

Abstract
We have synthesized the environment-sensitive fluorophores 2-cyano-6-dihexylaminoanthracene and 2-propionyl-6-dihexylaminoanthracene (Anthradan) starting from 2,6-diaminoanthraquinone. Anthradan is the benzologue of the well-known family of naphthalene 2-propionyl-6-dimethylaminonaphthalene (PRODAN) fluorophores. The additional spectral red shift of the anthracene avoids the autofluorescence of many biological systems and provides for more favorable excitation wavelengths for fluorescence applications. Furthermore, Anthradan exhibits polarity-sensitive emission comparable to that of PRODAN and displays high quantum yields in a range of solvents. Single molecules of these anthracene-containing fluorophores have been imaged in polymer hosts as a proof-of-principle.
Introduction
Fluorescence probes are widely used in photobiology, and the enhancement of function of these materials is actively sought.1 Especially attractive are molecules that exhibit a reporter function arising from some observable that reflects features or changes in the local environment. One of the best known among polarity-sensitive fluorophores is 2-propionyl-6-dimethylaminonaphthalene (PRODAN), which was originally introduced by Weber and Farris.2 This specific dye and many of its derivatives have subsequently been extensively explored as fluorescence probes for studying nanoenvironments in various chemical and biological systems. PRODAN is a push—pull charge-transfer chromophore with an electron-donating dimethylamino group and an electron-withdrawing propionyl group located at the 2,6-positions of naphthalene. Numerous modifications of PRODAN such as 6-acryloyl-2-dimethylaminonaphthalene (ACRYLODAN), 6-lauroyl-2-dimethylaminonaphthalene (LAURDAN), 2′-(N,N-dimethylamino)-6-naphthoyl-4-trans-cyclohexanoic acid (DANCA), and other related fluorophores have been introduced.3 However, one drawback of these dyes is that they all absorb in the blue, thus stimulating cellular autofluorescence4 that can diminish the signal/background ratio, thereby making ultrasensitive measurements more difficult in future cell applications.
Our continued interest in fluorescent dyes for single-molecule fluorescence spectroscopy5 in biologically relevant systems led us to explore the anthracene analogues of these naphthalene dyes. We anticipated that the push—pull structure of a 2,6-substituted anthracene would retain the useful properties found in PRODAN (i.e., polarity-induced shift of emission and sensitivity to local nanoenvironments) while shifting the absorption further to the red for better results in cellular imaging. No amine-donor anthracene chromophores have been reported with a donor—acceptor pair located only at the 2,6-positions. Here, we report the synthesis and physical properties of 2,6-substituted anthracene push—pull molecules that possess a modest change in solvent-dependent absorption but a large change in solvent-dependent fluorescence. The potential utility of these dyes is illustrated by a demonstration of direct single-molecule imaging.
Simple anthracene derivatives have been extensively used as fluorescent probes in biology, chemical sensors, and optoelectronic materials.6 For instance, Tocanne and co-workers studied methyl 8-(2-anthroyl)octanoate as a solvatochromic lipophilic probe.7 Coleman and Mortensen synthesized 1- and 2-anthrancenyl β-C2’-deoxyribosides to study the dynamic properties of DNA using ultrafast photophysical techniques.8 Sakamoto and co-workers synthesized anthracene-linked polythiazaalkane and polythiaalkane derivatives as silver-ion-selective fluoroionophores.9 The absorption and fluorescence of these simple anthracene derivatives is typically in the range of 350–400 nm. However, red-shifted 2,6-donor—acceptor-substituted anthracenes, in which the two groups are located at maximum distance across the acene, are rare. Only very recently, Ihmels and co-workers reported a 2,6-donor—acceptor-substituted anthracene with a moderate-strength methoxy donor group and a benzoxazole or oxazoline acceptor, which were derived from a carboxylic acid.10 These molecules exhibit moderate solvatochromism and were explored as acid-sensitive fluorescence probes. An anthracene with a 6-amino donor and a 2,3-dicarboxylic acid acceptor pair was prepared by building the anthracene core in several steps.11 Sun and Desper’s recent modification involved protection of 2,3-dimethylanthracene and subsequent nitration and reduction. However, the starting material was not commercially available and it had to be prepared in several steps.12 These methods are not useful for the 2,6-donor—acceptor-substituted anthracenes with an amino donor.
