

Abstract
Polypeptide growth factors activate common signal transduction pathways, yet they can induce transcription of different target genes. The mechanisms that control this specificity are not completely understood. Recently, we have described a fibroblast growth factor (FGF)-inducible response element, FiRE, on the syndecan-1 gene. In NIH 3T3 cells, the FiRE is activated by FGF-2 but not by several other growth factors, such as platelet-derived growth factor or epidermal growth factor, suggesting that FGF-2 activates signaling pathways that diverge from pathways activated by other growth factors. In this paper, we report that the activation of FiRE by FGF-2 requires protein kinase A (PKA) in NIH 3T3 cells. The PKA-specific inhibitor H-89 (N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide) blocked the FGF-2-induced activation of FiRE, the transcription of the syndecan-1 gene, and cell proliferation. Also, expression of a dominant-negative form of PKA inhibited the FGF-2-induced FiRE activation and the transcription of the syndecan-1 gene. The binding of activator protein-1 transcription-factor complexes, required for the activation of FiRE, was blocked by inhibition of PKA activity before FGF-2 treatment. In accordance with the growth factor specificity of FiRE, the activity of PKA was stimulated by FGF-2 but not by platelet-derived growth factor or epidermal growth factor. Furthermore, a portion of the PKA catalytic subunit pool was translocated to the nucleus by FGF-2. Noticeably, the total cellular cAMP concentration was not affected by FGF-2 stimulus. We propose that the FGF-2-selective transcriptional activation through FiRE is caused by the ability of FGF-2 to control PKA activity.
Keywords: activator protein-1, cAMP, enhancer, fibroblast growth factor-inducible response element (FiRE), syndecan-1
Growth factors exert their effects by activating intracellular signaling pathways that elicit specific changes in gene expression. The Ras/ERK pathway is the best-characterized signal-transduction pathway and one of the key pathways responsible for transmitting signals from growth factor-activated receptor tyrosine kinases to the nucleus (1). Many growth factors activate the same signaling pathways, although they can induce different responses on target cells, e.g., proliferation versus differentiation as well as transcription of different genes (2). The variance in transcriptional responses to different growth factors can be achieved by controlling the activity of the Ras/ERK pathway through interacting pathways or specific scaffold proteins (3–6).
We have previously characterized a far upstream fibroblast growth factor (FGF)-inducible response element (FiRE) (Fig. 1a) on the mouse syndecan-1 gene (7). After FGF-2 induction, FiRE binds several transcription factors, including two FGF-2-inducible activator protein-1 (AP-1) complexes, a 50-kDa FGF-2-inducible nuclear factor-1 (FIN-1), and an upstream stimulatory factor-1 (USF-1). In NIH 3T3 cells, FiRE is specifically activated by the FGF family members, with FGF-2 being the most potent activator, but not by epidermal growth factor (EGF) or platelet-derived growth factor (PDGF). In this study, we used this element as a tool to investigate the mechanisms responsible for the FGF specificity in signal transduction and subsequent transcription. Because FGFs, like most of the receptor tyrosine kinase-activating growth factors (including EGF and PDGF), activate the Ras/ERK pathway (8, 9), the involvement of additional signal transduction pathways that participate in the FGF-2 specific activation of FiRE was considered.
Figure 1.

