

Abstract
A combination of LC and MS was applied to an isogenic breast tumor metastasis model to identify proteins associated with a cellular phenotype. Chromatofocusing followed by nonporous-RP-HPLC/ESI-TOF MS was applied to cell lysates of a pair of monoclonal cell lines from the human breast carcinoma cell line MDA-MB-435 that have different metastatic phenotypes in immune-compromised mice. This method was developed to separate proteins based on pI and hydrophobicity. The high resolution and mass accuracy of ESI-TOF measurements provided a good correlation of theoretical MW and experimental Mr values of intact proteins measured in mass maps obtained in the pH range 3.8–6.4. The isolated proteins were digested by trypsin and analyzed by MALDI-TOF MS, MALDI-QIT-TOF MS, and monolith-based HPLC/MS/MS. The unique combination of the techniques provided valuable information including quantitation and modification of proteins. We identified 89 selected proteins, of which 43 were confirmed as differentially expressed. Metastasis-associated proteins included galectin-1, whereas annexin I and annexin II were associated with the nonmetastatic phenotype. In this study, we demonstrate that combining a variety of MS tools with a multidimensional liquid-phase separation provides the ability to map cellular protein content, to search for modified proteins, and to correlate protein expression with cellular phenotype.
Keywords: Cancer biomarkers, Electrospray ionization time-of-flight mass spectrometry, Liquid chromatography, Metastasis, Monolith
1 Introduction
Breast cancer is the second most common cause of cancer-related deaths among American women, largely due to the recurrence of disseminated disease. The ability to predict the metastatic behavior of a patient’s tumor and to eradicate or control recurrent disseminated malignancy remain major clinical challenges in oncology. Unfortunately, the multistep nature of metastasis poses difficulties in both design and interpretation of experiments to unveil the mechanisms causing the process. Studies on excised fixed human tissues are complicated by the variance of genetic background between individuals and by the cellular heterogeneity of a complex tissue mass. Thus, research into the mechanisms of metastasis requires appropriate models which are amenable to detailed genetic analysis. In order to facilitate the investigation of this complex multistep process, we have developed an experimental system in which the role of candidate metastasis genes can be screened and tested [1, 2]. Monoclonal cell lines M4A4 and NM2C5 are spontaneously occurring sublines of the MDA-MB-435 cell breast tumor cell line [3] which exhibit many phenotypic differences in growth, invasion, dissemination, and spontaneous metastatic efficiency from an orthotopic site. The common origin of these cell lines enables the comparative investigation of cellular and molecular events in the metastatic process in a stable and isogeneic model. We have performed several studies on these cell lines, comparing cellular and tumor morphology [1], dissemination patterns in vivo [2], cytogenetic composition [4], and selective and global transcriptional profiles [5, 6]. However, while molecular changes during phenotypic transformation clearly occur at the level of transcription, it is the protein complement that ultimately determines the cellular phenotype. Furthermore, changes occur not only in protein abundance, but also involve protein structural changes such as sequence variation or PTM. Thus, it is important to develop rapid, accurate, and reproducible protein mapping techniques for the study of tumor cell protein content since many of the changes that occur in tumor cell transformation may be manifested only in the proteome.
A 2-D liquid separation-mass mapping method has been developed in our laboratory for the analytical profiling of proteins in complex biological material [7–9]. The analytical approach fractionates proteins based upon pI in the first dimension by chromatofocusing (CF), and each fraction is subsequently separated based upon polarity of the protein in the second dimension by nonporous-RP-HPLC (NPS-RP-HPLC). The protein eluants from RP-HPLC are directed online to ESI TOF-MS to obtain an accurate and reproducible protein intact molecular weight. Purified proteins fractionated by means of the 2-D liquid separation are collected and stored for further analysis by peptide mapping using matrix-assisted laser desorption-ionization TOF MS (MALDI-TOF MS). The combination of these methods enables high accuracy protein profile comparisons between biological samples. The mass mapping method also provides the ability to identify protein expression patterns that are not based solely on abundance. The combination of peptide mapping with the highly accurate intact protein Mr data enables the elucidation of peptide-specific alterations. Thus, the mass mapping technique can provide comparative information on truncated or alternatively spliced protein isoforms, and on specific PTM of proteins in complex biological samples. Furthermore, in this study we employed monolithic capillary columns synthesized with silica or polymer [10–12]. These columns provide a number of distinct advantages derived from fast mass transfer kinetics [13] due to the lack of interstitial space, and which enable high resolution, high efficiency, and high recovery [14] in a rapid separation time. These unique characteristics are ideally suited for the analysis of complex proteomes [7, 15, 16].
In the present study, we applied the liquid-phase 2-D technology approach to investigate whether proteomic differences could be found that distinguished the metastatic M4A4 and nonmetastatic NM2C5 cell lines. Thirteen fractions were collected (pH range 3.8–6.4) containing a total of approximately 700 unique proteins from each cell line, with molecular weights ranging from 5 to 80 kDa. Candidate differentially expressed proteins were subsequently collected in the liquid phase and identified by ESI-TOF MS and MALDI-TOF-MS mass mapping. The monolithic LC/MS/MS proved useful in confirming protein identifications and elucidating important PTMs. Forty-three candidate proteins which displayed a more than two-fold difference in expression level in metastatic cancer cells compared to nonmetastatic cells, were identified. Twelve proteins were up-regulated and 15 proteins were down-regulated in the metastatic M4A4 sample. Furthermore, 16 proteins were detected as being uniquely expressed in either cell line. The proteomic profiling approach described here enables the identification of phenotype-associated proteins for subsequent functional analyses and disease biomarker development.
2 Materials and methods
2.1 Cell lines
Human mammary tumor cell lines M4A4 and NM2C5 were derived from breast tumor cell line MDA-MB-435 as described previously [1]. Cell lines were maintained as subconfluent monolayer cultures in RPMI 1640 medium (Gibco-BRL, NY) supplemented with 10% fetal calf serum at 37°C under 5% CO2/95% air. The medium was exchanged every third day, and passaging was routinely performed as cultures reached ~75% confluency. In order to limit any potential biological variability when performing a comparative profiling study of these tumor cell lines, analyses were performed on cultures passaged no more than ten times from frozen stock vials designated passage 1 at the time of initial monoclonal expansion, and cell lines were cultured under parallel conditions such that feeding and passaging regime, confluency, and all serum/media batches were exactly the same for both lines.
2.2 Sample preparation
Approximately 60–90 million cells were used for each preparation of CF. The cell pellets were allowed to thaw at room temperature and were quickly mixed with lysis buffer. The lysis buffer was composed of 6 M urea, 2 M thiourea, 2% w/v n-octyl β-D-glucopyranoside (OG), 5 mM TCEP, 1 mM PMFS (Sigma, St. Louis, MO), 10% v/v glycerol, and 10 mM tris (2-carboxyethyl) phosphine. After 30 min of lysis at room temperature, the sample was centrifuged at 35 000 rpm for 1 h at 4°C. The supernatant was recovered and total protein present was determined using a Bradford-based assay (BioRad, Hercules, CA).
2.3 CF
Prior to CF separation, the lysis buffer in the whole cell lysate was replaced by a start buffer (25 mM bis-tris propane, 6 M urea, 0.2% w/v OG, pH 6.4 ± 0.1) using a PD-10 column (Amersham Biosciences, Piscataway, NJ). The Bradford method was performed to determine the relative protein contents of the samples. Around 2.5 mg of buffer-exchanged sample was loaded onto a CF column (Eprogen, Darien, IL). The protein was eluted from the column by using a 0.2 mL/min of an eluting buffer, which contained 8% v/v polybuffer 74 (Amersham Biosciences ), 6 M urea, 0.2% w/v OG, pH 4.0 ± 0.1. Saturated iminodiacetic acid was used to adjust the pH of all CF buffers. Online pH measurement was monitored by a postdetector pH flow cell (Lazer Research Laboratories, Los Angeles, CA) and the eluent was collected from pH 6.4 to 3.8 every 0.2 pH unit intervals. CF fractions were stored at −80°C for further analysis.
