycerol gradients, and capping enzyme and GFP were identified by Western blotting of individual fractions using antibody to the myc tag on each of these proteins. The fractions corresponding to the 140-kDa complex from panel B were pooled and recovered using beads containing immobilized anti-myc monoclonal antibody. These were incubated with [α-32P]GTP, ATP, and 5′-P-RNA, and RNA recovered from this reaction was digested with P1 nuclease and separated on PEI-cellulose TLC to identify the product of the capping reaction.
To test if this complex contains both of the proteins needed to add GMP onto 5′-P-RNA, the gradients were repeated, and fractions corresponding to the 140-kDa complex (identified by Western blotting) were pooled and recovered by immunoprecipitation with beads containing covalently bound antibody to the myc tag on both proteins (Fig. 8C). After being washed, these were incubated as shown in Fig. 4 with [α-32P]GTP, ATP, and 5′-P-RNA, and the recovered RNA was digested with P1 nuclease. PEI-cellulose TLC (Fig. 4, bottom) shows that immunoprecipitated capping enzyme complex recovered from the gradient converted the 5′ end of substrate RNA to GpppA, and no product was formed by the same gradient fractions from GFP-expressing cells. These data indicate that cytoplasmic capping enzyme participates in a complex that contains the enzymes necessary to convert 5′-monophosphate RNA to a diphosphate capping substrate and add GMP onto this.
Because the size of the capping enzyme complex is sufficient to also contain cap methyltransferase, we examined its recovery with capping enzyme from nuclear and cytoplasmic extracts and its sedimentation in the gradient shown in Fig. 8C (see Fig. S3 in the supplemental material). Cap methyltransferase does not bind directly to capping enzyme, and only a faint signal was observed on an overexposed blot for protein recovered from nuclear extract. Although cap methyltransferase was present in cytoplasmic extracts, it did not sediment with the 140-kDa capping enzyme complex but instead was at its approximate size on the top of the gradient. Thus, any methylation that might occur after GMP addition is unlikely to occur in this complex.
Impact of cytoplasmic capping enzyme on cellular recovery from stress.
Because a definitive method for quantifying capped rather than uncapped mRNAs has yet to be described, we looked for biological evidence of cytoplasmic capping using the tetracycline-inducible lines described above. Sodium arsenite-induced oxidative stress results in a generalized inhibition of protein synthesis and causes mRNAs to accumulate in P bodies and mRNAs bound by 48S subunits to accumulate in stress granules (12). For the experiment shown in Fig. 9, each of the tetracycline-inducible cell lines described above was cultured overnight with 2 μg/ml of doxycycline to maximally induce recombinant protein expression and then treated for 40 min with or without sodium arsenite. Cell viability was determined 24 h later using a luminescence assay that quantifies the relative amount of ATP present in each culture. Arsenite stress reduced the viability of each of the cell lines by 20 to 25% compared to that of the unstressed controls, and this was unchanged in cells expressing GFP or wild-type cytoplasmic capping enzyme (mCE ΔNLS+NES). Importantly, arsenite was twice as toxic to cells that overexpress the K294A ΔNLS+NES form of capping enzyme, reducing viability to 50% of that of unstressed cells. These results indicate that the inactive form of cytoplasmic capping enzyme had a dominant-negative effect on the ability of cells to recover from stress. Since this enzyme differs from mCE ΔNLS+NES by only a single active site residue, we conclude that survival following stress is enhanced by cytoplasmic capping.
FIG. 9.

Impact of expressing cytoplasmic forms of capping enzyme on cellular recovery from stress. Cultures of U2OS cells stably transfected with plasmids expressing tetracycline-inducible GFP or K294A ΔNLS+NES (left) or GFP or mCE ΔNLS+NES (right) were cultured overnight without (−) or with (+) 2 μg/ml of doxycycline (Dox). The next day, one-half of each set was exposed for 40 min to 0.5 mM sodium arsenite, then washed, and placed into fresh medium. Cell viability was determined 24 h later using the CellTiter-Glo luminescent cell viability assay. Each bar represents the mean ± standard deviation for triplicate samples.