PKA is required for FGF-2-induced activation of FiRE and induction of cell proliferation. (a) Schematic model of the FGF-inducible response element (FiRE), located at −10 kb of the translation initiation site of murine syndecan-1 gene. When activated by FGF-2, FiRE binds AP-1, a 50-kDa FIN-1, and USF-1 transcription factors. (b) PKA-specific inhibitor H-89 inhibits the FGF-2-induced activation of FiRE in a concentration-dependent manner. NIH 3T3 cells were stably transfected with FiRE-CAT plasmid, serum-starved for 48 hr, and treated for 30 min with H-89 (1 μM or 10 μM) before overnight exposure to FGF-2 (10 ng/ml), followed by determination of CAT activity. Means and standard deviations of three independent experiments of three parallel wells are shown in each. (c) The expression of dominant-negative PKARΙ inhibits the FGF-2-induced activation of FiRE. NIH 3T3 cells bearing the p271FiRECAT construct were transfected by a construct encoding a dominant-negative form of PKARΙ (PKARΙ-mut). The production of PKARΙ-mut was induced by application of ZnSO4 (50 μM final concentration) into the culture medium 3 hr before a 12-hr FGF-2 stimulation. Means and standard deviations of three independent experiments are shown. Control represents CAT activity from non-growth factor-treated cells. The ZnSO4-induced production of the dominant-negative PKARΙ was verified with anti-PKARΙ antibody. PKARΙ-mut stably transfected cells were treated with 50 μM ZnSO4 for 12 hr, and protein lysates were collected, blotted, and detected with anti-PKARΙ. As a control, nontransfected NIH 3T3 cells (wt) were similarly treated with ZnSO4. The immunoblots show significant increase in total PKARΙ immunoreactivity in ZnSO4-treated stably transfected cells, whereas in nontransfected cells, ZnSO4 treatment had no effect on the amount of PKARΙ. (d) PKA-specific inhibitor H-89 inhibits the FGF-2-induced DNA synthesis of NIH 3T3 cells in a concentration-dependent manner. Serum-starved NIH 3T3 cells were pretreated with H-89 (1 μM or 10 μM) for 30 min before an 18-hr FGF-2 treatment, and the incorporated radioactivity was measured with a γ counter.
cAMP-dependent serine protein kinase, also called protein kinase A (PKA), is able to control the activation and duration of the Ras/ERK pathway (10). PKA is activated by hormonal stimuli, but its role on growth factor-induced transcription has been less well characterized. Because PKA is known to control the activation of the Ras/ERK pathway and because functional interactions between growth-factor receptors and PKA have been previously suggested (11), we investigated whether PKA could be involved in the FGF-2-induced activation of FiRE.
Materials and Methods
Materials.
The cell-permeant, PKA-specific inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX), the nondegradable cAMP analogue 8-bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP), the adenylate cyclase activator forskolin, and the heat-stable protein kinase inhibitor were purchased from Calbiochem. FGF-2, EGF, and PDGF were purchased from PeproTech (Rocky Hill, NJ) and used in 10 ng/ml concentrations. The monoclonal rabbit anti-mouse PKAC and monoclonal rabbit anti-mouse PKAR were purchased from Transduction Laboratories (Lexington, KY).
Cell Culture, Plasmids, and Transfections.
Mouse fibroblast cell line NIH 3T3 was cultured in DMEM supplemented with 5% FCS. Cells were serum-starved with 2% carboxymethyl-Sepharose-eluted FCS for 48 hr before growth factor and chemical treatments. Construction of the p271FiRECAT reporter plasmid (7) and PKARΙ-mut expression plasmid MT-REV have been previously described (12, 13). NIH 3T3 cells were transfected by using the calcium phosphate method (14), and transfected cells were selected with geneticin (500 μg/ml) and hygromycin-B (250 μg/ml).
Chloramphenicol Acetyltransferase (CAT) Assays, Cell Proliferation Assay, and Northern Analysis.
CAT assays were performed by the xylene-extraction method, and CAT activities were measured by liquid scintillation counting (7). For the cell-proliferation assay, cells were incubated for 2 to 4 hr with 0.25 μCi (1 Ci = 37 GBq) of 5-[125I]iodo-2′-deoxyuridine (5-[125I]IdUrd; Amersham Pharmacia), washed three times with PBS, and solubilized in 1 M NaOH. Radioactivity was measured by a γ counter (Wallac, Gaithersburg, MD). For Northern analysis, cells were lysed in 4 M guanidine isothiocyanate, and RNA was isolated with cesium chloride ultracentrifugation, run on 1% agarose gel, and transferred to Hybond-N nylon membrane (Amersham Pharmacia). The membranes were hybridized with random-primed, labeled, partial cDNA of the mouse syndecan-1 gene (PM-4).
Nuclear Extracts and Gel Mobility-Shift Assays.
The extraction of nuclear proteins and the conditions used in the binding reactions have been described previously (7). Double-stranded oligonucleotides containing the indicated transcription-factor binding sites were 5′-CGC TTG ATG AGT CAG CCG GAA-3′ (AP-1 consensus; Promega), 5′-CTG GGT CAT TGA TGA CTG TTG TGT GGG ATA CCT G-3′ (motif 5), 5′-AGG AGT GAG CCA TGC CAC C-3′ (motif 4), and 5′-TTG GCA CAC CTG GGA GGA TG-3′ (motif 2).
PKA Assay and Measurement of cAMP Concentration.