Results and Discussion
The synthetic approaches we pursued to prepare 2,6-donor—acceptor-substituted anthracenes all began with commercially available 2,6-diaminoanthraquinone 1 (Scheme 1). There are two key issues concerning the application of this precursor: (1) breaking the symmetry of the molecule which involves retaining one amine and converting the other amine to a different functional group (e.g., dialkylating one amine and converting the other to a bromide) and (2) reduction of the 9,10-anthraquinone system to anthracene. The second issue had already been dealt with in a variety of other substituted anthraquinones using either sodium borohydride13 or zinc in ammonia.14 Using either of these procedures, we successfully reduced 2,6-diamino-9,10-anthraquinone to 2,6-diaminoanthracene (2). However, all attempts to selectively modify only one of the two amines were unsuccessful. An attempt to carry out a selective Sandmeyer reaction (conversion of amine to bromide) on only one amine group failed at least in part due to solubility problems, resulting mainly in dibromide 3, and no monobromide 4 was isolated.
Scheme 1. Unsuccessful Approaches to Prepare Asymmetric 2,6-Disubstituted Anthraquinonesa,b.

a In all cases, the asymmetrical product (4, 6, 8, 9) was not produced in a useful yield. b(a) Zn, NH4OH; (b) 1 equiv of t-BuONO, 1 equiv of CuBr2, CH3CN; (c) 2.2 equiv of t-BuONO, 2.2 equiv of CuBr2, CH3CN; (d) dihexylamine, K2CO3, DMSO, 125–140 °C; (e) CuI, NH4OH.
We noticed that the commercially available 2,6-diaminoanthraquinone (1) had already been successfully converted to 2,6-dibromoanthraquinone (5) using the Sandmeyer reaction.15 Using the method described for this double Sandmeyer reaction, an effort to convert only one of the amino groups to a bromide by using 1 equiv or less of tert-butyl nitrite and cupric bromide resulted in only a mixture of 5 and the starting 1. Direct nucleophilic aromatic substitution of 5 with a secondary amine was not efficient and usually afforded an inseparable mixture. For example, reaction of 5 with dihexylamine in the presence of K2CO3 in DMSO at 125–140 °C afforded a mixture containing less than 5% of the desired 2-bromo-6-dihexylaminoanthraquinone (8). Other products included 2,6-bisdihexylaminoanthraquinone (7), but the mixture was too difficult to separate for any practical application. Amination of 5 with a CuI catalyst and an excess of ammonium hydroxide under pressure afforded only the starting amine 1.16 These failed attempts to obtain asymmetrically 2,6-disubstituted anthracenes and anthraquinones are summarized in Scheme 1.
One option to break the symmetry of the 2,6-disubstituted anthraquinone involves finding an intermediate that has similar or less solubility or reactivity than the starting substrate, so as to avoid preferential reactivity for the monofunctionalized product. In such a circumstance, at least a statistical product distribution might be obtained. We assume 2-bromo-6-fluoroanthraquinone (10) has solubility comparable to 5 and that the fluorine could be preferentially replaced by the dialkylamino functionality via aromatic nucleophilic substitution. It was reported that 5,6-dibromo-1,2-acenaphthenequinone was successfully converted to 5,6-difluoro-1,2-acenaphthenequinone by reaction with CsF in anhydrous DMSO,17 and we felt this approach might also be useful in the case of the 2,6-dibromoanthraquinone system at hand (Scheme 2). With 1.0 equiv of CsF in DMSO at 110 °C, the result was encouraging in that about 45% of the starting dibromide (5) was converted to 2-bromo-6-fluoroanthraquinone (10) accompanied by about 10% of 2,6-difluoroanthraquinone. An increase of the amount of CsF to 2 equiv, under the same conditions, consumed all the dibromide starting material but now gave about a 1:2 mixture of monofluoride and difluoride. We finally found that treatment of 5 with 1.3 equiv of CsF in anhydrous DMSO at 110 °C gave about a 2:1 ratio of 10 and 2,6-difluoroanthraquinone as detected by GC-MS (60% 2-bromo-6-fluoroanthraquinone, 30% 2,6-difluoroanthraquinone, and the remainder was the starting 5). No attempt was made to separate this mixture. Instead, the halogen exchange reaction mixture in DMSO was directly treated with N,N-dihexylamine and potassium carbonate at 70 °C to give, after chromatography, 2-bromo-6-dihexylaminoanthraquinone (8) in overall 18% yield for the two-step, one-pot reaction.