DISCUSSION
Mammalian cells have only one enzyme capable of adding a cap onto RNA, and the cotranscriptional capping of newly transcribed pre-mRNA is a well-characterized process. Nevertheless, in 1992, Lim and Maquat (15) noted that 5′-truncated forms of β-globin mRNA recovered from erythroid cells of transgenic mice expressing a nonsense-containing human gene were modified with a cap or caplike structure, and the current study sought to determine if these products are indeed capped. The case against cytoplasmic capping seemed straightforward. These shorter RNAs are generated in the cytoplasm (2, 28), and the downstream products of endonuclease cleavage have a 5′-monophosphate end. To be capped, the 5′-monophosphate end would have to be converted to a di- or triphosphate by a polynucleotide 5′-monophosphate kinase. The only reports of such an activity were published more than 30 years ago (23, 27). Given these points, we suspected that the previous study identified some other type of modification that behaved like a cap.
Several different approaches indicated that these 5′-truncated RNAs are indeed capped. These included selection with cap antibody and eIF4E (Fig. 1), competition for binding to eIF4E by m7GpppG but not ApppG, and loss of recovery after cap hydrolysis. In addition, these RNAs were resistant to degradation by a 5′-monophosphate-dependent exonuclease unless they were first treated with TAP or Dcp2 (Fig. 2). We considered the possibility that capping might be catalyzed by a cellular ortholog of the vesicular stomatitis virus L protein, a novel capping enzyme that forms a covalent complex with the 5′-GDP end of the viral transcript (20). However, there was no evidence for this in database searches, and Western blotting with affinity-purified capping enzyme antibody and use of a sensitive guanylylation assay identified capping enzyme in cytoplasmic extracts of MEL, HeLa, COS-1, U2OS, and Huh7 cells. The presence of a nondialyzable guanylylation inhibitor in cytoplasmic extracts made Western blotting the more reliable assay, and based on protein assays, five- to eightfold more cytoplasmic rather than nuclear extract was needed to see a similar signal, indicating that only a fraction of cellular capping enzyme is in the cytoplasm. This was confirmed by immunofluorescence (Fig. 7). Despite the large excess of cytoplasmic protein used to normalize the signal for capping enzyme, neither U2AF65 nor histone H4 was detected in any of our blots.
As noted above, for capping to occur in the cytoplasm, RNA with a 5′-monophosphate end would have to be converted to a di- or triphosphate by an unknown polynucleotide 5′-monophosphate kinase. hClp1, the first identified RNA kinase (33), does not seem a likely candidate, because it acts on RNA with a 5′-hydroxyl and only adds a single phosphate. We reasoned that the putative kinase would likely be in close association with cytoplasmic capping enzyme for the latter to function efficiently, and the results depicted in Fig. 4 show that a kinase activity recovered with immunoprecipitated cytoplasmic capping enzyme transfers the gamma phosphate from ATP onto RNA with a 5′-monophosphate terminus to create substrate for transfer of GMP covalently bound to capping enzyme. Importantly, immunoprecipitated cytoplasmic capping enzyme did not add GMP onto RNA with a 5′-hydroxyl terminus unless this was first treated with ATP and T4 polynucleotide kinase (Fig. 4 and 5). Because protein that lacked the triphosphatase domain but was otherwise active (211/597) was as effective as full-length capping enzyme in transferring GMP, the proximal product must have a 5′-diphosphate and not a triphosphate end.