The activity of PKA was measured with SignaTECT cAMP-Dependent Protein Kinase (PKA) Assay System (Promega), as recommended by the manufacturer. The changes in intracellular cAMP concentration were measured with cAMP EIA Kit from Cayman Chemicals (Ann Arbor, MI), following the manufacturer's instructions.
Western Blots and Immunofluorescence Staining.
For nuclear extracts, NIH 3T3 cells were plated on 16-cm dishes. Nuclear proteins were extracted by using a modification described by Lee et al. (15). Protein concentrations were measured by the Bradford reaction (16). Ten micrograms of protein was loaded onto an SDS/PAGE gel and transferred into a nitrocellulose membrane. For Western blots, primary antibody was diluted 1:1000. For immunofluorescence detection of PKAC, the cells were grown on plastic coverslips (Amersham Pharmacia). Cells were fixed with 4.0% paraformaldehyde/0.25% Triton X-100 for 10 min at room temperature followed by 10 min in methanol:acetone (1:1) at −20°C. The immunofluorescence staining was documented with a Leica true confocal system, four-dimensional confocal laser scanning microscope (Deerfield, IL).
Results
PKA Activity Is Required for FGF-2-Induced Activation of FiRE and Induction of Proliferation of NIH 3T3 Cells.
The mouse mesenchymal cell line NIH 3T3 was stably transfected with reporter gene constructs containing FiRE in front of a CAT reporter gene (p271FiRECAT) (7). To study the involvement of PKA in the FGF-2-induced activation of FiRE, the cells were pretreated with PKA-specific inhibitors, followed by overnight FGF-2 stimulation (10 ng/ml). A PKA-specific, cell-permeant inhibitor H-89 (17) markedly inhibited the FGF-2-induced FiRE activation (Fig. 1b). H-89 (1 μM) reduced the FGF-2-induced FiRE activity >60%, and the activity was completely inhibited with 10 μM H-89. Likewise, another PKA-specific inhibitor, KT5720 at 3 μM, inhibited the FGF-2-induced FiRE activity >50% (data not shown). The pretreatment of cells with PKA activators, nondegradable cAMP analogue 8-Br-cAMP, or phosphodiesterase inhibitor IBMX (18) before FGF-2 stimulation increased the FiRE activity >2.5-fold as compared with cells treated with FGF-2 alone (data not shown).
To further study the involvement of PKA, we used NIH 3T3 cells stably cotransfected with p271FiRECAT and a cDNA construct (MT-REV) that encodes a dominant-negative form of the PKA regulatory subunit-Ι (PKARΙ-mut) under the control of the inducible mouse metallothionein-1 promoter (12, 13). In PKARΙ-mut both cAMP binding sites are mutated to prevent cAMP binding. These mutations cause the catalytic subunits of PKA (PKAC) to be captured into stable, inactive complexes with the mutated regulatory subunits (13). Production of the PKARΙ-mut was induced by introducing ZnSO4 into the culture medium (Fig. 1c). The FGF-2-induced activation of FiRE was inhibited by PKARΙ-mut, similar to chemical PKA inhibitors. With 50 μM ZnSO4, the FGF-2-induced FiRE activation decreased 70% (Fig. 1c). As a negative control, a MT-REV construct that lacked the cDNA encoding the PKARΙ-mut, except for a residual <100 bp, (MT-REVdel) was used. In NIH 3T3 p271FiRECAT cells transfected with MT-REVdel, the FiRE activity was stimulated by FGF-2 equally well without ZnSO4 or with 50 μM ZnSO4 (data not shown).
Similarly, the FGF-2-induced proliferation of NIH 3T3 cells was blocked with PKA-specific inhibitor H-89 (Fig. 1d), as assayed by 5-[125I]IdUrd incorporation after an 18-hr FGF-2 induction. Taken together, these results imply that active PKA is required for FGF-2-induced activation of FiRE and, furthermore, for the FGF-2-induced proliferation of NIH 3T3 cells.
PKA Activity Is Required for FGF-2-Induced Transcription of Murine Syndecan-1 Gene.