2.4 NPS-RP-HPLC/ESI-TOF MS
The NPS-RP-HPLC experiment was performed on an ODSIII-E (4.6 mm × 33 mm) column packed with 1.5 μm C18 nonporous silica beads (Eprogen). The column was maintained at 65°C in a column heater (Timberline, Boulder, CO) to improve the resolution and the speed of separation. Each CF fraction was loaded for separation with the HPLC System Gold equipped with UV detector (Beckman Coulter, Fullerton, CA). The solvent system, 0.1% v/v TFA in water (solvent A) and 0.1% v/v TFA in ACN (solvent B), was used to generate a gradient elution profile as follows: 5–15% B in 1 min, 15–25% B in 2 min, 25–31% B in 3 min, 31–41% B in 10 min, 41–47% B in 3 min, 47–67% B in 4 min, and 67–100% B in 1 min. The flow rate was set at 0.5 mL/min, which was split postcolumn to produce a flow rate of 200 μL/min through a premixing chamber and of 300 μL/min through a UV detector. A syringe pump was set at 25 μL/min to deliver 10% formic acid (Sigma) to the premixing chamber and the final flow was delivered to the source of the ESI-TOF MS (LCT system, Micromass, Manchester, UK) for Mr analysis of intact proteins. The Beckman model 166 detector was set at 214 nm for detection of proteins. The proteins eluting from the column were collected using an automated fraction collector, controlled by an in-house designed DOS-based software program. Collected fractions were subjected to proteolytic digestion for MALDI-TOF MS analysis. For these experiments, capillary voltage was 3250 V, desolvation temperature was 350°C, source temperature was 150°C, desolvation gas flow 600 L/h, and maximum nebulizer gas flow. One MS spectrum was acquired per second. Prior to each experiment, the instrument was externally calibrated by direct infusion of NaI-CsI solution using a syringe pump. To assist quantitation, 1 μg of bovine insulin (Sigma) was added as an internal standard to each CF fraction prior to each run.
2.5 ESI-TOF data analysis
ESI-TOF data analysis was performed using MassLynx version 4.0 (Waters-Micromass). The MaxEnt 1 deconvolution algorithm was used to perform the deconvolution of the multiple-charged-ion umbrella of the ESI-TOF MS spectrum into a MaxEnt spectrum on a real-mass scale. The MaxEnt peaks were converted to the intensity bars where peak height was proportional to its peak area. For each separation, deconvoluted molecular weights were pooled into a single mass spectrum. They were converted to a single text file and imported into a single mass mapping program. Multiple separations could be combined to produce a virtual 1-D gel or mass map, to aid in comparisons of protein levels between different samples.
2.6 Tryptic digestion and MALDI sample preparation
The protein fractions collected from NPS-RP-HPLC were concentrated to 20 μL using a SpeedVac concentrator (Labconco, Kansas City, MO) operated at 45°C. A 30 μL of 50 mM ammonium bicarbonate (Sigma) at pH 7.8 was then added to neutralize the remaining TFA in the samples. The sample was vortexed and 0.5 μg of TPCK modified sequencing grade trypsin (Promega, Madison, WI) was added for digestion. The samples were vortexed again and incubated at 37°C for 18 h. The digestion was terminated by adding 10 μL of 10% v/v TFA to the digest. A 30 μL aliquot of tryptic digestion mixture was dried completely and reconstituted in 5 μL of deionized water for later monolith-based LC/MS/MS analysis. The rest of the tryptic digests was desalted and concentrated using a 2 μm C18 ZipTip (Millipore, Bedford, MA). The peptides were eluted in 5 μL of 60% v/v ACN with 0.5% v/v TFA. The samples were stored for MALDI-TOF MS and MALDI-QIT-TOF MS analysis.
2.7 MALDI-TOF MS
The matrix-standard solution was prepared by first diluting a saturated α-CHCA (Sigma) with three parts of 1% v/v TFA and 60% v/v ACN. Angiotensin I (1294 Da), adrenocorticotropic hormone (ACTH) chip 1–17 (2093 Da), and ACTH chip 18–39 (2465 Da) were added to the diluted matrix solution to serve as internal peptide standards. The final concentration of each internal standard was 50 fmol in each spot on the MALDI plate. A 1 μL aliquot of the peptide sample was spotted on to a Micromass 96-spot plate followed by 1 μL of matrix/internal standard solution on the top for cocrystallization.
PMFs of the digest sample were generated by a TofSpec 2E (Waters-Micromass) in reflectron mode, with 20 kV total acceleration and a pulse voltage of 2450 V at a 520 ns delayed extracting time. The peptide masses were measured from 500 to 4000 m/z and each final spectrum was the summed average of 80–150 spectra collected at a rate of four spectra per second. Using the internal standard signals, the peptide mass spectra were internally calibrated and consistently achieved a mass accuracy of 50 ppm or less. The Masslynx version 4.0 (Waters-Micromass) was used to process the calibrated spectra and the monoisotopic experimental masses were selected and submitted to MS-Fit (http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm) to search the NCBI/Swiss-Prot databases for protein identification. The search was performed using the following parameters; (i) species: Homo sapiens (ii) allowing one missed cleavage (iii) possible modifications: peptide N-terminal Gln to pyroGlu, oxidation of M, protein N-terminal acetylated, and phosphorylation of S, T, and Y (iv) MW ranged from 1000 to 100 000 and (v) pI range of protein was 3–10. The peptide mass tolerance was 50–100 ppm.
2.8 Monolithic HPLC MS/MS
Monolithic capillary columns (200 μm id × 60 mm length) were prepared by copolymerizing styrene and divinylbenzene according to the protocol described elsewhere [7]. The Ultra-Plus II MD Capillary Pump module (Micro-Tech Scientific, Vista, CA) with an in-house designed column heater was used for all chromatographic separations. The capillary column was directly mounted on a microinjector (model C4-1004-.5, Valco Instruments, Houston, TX) with a 500 nL internal sample loop and a microtight union with 5 nL swept volume (Upchurch Scientific, Oak Harbor, WA). The flow rate of the solvent delivery pump was set at 0.5 mL/min, which was split precolumn to produce a flow rate of 2.5 μL/min at 60°C through the monolithic capillary column. A mobile phase system of two solvents was used, where solvents A and B were composed of 0.05% formic acid (Sigma) in HPLC grade water (Fisher Scientific, Hanover Park, IL) and ACN, respectively. A linear gradient of 0–100% B in 18 min was applied. In order to sequence the tryptic peptides of interest, tandem MS experiments were performed using linear IT MS (LTQ, Thermo Finnigan, San Jose, CA), where the monolithic HPLC configuration described above was directly interfaced. The capillary transfer tube was set at 200°C and ESI voltage at 4.0 kV. A sheath gas flow of 12 arbitrary units was used. The ion activation was achieved by utilizing helium at a normalized collision energy of 35%. All MS/MS data obtained were analyzed using the TurboSequest feature of Bioworks 3.1 SR1 (Thermo Finnigan). By allowing up to two missed cleavages, peptide ions were automatically assigned with the Xcorr values to consider >3.5 for + 3 ions, >2.5 for + 2 ions, and >1.5 for +1 ions and the ΔCn of 0.1 or higher [17]. The search parameters were as follows: (i) species Homo sapiens, (ii) allowing two missed cleavage, (iii) possible modifications were peptide N-terminal Gln to pyroGlu, oxidation of M, protein N-terminal acetylated, and phosphorylation of S, T, and Y, (iv) peptide tolerance ± 1.5 Da, (v) MS/MS tolerance ± 1.5 Da, and (vi) peptide charge +1, +2, +3. Swiss-Prot database was used for MS/MS analysis.
2.9 MALDI-QIT-TOF MS
A Shimadzu-Biotech AXIMA-QIT MALDI quadrupole IT-TOF instrument (Shimadzu-Biotech, Manchester, UK) was used to perform MS/MS of selected peptides. Samples were prepared in an identical fashion to those for peptide fingerprinting. 2,5-dihydroxybenzoic acid (Sigma) was used as the matrix at a concentration of 20 mg/mL in 1% v/v TFA, 60% v/v ACN. The purified peptide sample (0.5 μL) was deposited on a MALDI target plate along with 0.5 μL of the matrix solution. The TOF instrument was externally calibrated using a mixture of Bradykinin fragment 1–7, Angiotensin II, P14R, ACTH fragment 18–39, and Insulin chain B (Sigma), typically yielding the mass tolerance of 30 ppm. The standard instrument settings for optimum transmission at medium mass were used to record all mass spectra in this work. Data acquisition and processing was performed using Kompact LAUNCHPAD™ software and the ion masses input was submitted to the Swiss-Prot database using MASCOT MS/MS ions search (http://www.matrixscience.com). The MASCOT search parameters were as follows: (i) species Homo sapiens, (ii) allowing one missed cleavage, (iii) possible modifications: peptide N-terminal Gln to pyroGlu, oxidation of M, protein N-terminal acetylated, and phosphorylation of S, T, and Y, (iv) peptide tolerance ± 1 Da, (v) MS/MS tolerance ± 0.9 Da, and (vi) peptide charge +1.