The recovery of a polynucleotide 5′-monophosphate kinase activity with immunoprecipitated cytoplasmic capping enzyme suggested that both proteins act together in a complex. Cytoplasmic capping enzyme sediments at 140 kDa on glycerol gradients, and while there was evidence of this on gradients from uncross-linked cells, it was more apparent when cells were treated briefly with formaldehyde just prior to lysis. Mili and Steitz (18) showed that formaldehyde cross-linking mitigates the rearrangement of RNP complexes, and we suspect that cross-linking prevents dissociation of the cytoplasmic capping enzyme complex during the 20 h of centrifugation. Importantly, the results depicted in Fig. 8C show that the immunoprecipitated 140-kDa fractions from the capping enzyme gradient add [32P]GMP onto 5′-monophosphate RNA, whereas the same fractions from a GFP gradient do not. These results support the identity of the 140-kDa complex as the functional unit seen in Fig. 4. There is no nuclear capping enzyme complex per se; rather, capping enzyme binds to the phosphorylated C-terminal domain of elongating polymerase II (3, 8). Despite numerous attempts with different extraction techniques, we saw no evidence for the 140-kDa complex in nuclei.
To facilitate our understanding and analysis of cytoplasmic capping, we developed tetracycline-inducible cell lines expressing wild-type protein and the inactive K294A form of capping enzyme, both also containing modifications that restrict these proteins to the cytoplasm (mCE ΔNLS+NES, K294A ΔNLS+NES). The fact that mCE ΔNLS+NES assembles into the same-size complex as endogenous capping enzyme indicates that this form of capping enzyme can be used to study the nature of this complex. The kinase that creates the capping substrate is one of the proteins in this complex, and work is in progress to identify this and any other proteins associated with cytoplasmic capping enzyme. The other protein, K294A ΔNLS+NES, differs in having an alanine instead of the active-site lysine. Its distribution does not change in response to stress (see Fig. S2 in the supplemental material), so its apparent dominant negative effect must be limited to the pool of cytoplasmic mRNAs whose cap status changes either in response to stress or during the recovery period after stress.
The findings presented here raise the question, why is there capping in the cytoplasm at all? Capping of β-globin mRNA decay intermediates may be coincidental and perhaps the result of their prolonged (12-h) half-lives in erythroid cells. Stable downstream cleavage products were also seen with other mRNAs following cleavage by an antisense oligonucleotide (29). Some (and perhaps most) stored or silenced mRNAs accumulate in P bodies (1, 6), and while 5′-3′ decay can occur in visible P bodies, this process is actually quite slow (19). Since Dcp1 and Dcp2 are concentrated in P bodies, some mRNAs may be stored in a decapped state and reactivated for translation by cytoplasmic capping. Until recently, there was little evidence for uncapped mRNAs. Jiao et al. (11) developed a two-step method of separating capped from uncapped mRNAs from different stages of Arabidopsis development and used microarrays to identify populations of uncapped mRNAs for much of the transcriptome. Importantly, the relative quantity of uncapped mRNAs was elevated for transcripts encoding kinases, transporters, and nucleotide-binding proteins, is dependent on structural elements in the 5′ untranslated region, and varied with different growth states. Our data suggest at least two possible roles for cytoplasmic capping. One possibility is that cytoplasmic capping serves a surveillance/repair function in which fortuitously decapped mRNAs are recapped for the return to the translating pool. Alternatively, some silenced mRNAs may be stored in an uncapped state and, analogous to cytoplasmic polyadenylation, are activated for translation by cytoplasmic capping.
ADDENDUM IN PROOF
A report published after acceptance of this paper describes newly identified populations of short, capped RNAs that arise from posttranscriptional modification of longer transcripts (K. Fejes-Toth et al., Nature 457:1028-1032, 2009). Their observations are consistent with the recapping of β-globin mRNA decay intermediates described here, and it is likely that the cytoplasmic capping enzyme complex described here is responsible for the posttranslational recapping described in that report.
Supplementary Material
Acknowledgments
We thank Mike Kiledjian and Aaron Shatkin (Rutgers University), Brent Graveley (University of Connecticut), Jerry Pelletier (McGill University), Jens Lykke-Andersen (University of Colorado), and David Price (University of Iowa) for reagents. We also thank Mike Kiledjian, Aaron Shatkin, Kiong Ho, and members of the Schoenberg lab for their helpful suggestions.