Analogously to FiRE, FGF-2 also activates transcription of the endogenous syndecan-1 gene (7). Therefore, we studied whether the changes in PKA activity had an effect on the FGF-2-induced transcriptional activation of the syndecan-1 gene. Serum-starved NIH 3T3 cells were treated for 30 min with H-89 before FGF-2 stimulation, followed by RNA isolation and Northern analysis. The inhibition of PKA activity by H-89 blocked the FGF-2-induced transcription of the syndecan-1 gene (Fig. 2a). Likewise, expression of PKARΙ-mut inhibited the transcription of the syndecan-1 gene (Fig. 2b). In nontransfected NIH 3T3 cells, ZnSO4 had no effect on the FGF-2-induced transcription of syndecan-1 (Fig. 2c). The results indicated that, similar to the activation of FiRE, the FGF-2-induced expression of the endogenous syndecan-1 depends on PKA.
Figure 2.

PKA is required for FGF-2-induced expression of the syndecan-1 gene (SYN-1). (a) Inhibition of PKA by H-89 blocks the FGF-2-induced transcription of the syndecan-1 gene. Wild-type NIH 3T3 cells (NIH 3T3wt) deprived of serum were treated for 30 min with H-89 (10 μM) before 8-hr FGF-2 stimulus, followed by Northern analysis of mouse syndecan-1 mRNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as a loading control. (b) Inhibition of PKA activity by expression of PKARΙ-mut inhibits the FGF-2-induced transcription of the syndecan-1 gene. Production of PKARΙ-mut was induced by application of ZnSO4 (50 μM final concentration) into the culture medium 3 hr before 8-hr FGF-2 (10 ng/ml) stimulus. (c) In nontransfected cells, ZnSO4 had no effect on the FGF-2-induced syndecan-1 transcription.
Inhibition of PKA Activity Blocks the Binding of AP-1 Complexes to FiRE.
To elucidate the mechanisms as to how PKA controls the FGF-2-induced activation of FiRE, we studied the effects of PKA inhibition on the binding of the FGF-2-inducible transcription factors on FiRE. Motifs N4 and N5 (Fig. 3a) both bind AP-1 complexes after FGF-2 induction, whereas motif N2 (E-box) constitutively binds USF-1 (7). Serum-starved NIH 3T3 cells were pretreated for 30 min with 10 μM H-89 before 2-hr FGF-2 treatment. Nuclear proteins were isolated and gel retardation assays were performed with specific double-stranded oligonucleotides for different transcription-factor binding motifs. Motifs N4 and N5 are required for FGF-2-induced FiRE activation in NIH 3T3 cells, whereas motif N2 is not required (7). Inhibition of PKA by H-89 before FGF-2 stimulus reduced the binding of AP-1 complexes on motifs N4 and N5 to control levels, whereas binding of USF-1 on motif N2 was not reduced (Fig. 3a). The inhibitory effect of PKA inhibition on AP-1 binding was not restricted to N4 and N5 motifs of FiRE, because the FGF-2-induced binding of AP-1 to the concensus oligonucleotide containing the TPA (12-O-tetradecanoylphorbol 13-acetate)-responsive element sequence was similarly reduced (Fig. 3b).
Figure 3.

Inhibition of PKA activity decreases the binding of FGF-2 inducible AP-1 complexes to FiRE. (a) An electrophoretic mobility-shift assay was performed with radiolabeled, double-stranded oligonucleotides encompassing USF-1 binding motif N2 (E-box) and AP-1 binding motifs (N4 and N5) of FiRE. Nuclear extracts from serum-starved NIH 3T3 cells (control), serum-starved cells treated with FGF-2 (10 ng/ml) for 2 hr, or cells treated with 10 μM PKA inhibitor H-89 for 30 min before FGF-2 treatment were used. Arrowheads indicate specific binding described previously (7). (b) Reduction in FGF-2-induced AP-1 binding by PKA inhibition is not restricted to FiRE. Pretreatment with 10 μM H-89 prevented FGF-2-induced AP-1 binding to the AP-1 concensus oligonucleotide containing the TPA (12-O-tetradecanoylphorbol 13-acetate)-responsive element.
FGF-2 Increases the Activity of PKA in NIH 3T3 Cells Without Detectable Change in Intracellular cAMP Concentration.