2.10 Western blotting
Cell pellets were lysed on ice for 15 min with lysis/extraction reagent (Sigma) supplemented with protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Protein concentration was determined using the BCA Protein Assay (Pierce Chemical, Rockford, IL). Ten micrograms of total cell protein extracts from each cell line, in duplicate, and Mark 12 MW standards (Invitrogen, Carlsbad, CA) were resolved by SDS-PAGE (4–12% Bis-Tris gel, Novex, Invitrogen) under reducing conditions and electroblotted onto a PVDF membrane (Immobilon-PSQ; Millipore). One set of the immobilized proteins was probed with 1 μ/mL of anti-annexin I antibody (BD Transduction Laboratories, San Jose, CA) and the duplicate set with a 1:5000 dilution of anti-β-actin polyclonal antibodies (Sigma). The binding of HRP-conjugated secondary antibodies was detected by ECL (Amersham Biosciences).
3 Results and discussion
The identification of differentially expressed proteins in cell extracts requires a highly reproducible fractionation and detection system. To meet these criteria, we combined a highly reproducible 2-D liquid-phase separation by CF and NPS-RP-HPLC approach [8, 9, 18] with the high mass accuracy of detection by MS or MS/MS. The flowchart in Fig. 1 shows the experimental overview from cell lyses through the protein identification. Briefly, soluble proteins from lysed cells are separated by their pI in the first dimension using CF. The second dimension employs an NPS-RP-HPLC where the proteins are separated according to their hydrophobicity. Eluent from the NPS-RP-HPLC step is split postcolumn for direct introduction to ESI-TOF MS and to a UV detector for fraction collection for offline analysis by PMF or MS/MS. The deconvolution of ESI-TOF MS data provides the intact masses and the relative quantitation of the proteins in the cells of interest. The protein fractions collected from NPS-RP-HPLC are enzymatically digested and analyzed by MALDI-TOF MS for protein identification by PMF. In cases of ambiguity of protein identifications by PMF, we performed MS/MS analysis using capillary monolith LC separation of tryptic digests of proteins isolated from the liquid separations, with online detection by linear IT MS. Occasionally, the tryptic peptide sequences that were not detected/or performed by the monolith-based LC/MS/MS methods were successfully sequenced by MALDI-QIT-TOF MS. Comparison of the experimental Mr value of intact protein with database information provides highly reliable protein identification and the presence of possible modifications.
Figure 1.

Experimental overview of the liquid-phase 2-D protein separation approach and mass spectrometric techniques in peptide fingerprinting and sequencing analysis for MDA-MB-435 metastatic cell lines.
3.1 Protein separation and quantitation by ESI-TOF MS
Figure 2a shows an example of a CF chromatogram obtained from a metastatic M4A4 cell lysate. The online monitored linear gradient was superimposed on the image and 13 fractions were collected in the pH range 6.4–3.8. The most acidic and basic fractions were not mapped since there were only a limited number of proteins detected. The second dimension of NPS-RP-HPLC produces a 2-D UV map, as shown in Fig. 2b. The horizontal axis is the fraction pH and the vertical axis is the relative % hydrophobicity. Each band represents a protein peak eluted from the RP-HPLC separation with grayscale intensity representing the relative intensity of each protein peak. The image offers the same advantages of differential expression profiling provided by a 2-D gel analysis but is obtained in digitized form. The 2-D UV maps from the two samples are readily viewed by in-house software for comparison, including differentially expressed and common proteins. Based on the image bands, approximately 700 proteins were collected for each cell line. An important aspect of 2-D liquid separation is the reproducibility achieved in the pH dimension by CF. In order to maintain accuracy, we routinely perform duplicate evaluations of protein content overview maps. Furthermore, three replicates of CF runs pH 6.5–4.0 were performed on the M4A4 samples to monitor reproducibility. Three samples containing 2.4, 2.4, and 2.3 mg, respectively, of M4A4 protein were loaded on to a CF column, and one pH fraction from each CF run was separated by RP-HPLC. Figure 3A shows that the RP-HPLC separation profiles were very similar, confirming a high level of reproducibility of 2-D liquid separation for intersystem comparisons via differential mapping. In addition, a quantitative analysis of the protein bands in Fig. 3A and the other pH fractions examined (data not shown) indicated that the error rate was less than 10% in these experiments.
Figure 2.
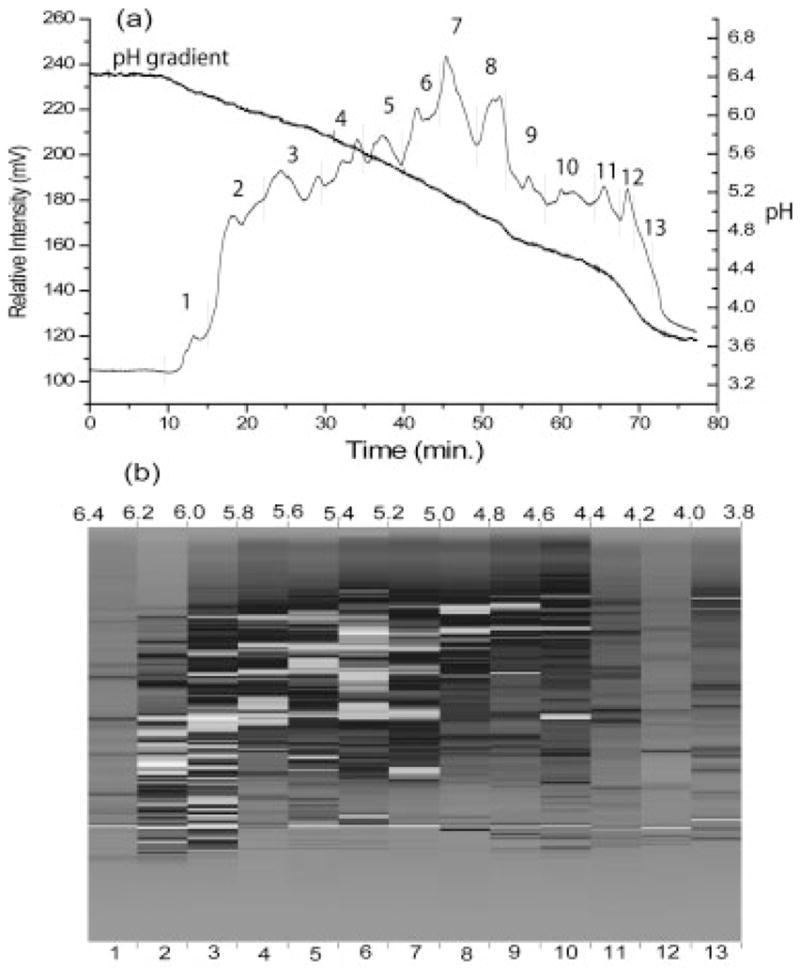
(a) A CF chromatogram of the metastatic M4A4 cell line. The UV detector was set at 280 nm and the gradient was originated from pH 6.4 to 3.8 for 75 min. The fractions were collected at every pH 0.2 unit interval. (b) A 2-D UV map of M4A4 sample. Each grayscale band represents the relative intensity of each protein, from gray (lowest intensity) to white (highest intensity).
Figure 3.