Scheme 2. Overall Synthesis of the Asymmetric Anthracenes 11–13 Starting from 2,6-Diaminoanthraquinone 1a.

a (a) t-BuONO, CuBr2, CH3CN; (b) CsF, DMSO, 110 °C; (c) R = hexyl, R2NH, DMSO, K2CO3, 70 °C (18% yield, b and c, two steps); (d) (1) NaBH4, i-PrOH, (2) HCl aq, (3) NaBH4, i-PrOH, (4) HCl aq (53%); (e) CuCN, DMF, reflux 12 h (83%); (f) CH3CH2MgCl, CuCl (cat.), reflux 24 h (77%).
Reduction of the 2-bromo-6-dihexylaminoanthraquinone was performed in 53% yield using the method of Klanderman involving successive sodium borohydride reduction and dehydration.13 The product, 2-bromo-6-dihexylaminoanthracene (11), was treated with cuprous cyanide in DMF to afford 2-cyano-6-(dihexylamino)anthracene, 12.18 Finally, 6-propionyl-2-(dihexylamino)anthracene (Anthradan) 13 was prepared by reaction of this cyano compound with ethylmagnesium chloride catalyzed with cuprous chloride followed by hydrolysis of the imine intermediate.
Absorption and Emission Properties
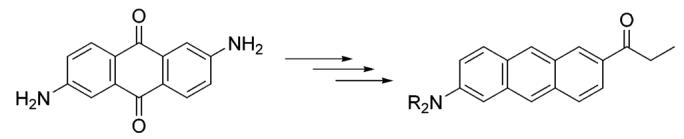
The absorption spectra of 11, 12, and Anthradan 13 in THF are all shown in Figure 1. The spectra are significantly different from those for anthroyl derivatives that have no push—pull substitution. The maximum long-wavelength absorption band of a 2-anthroyl chromophore is around 375–400 nm, and this band was assigned as the longitudinally polarized band with vibrational structure.19 Although the absorption of molecules 12 and 13 exhibits the same absorption with vibrational detail at the same wavelength, the most notable absorption feature of 12 and 13 is the appearance of a new broad band around 440–460 nm that is assigned as an intramolecular charge-transfer band. The longitudinally polarized band with vibrational features in the anthroyl derivative is diminished substantially. Bromide 11, which is the precursor of 12 and has the same 2,6-substitution pattern, has much less absorption intensity for the intramolecular charge-transfer band due to the lack of a strong electron-withdrawing group. The maximum charge-transfer molar extinction coefficients of 12 and 13 in THF are 1.03 × 104 and 1.21 × 104 L cm-1 mol-1, respectively.
Figure 1.
Comparison of the absorption spectra of 11, 12, and 13 in THF. All of these three anthracene derivatives have a common 2-dihexylamine donor but differ in the 6-substituent (Br, CN, and COCH2CH3, respectively).