To determine the effects of FGF-2 on the PKA activity, subconfluent (60–75%) serum-starved NIH 3T3 cells were treated with FGF-2 for 30 min and the activity of the total cellular PKA was determined by measuring the phosphorylation (nmol⋅min−1⋅mg−1) of a PKA-specific substrate, Kemptide (Promega) (19), in standard conditions. The activity of PKA increased 2-fold in FGF-2-treated cells, and the PKA-specific inhibitor H-89 completely inhibited the FGF-2-induced PKA activation (Fig. 4a). No increase of PKA activity was observed when cells were treated with PDGF or EGF. As a positive control to the activation of PKA, stimulation with 8-Br-cAMP increased PKA activity nearly 4-fold over the basal level (Fig. 4a). The specificity of the PKA-assay was verified by using a PKA-specific inhibitory protein, the heat-stable protein kinase inhibitor (20). Heat-stable protein kinase inhibitor (1 μM) reduced phosphorylation of the PKA substrate over 90% (data not shown).
Figure 4.

FGF-2 increases activity of PKA in NIH 3T3 cells without detectable change in intracellular cAMP concentration. (a) FGF-2 controls the activity of PKA. Serum-starved NIH 3T3 cells were treated with indicated growth factors (10 ng/ml) and PKA-activators 8-Br-cAMP (200 μM) for 30 min. H-89 (10 μM) was added 30 min before FGF-2. Phosphorylation of the PKA-specific substrate Kemptide was measured by liquid scintillation counting. Means and standard deviations of four independent experiments are shown. (b) FGF-2 induction does not increase the intracellular concentration of cAMP. NIH 3T3 cells were deprived of serum and treated with FGF-2 (10 ng/ml) or IBMX (1 mM) with forskolin (10 μM) for indicated times. Changes in intracellular cAMP concentration were measured with cAMP-specific ELISA.
The elevation of intracellular cAMP concentration is the most common and best known mechanism of PKA activation. The role of cAMP in the activation of PKA by FGF-2 was addressed by measuring total cellular cAMP levels after 5 to 60 min of FGF-2 stimulus. Interestingly, no change in the total cellular cAMP concentration in FGF-2-treated NIH 3T3 cells was detected (Fig. 4b).
FGF-2 Induces Relocalization of the PKA Catalytic Subunit.
PKAC subunits, complexed with PKARI in the inactive state, have been reported to be associated with cytoskeleton and different cytoplasmic membrane structures like Golgi stacks through anchoring proteins, e.g., AKAP79 and AKAP75 (21–24). When the PKA holoenzyme is activated, the PKAC dissociates from the PKAR and enters the nucleus, while the PKAR remains in the cytoplasm (25, 26). This relocalization is assumed to be essential for the PKA-induced transcriptional activation (27, 28). Therefore, we analyzed whether FGF-2 treatment induces relocalization of PKAC in NIH 3T3 cells. First, we measured the amount of PKAC in nuclear protein extracts by Western blotting. The nuclear proteins were extracted and equal amounts of protein were separated on SDS/PAGE and blotted onto a nitrocellulose filter, and PKAC was detected with an anti-PKAC antibody. After 60 min of FGF-2 stimulus, the amount of PKAC was strongly increased in the nuclear fraction (Fig. 5a). To further study the relocalization of PKAC on FGF-2 induction, the NIH 3T3 cells were grown on coverslips and treated with FGF-2 or the PKA activator 8-Br-cAMP, followed by staining with the anti-PKAC antibody and visualization by confocal microscopy (Fig. 5b). In serum-starved control cells, the PKAC was located within defined cytoplasmic areas. After treatment with FGF-2, the PKAC immunoreactivity scattered around nuclear membranes and was also found within the nucleus. Similarly, treatment of cells with 8-Br-cAMP resulted in partial translocation of the PKAC immunoreactivity into the nucleus, whereas the rest scattered around the nuclear membranes (Fig. 5b).
Figure 5.
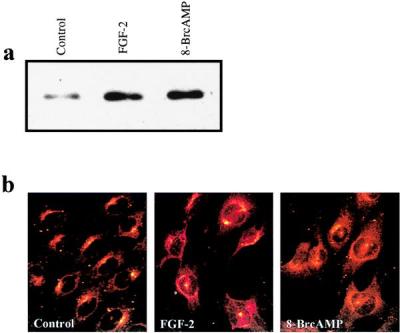
FGF-2 induces intracellular relocalization of the PKA catalytic subunit to the nuclear membranes and into the nucleus. (a) Quantification of PKAC in nuclear proteins of FGF-2-stimulated NIH 3T3 cells by Western blot analysis. Serum-starved NIH 3T3 cells were treated with FGF-2 (10 ng/ml) or PKA activator 8-Br-cAMP (200 μM) for 1 hr, and nuclear proteins were isolated. Ten micrograms of nuclear proteins was separated in 10% SDS/PAGE, blotted onto a nitrocellulose filter, and detected with anti-PKAC antibody. (b) Detection of PKAC in FGF-2 stimulated NIH 3T3 cells by indirect immunofluorescence confocal microscopy. Serum-starved NIH 3T3 cells were treated with FGF-2 or PKA-activator 8-Br-cAMP (200 μM) for 1 hr, fixed, and stained with anti-PKAC.