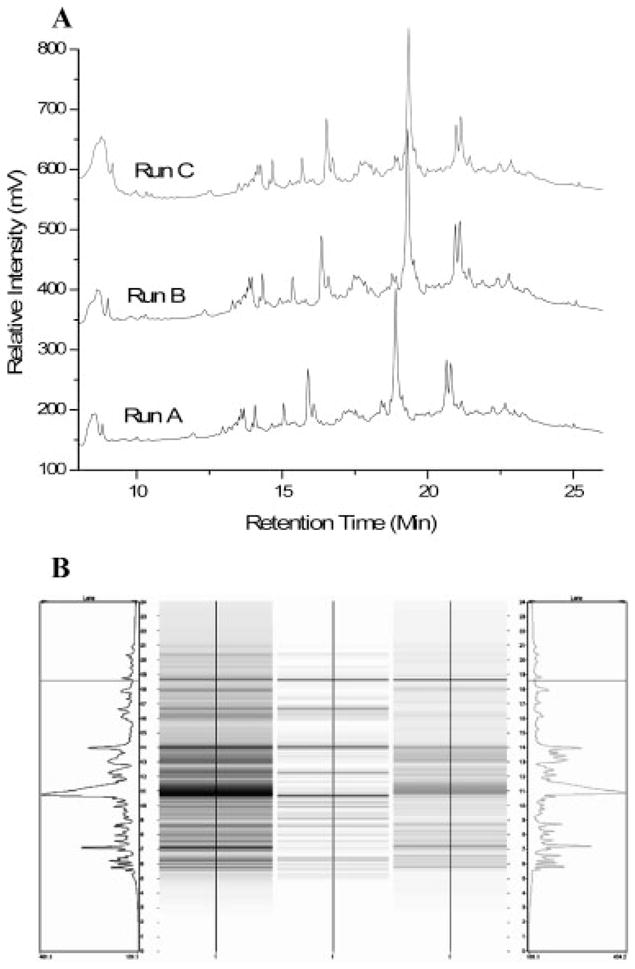
(A) Reproducibility study of 2-D liquid separation. Three batches of samples from the metastatic cell line M4A4 were separated by CF. Spectra depict a single pH fraction from each run selected for RP-HPLC analysis. (B) Representative differential map (center) for comparison of protein expression in metastatic cells (left) and nonmetastatic cells (right).
Another important feature of the use of liquid methods for separation of proteins is the capability to directly interface with MS. In this study, protein Mr was detected by online analysis using the LC/ESI-TOF-MS system. In the resulting data, the experimental Mr of the proteins closely matched the database MW of the corresponding proteins, as shown in Fig. 4. The intact proteins of galectin-1 and 60S ribosomal protein L27 of the M4A4 sample were detected with a high mass accuracy of 70 ppm. Although the mass accuracy is dependent on the protein umbrella spectra, this method provides much improved attainable mass accuracy (<0.1% or 1000 ppm) compared to the 5–10% typically achieved by 2-DE [19]. Additionally, the deconvoluted spectra of intact proteins occasionally provide additional information on the structure of specific proteins. For example, the deconvoluted spectrum of galectin-1 (Fig. 4A) suggested different isoforms with possible acetylations. The different isoforms have close pI and MW values as calculated from their protein sequences using Compute pI/MW tool (http://ca.expasy.org/tools/pi_tool.html). Our analysis showed that these isoforms of galectin-1 have close retention times and, hence, similar hydrophobicities. Acetylation at the N-terminus of the first amino acid in the galectin-1 sequence has been reported previously [20]. Further work on the identification of additional potential modifications of galectin-1 is in progress. The ability to obtain highly accurate Mr values revealed acetylated isoforms of a number of other proteins present in both cell lines (data not shown). In addition to providing accurate Mr and structural information, ESI-TOF MS also plays a role in accurate quantitation [21]. The resulting protein umbrella from the TOF signature is processed by Max-Ent software to create a selected ion chromatogram (SIC) for each detected protein [19]. The deconvoluted molecular spectrum peak area is representative of the accurate measurement of the entire amount of the protein present in the sample. To aid in quantitative sample comparisons, 1 μg of bovine insulin internal standard is added to the CF fraction prior to loading onto NPS-RP column. In this study, we examined the proteins in the cell lysates from MDA-MB-435 sublines with molecular weights ranging from 5 to 80 kDa, based on ESI-TOF measurements. Figure 5 shows representative 2-D mass maps obtained from the online LC/MS of CF fractions from cell lines M4A4 (metastatic) and NM2C5 (nonmetastatic). Approximately 70–80 unique proteins Mr were detected for each CF fraction. The image is similar to that of a 2-D gel where the vertical axis is Mr and the horizontal axis is pH. The intensities of protein bands correlate to the relative protein expression levels detected.
Figure 4.

Deconvoluted spectra show (a) Mr of galectin-1. (b) Mr of 60S ribosomal protein L27. The database molecular weights of galectin-1 and 60S ribosomal protein are 14 716 and 15 667 Da, respectively. The deconvoluted spectra show good correlation of experimental molecular masses and database masses of corresponding proteins. The mass accuracy is better than 70 ppm.
Figure 5.

Combined 2-D mass maps for CF fractions covering the pH range 5.4–6.2 of M4A4 (right) and NM2C5 (left) cells. The pH of each fraction is plotted vs. the Mr of the proteins detected in that fraction. The arrows indicate examples of differentially expressed proteins subsequently identified by MALDI-TOF MS and online monolith-LC/MS/MS. The mass maps are shown in relative value. The original data can be recovered to give a detailed view using ProteoVue software.
3.2 Identification of separated proteins by MALDI-TOF MS
To study and identify proteins collected by RP-HPLC, a MALDI-TOF-MS technique was employed. The isolated protein fractions underwent tryptic digestion and SPE prior to MALDI analysis. The profiles of tryptic peptides present in the fraction were measured using MALDI-TOF MS, in a process referred to as PMF. The monoisotopic peaks were selected and submitted to the NCBI/Swiss-Prot database in MS-Fit. Certain established criteria were required for enhancing confidence and accuracy in the search results of which a MOWSE score of at least 1000, percent coverage of at least 20% and proteins ranged in the top four and five minimum peptide matches were accepted for protein identifications (IDs). In addition, the molecular weight of the assigned protein must fall within ± 250 Da of the experimental Mr for database identification. After 2-D separation, we compared the UV maps of M4A4 and NM2C5 cell lines to view relative differences in protein band intensities. This provides an initial comparison of protein expression. Of the approximately 700 protein bands visualized on each UV map, 250 protein bands were selected for further analysis on the basis of two criteria: (i) proteins revealed to be differentially expressed by mass mapping and (ii) abundant proteins present equally in both cell lines. Of the 250 proteins selected for identification, 65 (26%) proteins were positively identified solely by the MALDI fingerprinting-mass mapping method. The MALDI percent coverage of identified proteins ranged between 13 and 81%.