This work was supported by PHS grants R01 GM38277 and R21 DK067035 to D.R.S. Y.O. was supported by a postdoctoral fellowship from the American Heart Association, Great Rivers division, and N.L.K. was supported by NIH grants R01 AI033600 and R01 AR051472. Support for shared facilities was provided by PHS grant P30 CA16058 from the National Cancer Institute to the Ohio State University Comprehensive Cancer Center.
Author contributions were as follows. Y.O. engineered the cell lines and designed and performed all of the biochemical experiments. N.L.K. designed and performed all of the microscopy experiments. D.R.S. designed experiments, oversaw and coordinated all of the experiments, prepared the graphics, and drafted the manuscript. All three authors participated in data analysis and the writing of this paper. We certify that all authors have agreed to the content of this paper.
Footnotes
Published ahead of print on 17 February 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bhattacharyya, S. N., R. Habermacher, U. Martine, E. I. Closs, and W. Filipowicz. 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 1251111-1124. [DOI] [PubMed] [Google Scholar]
- 2.Bremer, K. A., A. Stevens, and D. R. Schoenberg. 2003. An endonuclease activity similar to Xenopus PMR1 catalyzes the degradation of normal and nonsense-containing human beta-globin mRNA in erythroid cells. RNA 91157-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho, E. J., T. Takagi, C. R. Moore, and S. Buratowski. 1997. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 113319-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cougot, N., S. Babajko, and B. Seraphin. 2004. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 16531-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eberle, A. B., S. Lykke-Andersen, O. Muhlemann, and T. H. Jensen. 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 1649-55. [DOI] [PubMed] [Google Scholar]
- 6.Eulalio, A., I. Behm-Ansmant, and E. Izaurralde. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 89-22. [DOI] [PubMed] [Google Scholar]
- 7.Garneau, N. L., J. Wilusz, and C. J. Wilusz. 2007. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8113-126. [DOI] [PubMed] [Google Scholar]
- 8.Ho, C. K., V. Sriskanda, S. McCracken, D. Bentley, B. Schwer, and S. Shuman. 1998. The guanylyltransferase domain of mammalian mRNA capping enzyme binds to the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2739577-9585. [DOI] [PubMed] [Google Scholar]
- 9.Hosoda, N., Y. K. Kim, F. Lejeune, and L. E. Maquat. 2005. CBP80 promotes interaction of Upf1 with Upf2 during nonsense-mediated mRNA decay in mammalian cells. Nat. Struct. Mol. Biol. 12893-901. [DOI] [PubMed] [Google Scholar]
- 10.Huntzinger, E., I. Kashima, M. Fauser, J. Sauliere, and E. Izaurralde. 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 142609-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao, Y., J. L. Riechmann, and E. M. Meyerowitz. 2008. Transcriptome-wide analysis of uncapped mRNAs in Arabidopsis reveals regulation of mRNA degradation. Plant Cell 202571-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kedersha, N., G. Stoecklin, M. Ayodele, P. Yacono, J. Lykke-Andersen, M. J. Fitzler, D. Scheuner, R. J. Kaufman, D. E. Golan, and P. Anderson. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169871-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwak, J. E., L. Wang, S. Ballantyne, J. Kimble, and M. Wickens. 2004. Mammalian GLD-2 homologs are poly(A) polymerases. Proc. Natl. Acad. Sci. USA 1014407-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim, S., J. J. Mullins, C. M. Chen, K. W. Gross, and L. E. Maquat. 1989. Novel metabolism of several β°-thalassemic β-globin mRNAs in the erythroid tissues of transgenic mice. EMBO J. 82613-2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim, S. K., and L. E. Maquat. 1992. Human beta-globin mRNAs that harbor a nonsense codon are degraded in murine erythroid tissues to intermediates lacking regions of exon I or exons I and II that have a cap-like structure at the 5′ termini. EMBO J. 113271-3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim, S. K., C. D. Sigmund, K. W. Gross, and L. E. Maquat. 1992. Nonsense codons in human beta-globin mRNA result in the production of mRNA degradation products. Mol. Cell. Biol. 121149-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maquat, L. E. 2004. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 589-99. [DOI] [PubMed] [Google Scholar]