Discussion
In this paper, we report, by using both chemical inhibitors and a dominant inhibitory form of PKARI, that the FGF-2-induced transcription of the syndecan-1 gene in NIH 3T3 fibroblasts requires active PKA. We have shown that the binding of FGF-2-inducible AP-1 complexes on FiRE is decreased when PKA activity is inhibited before FGF-2 induction. Furthermore, we have demonstrated that FGF-2 is able to control the enzymatic activity of PKA as well as the intracellular compartmentalization of PKAC in these cells.
Considering the Ras/ERK pathway as the main pathway for growth factor-initiated signal transduction leading to the phosphorylation of transcription factors, the role of PKA in FGF-induced signaling may be to adjust the activity of the Ras/ERK pathway. Strict control over the kinase activity as well as over the duration of the activity may be mandatory to induce proper phosphorylation of transcription factors. PKA can control the ERK activity by directly phosphorylating the members of the Raf-family and by activating upstream signaling molecules, such as Rap-1 (29, 30). The FGF-2-induced PKA activity may, therefore, adjust ERK activity to a level required for appropriate transcription-factor activation. Recently it was shown (31, 32) that sustained and high-intensity activity of Raf-1 leads to cell-cycle arrest by increasing the induction of p21Cip1 and the inhibition of cyclin-dependent kinase activity. Therefore, it seems possible that inhibition of PKA activity before FGF-2 induction leads to sustained activation of Raf-1 and ERKs, which subsequently increases the amount of p21Cip1 and blocks the cell proliferation. This result would support the view of PKA as the regulator of growth factor-induced activation of the Ras/ERK pathway.
Differential activation of the Ras/ERK pathway can change the composition of an AP-1 complex. Transient activation of the Ras/ERK pathway induces short-term expression of c-Fos and Fos-B, whereas sustained activation of the Ras/ERK pathway leads to elevated expression of Fra1, Fra2, c-Jun, and JunB (33). The duration of Ras/ERK-pathway activation may determine the repertoire of Fos and Jun proteins available, leading to qualitative changes in specific gene expression (34). Alterations in the composition of AP-1 dimers and the availability of different Fos and Jun proteins are likely to change the transcriptional activity of these complexes.
PKA may directly phosphorylate FGF-2-inducible transcription factors that are present in the activated FiRE complex, namely FIN-1 and AP-1. Interestingly, the binding of FIN-1 is not induced by EGF (35), but is activated by inducing PKA with 8-Br-cAMP or IBMX (data not shown). PKA could also control the transactivation capacity of AP-1 complexes by directly phosphorylating the Fos-family members (36, 37) or by regulating the nuclear entry of c-Fos (37, 38) by phosphorylating proteins that control the nuclear translocation of transcription factors.
FiRE contains two AP-1 binding sites that are both required for its FGF-2-induced activation (7). In this paper, we have shown that PKA modifies the binding of AP-1 to the FiRE and, therefore, reduces the transcriptional activity of the element and the subsequent up-regulation of the syndecan-1 gene. Noticeably, our data (J.-P.P., unpublished data) indicate that the effects of PKA on AP-1 activity are caused by direct regulation of DNA binding, assumedly by phosphorylation rather than regulation of transcription of the AP-1 complexes. Taken together, the data indicate that PKA controls the AP-1-mediated, FGF-2-specific transcription by regulating the growth factor-induced DNA binding of AP-1 complexes.