3.3 Monolith-based HPLC/MS/MS
Obtaining high protein sequence coverage is particularly important in a bottom-up proteomics approach for confident protein identification. As shown in Tables 1 and 2, sufficiently high protein sequence coverage obtained for most of the proteins identified by PMF analysis strongly indicates highly confident protein identification, supported also by closely matching intact protein MW values. However, several proteins were identified with a relatively low sequence coverage that can lead to ambiguous identifications. This may be due to several factors associated with the MALDI-MS experiment, including peptide losses during sample clean-up procedures, varying ionization efficiencies of peptides, and dependence on the choice of matrices [22]. Sequencing analysis can provide unambiguous information on individual peptide sequences to confirm their presence in proteins. It can also become an additional means to further confirm the protein identifications already obtained through PMF analysis. In this experiment, a monolithic capillary HPLC was chosen for online interface with a linear IT MS for sequencing analysis. The use of monolithic capillary columns synthesized with silica or polymer [10–12] has gained popularity as successfully demonstrated in the analysis of various biological molecules [7, 15, 16]. Compared to C18 silica packed columns, a monolithic column provides a number of distinct advantages derived from fast mass transfer kinetics [13] due to the lack of interstitial space to result in high resolution, high efficiency, and high recovery [14] in a rapid separation time. The fast separation speed, among many other advantages over packed columns, is particularly well suited for the rapid analysis of large numbers of proteins required in the current study. Furthermore, its high recovery is another advantage that helps to detect peptides of low abundance or low ionization efficiency. This leads to the analysis of the proteins of low abundance that are known to play important roles in proteomes where a large dynamic range presents a great challenge. All the proteins shown in Table 3 were successfully analyzed by monolithic LC/MS/MS for tryptic digests by applying data filtering criteria of high stringency. It is interesting to note that the sequencing analysis based on monolithic LC/MS/MS successfully identified the proteins that went undetected by MALDI-based PMF analysis. For example, ESI-TOF MS indicated the presence of the protein that matched the size of ca. 50 kDa protein, which was, however, not identified by MALDI-TOF MS. This protein was later identified as septin-7 (Table 3) by the monolithic LC/MS/MS through successful sequencing of five fully tryptic peptides. The accumulating evidence suggests the involvement of septins in human neoplasia [23]. Although this only represents 12% of this protein sequence, it is still considered reliable, as two or more fully or partially sequenced peptides are generally considered to be sufficient for confident identification [24]. A sequencing analysis is also useful for detailed characterization of modifications or variations of proteins to study their significance in progression of various diseases. In this experiment, monolithic LC/MS/MS successfully sequenced peptides containing PTMs in several proteins. Annexin I (annexin A1), previously identified with 39% of sequence coverage through PMF analysis, was successfully sequenced to result in 12 peptides, corresponding to 41% of sequence coverage to further confirm that it was correctly identified. Annexin I is a calcium/phospholipid-binding protein involved in exocytosis and regulates phospholipase A2 activity [25]. Interestingly, the monolithic HPLC/MS/MS also characterized one of its PTMs, N-terminal acetylation that has been reported in previous studies [26]. Figure 6 shows an annotated MS/MS spectrum obtained for one of the tryptic peptides from annexin I, AMVSEFLK (amino acid no. 1–8), where 42 Da mass shift, a signature of acetylation, was clearly observed from each of the fragment type b ions compared to the predicted fragmentation of unmodified sequence, but not from y ions, suggesting that the modification is present at its N-terminal end. The N-terminal acetylation of annexin I was observed in both cell lines. Another protein, histone H1.2, was also found to be acetylated at the N-terminus through sequencing analysis. The acetylation of SETAPAAPAAAPPAEK in mature forms of histone H1.2 has been reported [27]. The monolithic LC/MS/MS proved very useful in elucidating and confirming protein identifications, and identifying important PTMs. In all, 65 proteins were successfully identified solely by MALDI-TOF MS, when the search results of the spectra met the criteria discussed in the experimental Section 2.7. The Swiss-Prot database was primarily considered for database searching. Accession numbers and protein identities in Tables 1–3 were labeled as annotated in the Swiss-Prot database unless noted otherwise. These proteins were identified with high sequence coverage and high MOWSE scores, which resulted in high confidence identifications. The remaining spectra without positive identification by PMF were selected for analysis by LC/MS/MS or MALDI-QIT-TOF MS. Thirty proteins were positively identified by LC/MS/MS and 12 proteins were positively identified by MALDI-QIT-TOF MS. The selection of proteins for LC/MS/MS and/or MALDI-QIT-TOF MS was to confirm a number of differentially expressed proteins and to investigate potential PTMs. More than 180 peptides were sequenced in this work, of which 120 peptides were matched to 32 unique proteins. For MS/MS analysis, the Swiss-Prot database was used for searching protein IDs, and peptide N-terminal Gln to pyroGlu, oxidation of M, protein N-terminal acetylated, and phosphorylation of S, T, and Y were considered for PTM searching. Of the approximately 250 protein bands revealed over the limited pH range, 89 (36%) proteins were positively identified by MS or MS/MS.
Table 1.
Proteins identified as differentially expressed (>2-fold change) in metastatic M4A4 and nonmetastatic NM2C5 cell lines using a 2-D liquid-phase method and MS and/or MS/MS
| Identity | Accession number | Database MW/pI | Experimental |
|||
|---|---|---|---|---|---|---|
| M4A4 Mr | NM2C5 Mr | MALDI % Cov. | Fold change | |||
| Up-regulated in M4A4 cells | ||||||
| Pyruvate kinase, isozymes M1/M2 | P14618 | 57937/8.0 | 57939 | 57925 | 26 | 34.6 |
| 60S Ribosomal protein L24 (ribosomal protein L30) | P83731 | 17779/11.3 | 17783 | 17781 | 33 | 4.9 |
| 40S Ribosomal protein S16 | P62249 M | 16445/10.2 | 16315 | 16316 | 47 | 4.2 |
| 28 kDa Heat- and acid-stable phosphoprotein | Q13442 | 20630/8.8 | 20660 | 20664 | 35 | 4.0 |
| Hematological and neurological expressed gene 1-like protein variant a; ST1a | 19070523 Ma) | 16015/5.5 | 16007 | 16005 | 37 | 3.7 |
| Elongation factor Tu, truncated | P49411 | 49542/7.3 | 45095 | 45085 | 33 | 2.8 |
| NADH-ubiquinone oxidoreductase 13 kDa-B subunit | Q16718 | 13459/5.8 | 13372 | 13371 | 40 | 2.8 |
| T-complex protein 1, eta subunit | Q99832 | 59367/7.6 | 59436 | 59468 | 20 | 2.5 |
| Nuclease sensitive element-binding protein 1 | P67809 M | 35924/9.9 | 35839 | 35838 | 55 | 2.3 |
| Superoxide dismutase [Cu-Zn] | P00441 | 15936/5.7 | 15891 | 15894 | 61 | 2.1 |
| 60S Ribosomal protein L4 | P36578 | 47698/11.1 | 47658 | 47655 | 13 | 2.1 |
| 40S Ribosomal protein S9 | P46781 | 22592/10.7 | 22463 | 22461 | 21 | 2.1 |
| Up-regulated in NM2C5 cells | ||||||
| Annexin A2 | P07355 | 38604/7.6 | 38517 | 38519 | 37 | 14.6 |
| Annexin A1 | P04083 | 38715/6.6 | 38673 | 38686 | 39 | 14.5 |
| 60 kDa Heat-shock protein, truncated | P10809 | 61055/5.7 | 57966 | 57967 | 47 | 11.1 |
| Heat-shock cognate 71-kDa protein (HSC70/HSP73) | P11142 | 70899/5.4 | 70960 | 70964 | 35 | 4.8 |
| T-Complex protein 1, β subunit | P78371 | 57489/6.0 | 57486 | 57493 | 43 | 4.3 |
| Heat-shock 70 kDa protein 1 (HSP70-1) | P08107 | 70053/5.5 | 70068 | 70069 | 38 | 4.1 |
| ATP synthase alpha chain, truncated | P25705 | 59751/9.2 | 55196 | 55210 | 20 | 4.0 |
| Alpha-endosulfine | O43768 | 13389/6.6 | 13299 | 13300 | 52 | 3.8 |
| Tumor protein D54 (hD54) (D52-like 2) | O43399 | 22238/5.3 | 22360 | 22278 | 58 | 3.5 |
| ATP synthase β chain, truncated | P06576 | 56560/5.3 | 51827 | 51844 | 36 | 3.3 |
| Platelet-activating factor acetylhydrolase IB b subunit | P68402 M | 25569/5.6 | 25527 | 25524 | 31 | 2.8 |
| Myosin light polypeptide 6 | P60660 | 16930/4.6 | 16875 | 16874 | 51 | 2.7 |
| Ubiquitin-conjugating enzyme E2 variant 2 | Q15819 | 16363/7.8 | 16275 | 16276 | 45 | 2.7 |
| Putative RNA-binding protein 3 | P98179 | 17171/8.9 | 17213 | 17217 | 45 | 2.6 |
| Calcyclin-binding protein | Q9HB71 | 26210/8.3 | 26123 | 26126 | 29 | 2.4 |
Accession number in NCBI database.
Table 2.