- 18.Mili, S., and J. A. Steitz. 2004. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 101692-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray, E. L., and D. R. Schoenberg. 2007. A+U-rich instability elements differentially activate 5′-3′ and 3′-5′ mRNA decay. Mol. Cell. Biol. 272791-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogino, T., and A. K. Banerjee. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 2585-97. [DOI] [PubMed] [Google Scholar]
- 21.Piccirillo, C., R. Khanna, and M. Kiledjian. 2003. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA 91138-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richter, J. D., and N. Sonenberg. 2005. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433477-480. [DOI] [PubMed] [Google Scholar]
- 23.Schibler, U., and R. P. Perry. 1976. Characterization of the 5′ termini of hn RNA in mouse L cells: implications for processing and cap formation. Cell 9121-130. [DOI] [PubMed] [Google Scholar]
- 24.Schoenberg, D. R., and K. S. Cunningham. 1999. Characterization of mRNA endonucleases. Methods Companion Methods Enzymol. 1760-73. [DOI] [PubMed] [Google Scholar]
- 25.Shatkin, A. J., and J. L. Manley. 2000. The ends of the affair: capping and polyadenylation. Nat. Struct. Mol. Biol. 7838-842. [DOI] [PubMed] [Google Scholar]
- 26.Sheth, U., and R. Parker. 2006. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell 1251095-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer, E., D. Loring, J. Hurwitz, and G. Monroy. 1978. Enzymatic conversion of 5′-phosphate-terminated RNA to 5′-di- and triphosphate-terminated RNA. Proc. Natl. Acad. Sci. USA 754793-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stevens, A., Y. Wang, K. Bremer, J. Zhang, R. Hoepfner, M. Antoniou, D. R. Schoenberg, and L. E. Maquat. 2002. Beta-globin mRNA decay in erythroid cells: UG site-preferred endonucleolytic cleavage that is augmented by a premature termination codon. Proc. Natl. Acad. Sci. USA 9912741-12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thoma, C., P. Hasselblatt, J. Kock, S. F. Chang, B. Hockenjos, H. Will, M. W. Hentze, H. E. Blum, F. von Weizsacker, and W. B. Offensperger. 2001. Generation of stable mRNA fragments and translation of N-truncated proteins induced by antisense oligodeoxynucleotides. Mol. Cell 8865-872. [DOI] [PubMed] [Google Scholar]
- 30.Tucker, M., and R. Parker. 2000. Mechanisms and control of mRNA decapping in Saccharomyces cerevisiae. Annu. Rev. Biochem. 69571-595. [DOI] [PubMed] [Google Scholar]
- 31.Unterholzner, L., and E. Izaurralde. 2004. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 16587-596. [DOI] [PubMed] [Google Scholar]
- 32.Wang, Z., X. Jiao, A. Carr-Schmid, and M. Kiledjian. 2002. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc. Natl. Acad. Sci. USA 9912663-12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weitzer, S., and J. Martinez. 2007. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature 447222-226. [DOI] [PubMed] [Google Scholar]
- 34.Yue, Z., E. Maldonado, R. Pillutla, H. Cho, D. Reinberg, and A. J. Shatkin. 1997. Mammalian capping enzyme complements mutant Saccharomyces cerevisiae lacking mRNA guanylyltransferase and selectively binds the elongating form of RNA polymerase. Proc. Natl. Acad. Sci. USA 9412898-12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.