Because no change in the total cellular cAMP concentration in FGF-2-treated NIH 3T3 cells was detected, it might be that FGF-2 changes the intracellular distribution of cAMP without altering the total cAMP concentration, thus leading to local increase of cAMP concentration as well as to local activation of PKA (39). Moreover, FGF-2 might induce PKA activation by a mechanism that does not require changes in the intracellular cAMP concentration or localization of the preexisting cAMP pool. PKA holoenzyme or PKAC can be associated with different proteins, and these complexes may be disrupted and active PKAC may be released in response to FGF-2. Disruption of a complex formed by IκB, NF-κB, and PKAC has been described in the mitogen-regulated activation of NF-κB (40). Interestingly, Tortora and coworkers (11) have shown that after EGF induction, PKAR1 is bound to the activated EGF receptor through GRB2. Similar formation and disruption of FGF receptor-PKA complexes might function in the FGF-2-induced activation of PKA.
The findings described in this paper imply mechanisms by which the FGF-2-specific transcription may be achieved. We propose that the ability of FGF-2 to control the PKA activity may determine the FGF-2-specific induction of AP-1-driven genes that are not activated by other growth factors.
Acknowledgments
We thank Dr. Stanley McKnight for the mutant PKA cDNA. Dr. Klaus Elenius and Dr. Anri Tienhaara are acknowledged for critical reading of the manuscript. We are grateful to Katri Hiilesvuo, Taina Kalevo-Mattila, Anni Kieksi, and Susanna Pyökäri for technical help. This work was financially supported by the Academy of Finland, the Finnish Cancer Union, the Juselius Foundation, Maud Kuistila Memorial Foundation, and Emil Aaltonen Foundation.
Abbreviations
- FGF
fibroblast growth factor
- FiRE
FGF-inducible response element
- PDGF
platelet-derived growth factor
- EGF
epidermal growth factor
- PKA
protein kinase A
- PKARΙ, PKA regulatory subunit-Ι
PKARΙ-mut, dominant-negative form of PKARΙ
- PKAC
PKA catalytic subunit
- AP-1
activator protein-1
- FIN-1
FGF-2-inducible nuclear factor-1
- USF-1
upstream stimulatory factor-1
- H-89
N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide
- 8-Br-cAMP
8-bromoadenosine 3′,5′-cyclic monophosphate
- IBMX
3-isobutyl-1-methylxanthine
- CAT
chloramphenicol acetyltransferase
References
- 1.Karin M, Hunter T. Curr Biol. 1995;5:747–757. doi: 10.1016/s0960-9822(95)00151-5. [DOI] [PubMed] [Google Scholar]
- 2.Marshall C J. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 3.Zanke B W, Rubie E A, Winnett E, Chan J, Randall S, Parsons M, Boudreau K, McInnis M, Yan M, Templeton D J, Woodgett J R. J Biol Chem. 1996;271:29876–29881. doi: 10.1074/jbc.271.47.29876. [DOI] [PubMed] [Google Scholar]
- 4.Dickens M, Rogers J S, Cavanagh J, Raitano A, Xia Z, Halpern J R, Greenberg M E, Sawyers C L, Davis R J. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 5.Whitmarsh A J, Yang S-H, Su M S-S, Sharrocks A D, Davis R J. Mol Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaeffer H J, Catling A D, Eblen S T, Collier L S, Krauss A, Weber M J. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 7.Jaakkola P, Vihinen T, Määttä A, Jalkanen M. Mol Cell Biol. 1997;17:3210–3219. doi: 10.1128/mcb.17.6.3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umbhauer M, Marshall C J, Mason C S, Old R W, Smith J C. Nature (London) 1995;376:58–62. doi: 10.1038/376058a0. [DOI] [PubMed] [Google Scholar]
- 9.Besser D, Presta M, Nagamine Y. Cell Growth Diff. 1995;6:1009–1017. [PubMed] [Google Scholar]
- 10.Yao H, York R D, Misra-Press A, Carr D W, Stork P J. J Biol Chem. 1998;273:8240–8247. doi: 10.1074/jbc.273.14.8240. [DOI] [PubMed] [Google Scholar]
- 11.Tortora G, Damiano V, Bianco C, Baldassarre G, Bianco A R, Lanfrancone L, Pelicci P G, Ciardiello F. Oncogene. 1997;14:923–928. doi: 10.1038/sj.onc.1200906. [DOI] [PubMed] [Google Scholar]