Proteins detected either in M4A4 or NM2C5 cell lines by MS and/or MS/MS
| Identity | Accession number | Database MW/pI | Experimental |
|||
|---|---|---|---|---|---|---|
| M4A4 Mr | NM2C5 Mr | MALDI% Cov. | INa) | |||
| Detected only in M4A4 cells | ||||||
| Galectin-1 | P09382 | 14716/5.3 | 14715 | nd | 58 | 29.1 |
| Triosephosphate isomerase | P60174 | 26670/6.4 | 26541 | nd | 56 | 29.1 |
| Nucleoside diphosphate kinase B | P22392 | 17298/8.5 | 17213 | nd | 48 | 24.0 |
| 54 kDa Nuclear RNA- and DNA-binding protein | Q15233 | 54101/9.0 | 54277 | nd | 38 | 12.9 |
| Phosphoglycerate mutase 1 | P18669 | 28804/6.7 | 28718 | nd | 38 | 10.7 |
| Phosphoglycerate kinase 1 | P00558 | 44615/8.3 | 44591 | nd | 36 | 8.0 |
| Peroxiredoxin 2 | P32119 | 21892/5.7 | 21811 | nd | 39 | 7.2 |
| 39S Ribosomal protein L13, mitochondrial (L13mt) | Q9BYD1 | 20692/9.2 | 20604 | nd | 81 | 5.3 |
| GST P | P09211 | 23356/5.4 | 23269 | nd | 34 | 2.4 |
| My016 protein | Q9H3K6 | 10117/6.1 | 10162 | nd | 61 | 1.8 |
| Tubulin β-5 chain (tubulin 5 β) | P04350 | 49631/4.8 | 49409 | nd | 21 | 0.9 |
| Dihydropteridine reductase | P09417 | 25804/6.9 | 25743 | nd | 22 | 0.2 |
| Detected only in NM2C5 cells | ||||||
| Stress-induced-phosphoprotein 1 (STI1) | P31948 | 62640/6.4 | nd | 62866 | 15 | 3.6 |
| Heat-shock 27 kDa protein | P04792 | 22783/6.0 | nd | 22864 | 63 | 2.6 |
| Mannose-6-phosphate receptor-binding protein 1 | O60664 | 47033/5.3 | nd | 46954 | 49 | 2.4 |
| Eukaryotic translation initiation factor 4H | Q15056 | 27254/6.9 | nd | 27336 | n/a | 0.5 |
IN, normalized intensity. For quantitation of each protein, the peak area of proteins were normalized against the peak area of bovine insulin internal standard
nd = not detected
n/a = not applicable
Table 3.
Proteins undetected by PMF analysis but successfully analyzed by monolithic LC/MS/MS
| Protein ID | Accession number | Database MW | Experimental |
||
|---|---|---|---|---|---|
| M4A4 Mr | NM2C5 Mr | Monolith-LC/MS/MS no. of sequenced peptides | |||
| Histone H1.2 | P16403 | 21221 | 21309 | 21310 | 3 |
| Histone H2A type 1 | P28001 | 14004 | 14004 | 14004 | 3 |
| Histone H3.1 | P68431 | 15273 | 15314 | 15313 | 2 |
| Septin-7 | Q16181 | 50649 | 50682 | 50681 | 5 |
| Eukaryotic translation initiation factor 4H | Q15056 | 27254 | nd | 27336 | 3 |
nd = not detected
Figure 6.

Acetylation of annexin I as detected by online monolithic LC/MS/MS. Comparison of predicted (unmodified) vs. observed fragment ions for a tryptic peptide sequence, AMVSEFLK, derived from annexin I.
3.4 Differentially expressed proteins
In the initial analysis, the 2-D UV maps provide an image of the proteins expressed in the cell line samples. The 2-D liquid map shown in Fig. 2B is based upon hydrophobicity. Two maps (identical pH lanes) can be directly compared using DeltaVue software, where the center lane displays the difference of protein expression between the same pH fractions for the respective samples. Figure 3B depicts the differential maps produced using DeltaVue for two specific pH fractions comparing M4A4 and NM2C5 cell samples. A protein with an increased level of expression in the metastatic sample, relative to the nonmetastatic sample, will show up in shades of grayscale in the differential map. When there are no changes in expression level between the two samples, the differential map will display these bands in white. The UV intensity gives an initial estimate on the expression level of each protein. However, there is often more than one protein in a peak, and then the UV intensity of that peak is not truly quantitative. In this case, further fractionation is needed. To solve this problem, ESI-TOF MS was used to separate selected proteins and quantitation was achieved using an internal standard. The combination of UV maps and mass maps provides reproducible results for quantitative study. Forty-three candidate proteins were confirmed to be differentially expressed more than twofold in either direction in metastatic tumor cells compared to isogenic, nonmetastatic tumor cells. Tables 1 and 2 summarize the comparison of experimental masses and the theoretical intact Mr, the relative abundance and the percent sequence coverage of the identified differentially expressed proteins. As shown in Table 1, 12 proteins were up-regulated and 15 proteins were down-regulated in the metastatic M4A4 sample. The observed fold changes fell between 2.1- and 4.9-fold for most of the identified candidates, however, a few identified proteins were found to show a >10-fold difference in expression level. A total of approximately 700 unique proteins were isolated within this study, and of those, 43 proteins were ultimately identified and deemed to be >2-fold differentially expressed between M4A4 and NM2C5. In our previous RNA-based microarray analyses of these paired, isogenic cell lines we found that 3 to 7% of the detected genes were designated as differentially expressed (>2-fold change). In line with this, the proteomic profiling revealed that ~6% of the total proteins identified in this study were differentially expressed.
A number of proteins increased in the M4A4 cells are indicative of high metabolic activity, including pyruvate kinase, and triose isomerase. High levels of four ribosomal proteins also indicate a higher synthetic rate in M4A4 that may drive or facilitate cell proliferation. These findings are in line with the fact that M4A4 cells grow more quickly than NM2C5 in vitro and in vivo, and may confer an advantage to M4A4 in establishing secondary tumor deposits. The annexins (ANXA1 and ANXA2) were found to be very highly expressed in the nonmetastatic NM2C5 cell line (~14-fold relative to M4A4). These proteins belong to a family of Ca2+-dependent phospholipid-binding protein implicated in the regulation of phagocytosis, cell signaling, and proliferation [28]. Relatively, little is known about annexin I (lipocortin I) expression in human cancer, but a previous study reported that annexin I was up-regulated in a highly metastatic lung carcinoma cell line compared to its expression in a less metastatic counterpart [29]. Studies have reported reduced ANXA1 expression in advanced head and neck cancers, particularly in tumors with nodal metastases [30]. A proteomic analysis of the tumor tissue–blood interface identified annexin I as a selective, tumor-associated endothelial cell target in solid tumors. Subsequent radio-immunotherapy targeted to annexin I was shown to destroy tumors and increase animal survival [31]. In addition, annexin II expression is observed to be down-regulated in prostate cancer and in human osteosarcoma metastases [32, 33]. The results from these studies support the association of increased annexin I and annexin II expression with a less aggressive nonmetastatic phenotype. Annexin I is also an endogenous anti-inflammatory mediator that operates to counteract the process of leukocyte extravasation [34]. As annexin I was more highly expressed in nonmetastatic NM2C5 cells (Fig. 7), it is feasible that it affects the ability of these cells to extravasate at distal sites, a process thought to be essential in metastatic sufficiency. This potential role for annexin I fits with our previous work regarding the clinical relevance of our metastasis model. Through green fluorescent protein labeling of the cell lines [2], we have demonstrated the ability of disseminated, nonmetastatic NM2C5 cells to survive and remain dormant in the tissues of a murine host for extended periods of time. Their failure to form metastases was not attributable to any intrinsic loss of proliferative capacity, as demonstrated by the ability of the cells to grow exponentially and indefinitely when retrieved from the lung tissue and recultured in vitro or reinoculated into the mammary gland. Therefore, NM2C5 cells remain immortal but are unable to complete an essential step in the metastatic cascade. This dormancy phenomenon is observed in human patients with breast cancer, whereby cells disseminated prior to removal of the primary tumor can be “reactivated” and form metastases several years later, and this is the major reason for the use of postsurgical, adjuvant treatment. This study has encouraged us to pursue a possible role for annexin I with respect to tumor cell dormancy and the metastatic efficiency of NM2C5 cells.
Our proteomic analysis also revealed that 16 proteins were uniquely expressed in either cell line (Table 2). While trace amounts of the protein may be undetected in a given sample, by using this method we can detect as little as 50 pg (~100 fmol) of a 40 kDa protein [21] which is comparable to silver staining methods. The majority of these factors were observed to be uniquely expressed at very high levels in the metastatic M4A4 cells. Galectin-1 and nucleoside diphosphate kinase B (NM23 · H2) were highly expressed in the M4A4 sample. The role of galectin-1 in cancer progress has been widely investigated and proposed as a potential biomarker of metastatic tumors [35, 36], as well as a target for anticancer drugs [37]. This protein is a member of the β-galactoside-binding proteins, reported to act as an autocrine negative growth factor that regulates cell proliferation [38] and is implicated in modulating cell–cell and cell–matrix interactions [39]. Gal-1 is expressed in many malignant and normal tissues, but a correlation of increased Gal-1 has been in breast carcinomas with metastatic propensity [40]. Furthermore, Gal-1 has recently been shown to be regulated coordinately with ERBB2, a protein overexpressed owing to gene amplification in 25–30% of breast carcinomas with a poor clinical prognosis [41]. NM23·H2 is a multifunctional enzyme which can cleave DNA and regulate transcription of specific genes through multiple mechanisms, and thus can have profound effects on the cellular phenotype dependant on context [42]. While reduced expression of NM23.H1 has been reported to be concordant with the presence of lymph node metastasis or local invasiveness of the primary tumor, this is not so for NM23.H2 [43, 44]. The expression of certain groups of heat-shock proteins has been described in the literature as a prognostic factor in many tumors. As molecular chaperones, these proteins play an important role in multiple cellular mechanisms. Here we identified several heat-shock proteins as being differentially expressed, notably HSP70. The heat-shock cognate 71-kDa protein (HSC70/HSP73) was found to be elevated five-fold in NM2C5 cells. This protein has been reported to have an important role in HCV-related hepatocellular carcinoma [45] and in the context of metastasis, a recent report has shown that HSP73 binds the chemokine receptor CXCR4 [46], a surface molecule with a well-documented role in metastatic sufficiency in a number of cancers [47], including breast [48, 49]. Although CXCR4 itself is not differentially expressed in our model [6], HSP73 may act downstream to differentially regulate the CXCR4 signaling pathway. Recent, large-scale microarray analyses of human tissue specimens are building consensus genetic profiles of breast tumors and have begun to define expression profiles which purport to predict the likelihood of breast cancer metastases and clinical outcome [50]. It is interesting that some of the proteins identified as being associated with metastatic sufficiency in this study are present in a proposed 70-gene breast prognosis signature. Members of the GST family, highly expressed in M4A4, and the ubiquitination pathway, down-regulated in M4A4 are regulated in the same direction in the proposed prognosis signature.
We have previously subjected the breast metastasis model to genomic profiling approaches. Both PCR-based [5] and oligonucleotide microarray-based global expression [6] analyses identified a number of genes as being differentially expressed in the NM2C5 and M4A4 cell lines, relative to one another. Both annexins I and II expression was identified at the transcriptional level to be 3- to 5-fold increased in NM2C5 cells. Our proteomic analysis confirmed the differential expression of these proteins, and in this case, at higher levels than genomic analyses suggested. Conversely, the marked differential expression of the heat-shock protein family was missed using genomic analyses alone, suggesting that the expression of these proteins is primarily regulated at the translational level. Proteomic and genomic analyses have different advantages and the combination of both disciplines is likely the most fruitful approach. Genomic profiling certainly has the ability to query many more targets in a single experiment, and supposed whole-genome coverage is feasible with this platform. However, it is clear that the functional endpoint of the genetic blueprint lies at the protein level, and that the levels of messenger RNA and protein do not always correlate well. This latter fact means that global transcriptional approaches require considerable validation at protein levels. Most importantly, the pivotal regulatory points of translation and PTM of proteins such as proteolytic processing, phosphorylation, or glycosylation can only be found using a multidimensional proteomics approach such as that employed in this study. These PTMs can influence the stability, activity, localization, and structural conformation of the protein. Thus, while genomic approaches can rapidly create gene lists for subsequent analysis, it is important to identify global changes in protein expression in disease states, including metastasis. Continued improvements in MS techniques should only accelerate the pace of discovery in this field.
4 Concluding remarks
Breast cancer has a great propensity to develop distant metastasis, and micrometastases may already be present at the time of primary tumor detection. Therefore, much effort is now being directed at finding an early marker in the primary tumor that could indicate its metastatic potential. In an effort to establish a methodological framework for analysis of molecules and mechanisms involved in this complex multistep process, we have developed a well-defined experimental system in which the role of candidate genes can be screened and tested. The unique advantage of the MDA-MB-435 metastasis model is the ability to profile cells of opposing metastatic phenotype that originate from a common genetic background. These cell lines constitute a stable and accessible model for the identification of genes involved in the multistep process of breast tumor metastasis. Manipulation of candidate genes in these cells and subsequent analysis of upstream and downstream effects will permit evaluation of their functional significance in the geometric progression of breast cancer.
In this study, we have identified a number of proteins that may participate in the processes associated with the metastasis of breast cancer, including metabolism, cell motility and invasion, and signal transduction. These findings demonstrate that the mass mapping technique is a powerful tool for the detection and identification of proteins in complex biological samples and which are specifically associated with a cellular phenotype. We have further developed multidimensional liquid-phase separation for protein separation and show the capabilities of interfacing this technique to multiple mass spectrometric methods. CF followed by NPS-RP-HPLC/ESI-TOF MS provides the means of accurate interlysate comparison based upon the high resolution and mass accuracy of Mr. It is important to develop rapid, accurate, and reproducible protein mapping techniques for the study of tumor cell protein content since many of the changes that occur in tumor cell transformation may be manifested only in the proteome. The sequence and structure of expressed proteins, elucidated by monolith-based HPLC coupled with a linear IT MS, revealed that many of the proteins in these cell lines displayed substantial PTMs and enabled us to detect and identify several metastasis-associated proteins that we had not found previously using genomic screening approaches. The multidimensional proteomic approach led to the identification of 24 proteins that were overexpressed in metastatic cells relative to their isogenic, nonmetastatic counterparts. Although further analysis will be required to fully characterize these proteins and their role in specific cellular phenotypes, our findings demonstrate that the combination of multidimensional liquid-phase separation to MS or MS/MS can accurately identify potential protein biomarkers of disease in complex biological materials.
The liquid separation-mass mapping method provides a means of separating and mapping hundreds of proteins which can be displayed in a 2-D image and where each protein can be tagged according to an exact Mr, pI, and hydrophobicity. The ability to obtain an accurate Mr value is an essential piece of information to confirm the identity of proteins. This improved accuracy will greatly facilitate the establishment of mass mapping databases for online query. Through comparison of such maps we will be able to identify subtle changes in human cell protein profiles that will, in turn, reveal trends in protein expression changes that correlate with disease status. Ultimately, the ability to identify those proteins involved in the progression of carcinoma will most readily lead to clinically useful markers for diagnosis and prognosis, and provide novel targets for in vivo imaging and therapeutic targeting.
Figure 7.

Western blot of annexin I expression in the metastatic M4A4 cell line, and the isogenic, nonmetastatic NM2C5 cell line. Estimation of equal lane loading was achieved using an anti-β-actin antibody.
Acknowledgments
This work was supported in part by the National Cancer Institute under grant RO1 CA108597 (SG). We also acknowledge the National Science Foundation under grant DBI 99874 for funding of the MALDI-TOF MS instrument used in this work. We would also like to thank Dr. Christian G. Huber (Saarland University, Germany) for supplying the monolithic columns.