- 12.Tasken K, Andersson K B, Erikstein B K, Hansson V, Jahnsen T, Blomhoff H K. Endocrinology. 1994;135:2109–2119. doi: 10.1210/endo.135.5.7956934. [DOI] [PubMed] [Google Scholar]
- 13.Clegg C H, Correll L A, Cadd G G, McKnight G S. J Biol Chem. 1987;262:13111–13119. [PubMed] [Google Scholar]
- 14.Chen C, Okayama H. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K A W, Bindereif A, Green M R. Gene Anal Tech. 1988;5:22–31. doi: 10.1016/0735-0651(88)90023-4. [DOI] [PubMed] [Google Scholar]
- 16.Bradford M M. Anal Biochem. 1976;72:248–252. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 17.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 18.Morgan A J, Murray K J, Challiss R A. Biochem Pharmacol. 1993;45:2373–2380. doi: 10.1016/0006-2952(93)90216-j. [DOI] [PubMed] [Google Scholar]
- 19.Kemp B E, Graves D J, Benjamini E, Krebs E G. J Biol Chem. 1977;252:4888–4894. [PubMed] [Google Scholar]
- 20.Fantozzi D A, Harootunian A T, Wen W, Taylor S S, Feramisco J R, Tsien R Y, Meinkoth J L. J Biol Chem. 1994;269:2676–2686. [PubMed] [Google Scholar]
- 21.Ndubuka C, Li Y, Rubin C S. J Biol Chem. 1993;268:7621–7624. [PubMed] [Google Scholar]
- 22.Klauck T M, Faux M C, Labudda K, Langeberg L K, Jaken S, Scott J D. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 23.Nigg E A, Schafer G, Hilz H, Eppenberger H M. Cell. 1985;41:1039–1051. doi: 10.1016/s0092-8674(85)80084-2. [DOI] [PubMed] [Google Scholar]
- 24.Rios R M, Celati C, Lohmann S M, Bornens M, Keryer G. EMBO J. 1992;11:1723–1731. doi: 10.1002/j.1460-2075.1992.tb05224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meinkoth J L, Ji Y, Taylor S S, Feramisco J R. Proc Natl Acad Sci USA. 1990;87:9595–9599. doi: 10.1073/pnas.87.24.9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagiwara M, Brindle P, Harootunian A, Armstrong R, Rivier J, Vale W, Tsien R, Montminy M R. Mol Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassano S, Gallo A, Buccigrossi V, Porcellini A, Cerillo R, Gottesman M E, Avvedimento E V. J Biol Chem. 1996;271:29870–29875. doi: 10.1074/jbc.271.47.29870. [DOI] [PubMed] [Google Scholar]
- 28.Feliciello A, Giuliano P, Porcellini A, Garbi C, Obici S, Mele E, Angotti E, Grieco D, Amabile G, Cassano S, et al. J Biol Chem. 1996;271:25350–25359. doi: 10.1074/jbc.271.41.25350. [DOI] [PubMed] [Google Scholar]
- 29.Erhardt P, Troppmair J, Rapp U R, Cooper G M. Mol Cell Biol. 1995;15:5524–5530. doi: 10.1128/mcb.15.10.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vossler M R, Yao H, York R D, Ming-Gui P, Rim C S, Stork P J S. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 31.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Mol Cell Biol. 1997;17:5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sewing A, Wiseman B, Lloyd A, Land H. Mol Cell Biol. 1997;17:5588–5597. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook S J, Aziz N, McMahon M. Mol Cell Biol. 1999;19:330–341. doi: 10.1128/mcb.19.1.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Xie Z, Scott R E. J Cell Biol. 1996;135:1151–1162. doi: 10.1083/jcb.135.4.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaakkola P, Määttä A, Jalkanen M. Oncogene. 1998;17:1279–1286. doi: 10.1038/sj.onc.1202002. [DOI] [PubMed] [Google Scholar]
- 36.Tratner I, Ofir R, Verma I M. Mol Cell Biol. 1992;12:998–1006. doi: 10.1128/mcb.12.3.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gauthier-Rouviere C, Vandromme M, Lautredou N, Cai Q Q, Girard F, Fernandez A, Lamb N. Mol Cell Biol. 1995;15:433–444. doi: 10.1128/mcb.15.1.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandromme M, Gauthier-Rouviere C, Lamb N, Fernandez A. Trends Biochem Sci. 1996;21:59–64. [PubMed] [Google Scholar]
- 39.Houslay M D, Milligan G. Trends Biochem Sci. 1997;22:217–224. doi: 10.1016/s0968-0004(97)01050-5. [DOI] [PubMed] [Google Scholar]
- 40.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]