Abbreviations
- ACTH
adrenocorticotropic hormone
- CF
chromatofocusing
- NPS-RP-HPLC
nonporous-RP-HPLC
- OG
n-octyl β-D-glucopyranoside
References
- 1.Urquidi V, Sloan D, Kawai K, Agarwal D, et al. Clin Cancer Res. 2002;8:61–74. [PubMed] [Google Scholar]
- 2.Goodison S, Kawai K, Hihara J, Jiang P, et al. Clin Cancer Res. 2003;9:3808–3814. [PubMed] [Google Scholar]
- 3.Cailleau R, Olive M, Cruciger QV. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 4.Goodison S, Viars C, Urquidi V. Cancer Genet Cytogenet. 2005;156:37–48. doi: 10.1016/j.cancergencyto.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Sloan DD, Nicholson B, Urquidi V, Goodison S. Am J Pathol. 2004;164:315–323. doi: 10.1016/S0002-9440(10)63121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodison S, Yuan J, Sloan D, Kim R, et al. Cancer Res. 2005;65:6042–6053. doi: 10.1158/0008-5472.CAN-04-3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Premstaller A, Oberacher H, Huber CG. Anal Chem. 2000;72:4386–4393. doi: 10.1021/ac000283d. [DOI] [PubMed] [Google Scholar]
- 8.Chong B, Yan F, Lubman DM, Miller FR. Rapid Commun Mass Spectrom. 2001;15:291–296. doi: 10.1002/rcm.227. [DOI] [PubMed] [Google Scholar]
- 9.Yan F, Subramanian B, Nakeff A, Barder TJ, et al. Anal Chem. 2003;75:2299–2308. doi: 10.1021/ac020678s. [DOI] [PubMed] [Google Scholar]
- 10.Motokawa M, Kobayashi H, Ishizuka N, Minakuchi H, et al. J Chromatogr A. 2002;961:53–63. doi: 10.1016/s0021-9673(02)00133-4. [DOI] [PubMed] [Google Scholar]
- 11.Gu B, Armenta JM, Lee ML. J Chromatogr A. 2005;1079:382–391. doi: 10.1016/j.chroma.2005.02.088. [DOI] [PubMed] [Google Scholar]
- 12.Svec F. J Sep Sci. 2004;27:747–766. doi: 10.1002/jssc.200401721. [DOI] [PubMed] [Google Scholar]
- 13.Gritti F, Piatkowski W, Guiochon G. J Chromatogr A. 2003;983:51–71. doi: 10.1016/s0021-9673(02)01648-5. [DOI] [PubMed] [Google Scholar]
- 14.Ikegami TF, Dicks E, Kobayashi H, Morisaka H, et al. J Sep Sci. 2004;27:1292–1302. doi: 10.1002/jssc.200401921. [DOI] [PubMed] [Google Scholar]
- 15.Oberacher H, Krajete A, Parson W, Huber CG. J Chromatogr A. 2000;893:23–35. doi: 10.1016/s0021-9673(00)00731-7. [DOI] [PubMed] [Google Scholar]
- 16.Lee D, Svec F, Frechet JM. J Chromatogr A. 2004;1051:53–60. doi: 10.1016/j.chroma.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 17.Zhu W, Reich CI, Olsen GJ, Giometti CS, Yates JR., III J Proteome Res. 2004;3:538–548. doi: 10.1021/pr034109s. [DOI] [PubMed] [Google Scholar]
- 18.Wall DB, Parus SJ, Lubman DM. J Chromatogr B Biomed Sci Appl. 2001;763:139–148. doi: 10.1016/s0378-4347(01)00382-6. [DOI] [PubMed] [Google Scholar]
- 19.Hamler RL, Zhu K, Buchanan NS, Kreunin P, et al. Proteomics. 2004;4:562–577. doi: 10.1002/pmic.200300606. [DOI] [PubMed] [Google Scholar]
- 20.Gevaert K, Goethals M, Martens L, Van Damme J, et al. Nat Biotechnol. 2003;21:566–569. doi: 10.1038/nbt810. [DOI] [PubMed] [Google Scholar]
- 21.Lubman DM, Kachman MT, Wang H, Gong S, et al. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;782:183–196. doi: 10.1016/s1570-0232(02)00551-2. [DOI] [PubMed] [Google Scholar]
- 22.Zhu K, Miller FR, Barder TJ, Lubman DM. J Mass Spectrom. 2004;39:770–780. doi: 10.1002/jms.650. [DOI] [PubMed] [Google Scholar]
- 23.Russell SE, Hall PA. Br J Cancer. 2005;93:499–503. doi: 10.1038/sj.bjc.6602753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veenstra TD, Conrads TP, Issaq HJ. Electrophoresis. 2004;25:1278–1279. doi: 10.1002/elps.200490007. [DOI] [PubMed] [Google Scholar]
- 25.Parente L, Solito E. Inflamm Res. 2004;53:125–132. doi: 10.1007/s00011-003-1235-z. [DOI] [PubMed] [Google Scholar]
- 26.Hall SC, Smith DM, Masiarz FR, Soo VW, et al. Proc Natl Acad Sci USA. 1993;90:1927–1931. doi: 10.1073/pnas.90.5.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohe Y, Hayashi H, Iwai K. J Biochem. 1989;106:844–857. doi: 10.1093/oxfordjournals.jbchem.a122941. [DOI] [PubMed] [Google Scholar]
- 28.Roviezzo F, Getting SJ, Paul-Clark MJ, Yona S, et al. J Physiol Pharmacol. 2002;53:541–553. [PubMed] [Google Scholar]
- 29.Jiang D, Ying W, Lu Y, Wan J, et al. Proteomics. 2003;3:724–737. doi: 10.1002/pmic.200300411. [DOI] [PubMed] [Google Scholar]
- 30.Garcia Pedrero JM, Fernandez MP, Morgan RO, Herrero Zapatero A, et al. Am J Pathol. 2004;164:73–79. doi: 10.1016/S0002-9440(10)63098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh P, Li Y, Yu J, Durr E, et al. Nature. 2004;429:629–635. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- 32.Liu JW, Shen JJ, Tanzillo-Swarts A, Bhatia B, et al. Oncogene. 2003;22:1475–1485. doi: 10.1038/sj.onc.1206196. [DOI] [PubMed] [Google Scholar]
- 33.Gillette JM, Chan DC, Nielsen-Preiss SM. J Cell Biochem. 2004;92:820–832. doi: 10.1002/jcb.20117. [DOI] [PubMed] [Google Scholar]
- 34.Perretti M, Gavins FN. News Physiol Sci. 2003;18:60–64. doi: 10.1152/nips.01424.2002. [DOI] [PubMed] [Google Scholar]
- 35.Glinsky VV, Huflejt ME, Glinsky GV, Deutscher SL, Quinn TP. Cancer Res. 2000;60:2584–2588. [PubMed] [Google Scholar]
- 36.Moiseeva EV, Rapoport EM, Bovin NV, Miroshnikov AI, et al. Breast Cancer Res Treat. 2005;91:227–241. doi: 10.1007/s10549-005-0289-8. [DOI] [PubMed] [Google Scholar]
- 37.Rabinovich GA. Br J Cancer. 2005;92:1188–1192. doi: 10.1038/sj.bjc.6602493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldini A, Gress T, Patel K, Muresu R, et al. Genomics. 1993;15:216–218. doi: 10.1006/geno.1993.1039. [DOI] [PubMed] [Google Scholar]
- 39.Hughes RC. Biochimie. 2001;83:667–676. doi: 10.1016/s0300-9084(01)01289-5. [DOI] [PubMed] [Google Scholar]
- 40.Andre S, Kojima S, Yamazaki N, Fink C, et al. J Cancer Res Clin Oncol. 1999;125:461–474. doi: 10.1007/s004320050303. [DOI] [PubMed] [Google Scholar]
- 41.Mackay A, Jones C, Dexter T, Silva RL, et al. Oncogene. 2003;22:2680–2688. doi: 10.1038/sj.onc.1206349. [DOI] [PubMed] [Google Scholar]
- 42.Postel EH. J Bioenerg Biomembr. 2003;35:31–40. doi: 10.1023/a:1023485505621. [DOI] [PubMed] [Google Scholar]
- 43.Caligo MA, Cipollini G, Berti A, Viacava P, et al. Int J Cancer. 1997;74:102–111. doi: 10.1002/(sici)1097-0215(19970220)74:1<102::aid-ijc18>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 44.Ouatas T, Salerno M, Palmieri D, Steeg PS. J Bioenerg Biomembr. 2003;35:73–79. doi: 10.1023/a:1023497924277. [DOI] [PubMed] [Google Scholar]
- 45.Ding SJ, Li Y, Tan YX, Jiang MR, et al. Mol Cell Proteomics. 2004;3:73–81. doi: 10.1074/mcp.M300094-MCP200. [DOI] [PubMed] [Google Scholar]
- 46.Ding Y, Li M, Zhang J, Li N, et al. Mol Pharmacol. 2006;69:1269–1279. doi: 10.1124/mol.105.020271. [DOI] [PubMed] [Google Scholar]
- 47.Miwa S, Mizokami A, Keller ET, Taichman R, et al. Cancer Res. 2005;65:8818–8825. doi: 10.1158/0008-5472.CAN-05-0540. [DOI] [PubMed] [Google Scholar]
- 48.Smith MC, Luker KE, Garbow JR, Prior JL, et al. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 49.Liang Z, Yoon Y, Votaw J, Goodman MM, et al. Cancer Res. 2005;65:967–971. [PMC free article] [PubMed] [Google Scholar]
- 50.Weigelt B, Hu Z, He X, Livasy C, et al. Cancer Res. 2005;65:9155–9158. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]