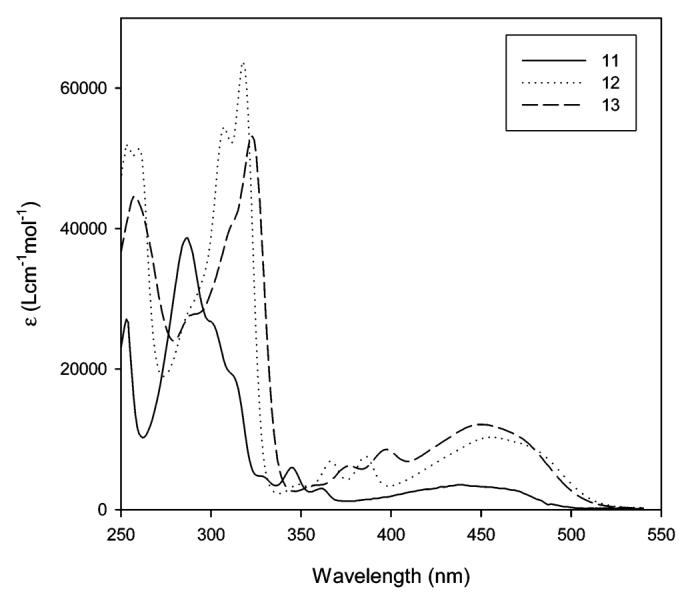
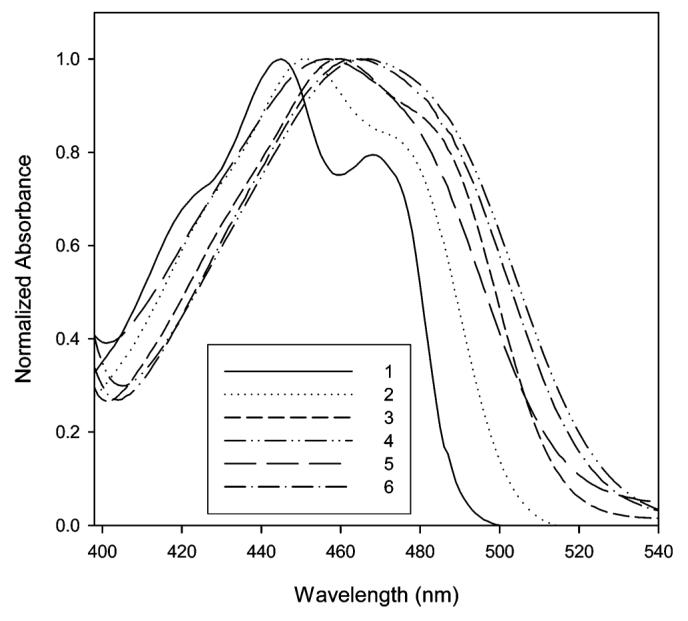
The absorption and emission maxima of PRODAN, 12, and Anthradan 13 in a variety of solvents spanning a wide range of polarity including nonpolar, polar, and polar aprotic solvents are found in Table 1. Examination of structure 13 reveals that it is quite similar to PRODAN, with the exception of the extension of the conjugation from naphthalene in PRODAN to anthracene in 13 and the different dialkyl groups on the amine donor. The dihexylamine group was employed with Anthradan to enhance solubility in ordinary organic solvents and will not significantly influence the absorption and emissions relative to the dimethylamino group as found in PRODAN. Similarly, structure 12 is an anthracene analogue of the classic 4-dimethylaminobenzonitrile (DMABN) which was the first example of the twisted intramolecular charge-transfer (TICT) state;20 thus, we anticipated that the emission of 12 and 13 might strongly depend on solvent polarity. The influence of selected solvents on the absorption of 12 and 13 is shown in Figures 2 and 3, respectively. The absorptions of both 12 and 13 were found to not be very sensitive to changes in the polarity of the solvent, and only moderate absorption solvatochromism was observed. In the low-polarity solvents hexane and toluene, a charge-transfer band with some structure features was observed, whereas in all other higher-polarity solvents, only a single broad charge-transfer band was observed. The absorption maximum of 12 changes from 445 (hexane) to 467 nm (DMSO). Polar protic solvents have no specific effect on the absorption of 12. The absorption maximum of 13 ranges from 439 (hexane) to 464 nm (ethylene glycol). Anthradan (13) and PRODAN are quite similar in their response to solvents, except Anthradan absorbs at approximately 100-nm longer wavelengths.
Table 1.
Absorption and Emission of PRODAN, 12, and 13 in Solvents with a Range of Δf (Orientational Polarizability)a
| fluorophore |
PRODANb |
12 |
13 |
||||
|---|---|---|---|---|---|---|---|
| solvent | Δf | λabs | λem | λabs | λem | λabs | λem |
| hexane | 0.0001 | 341c | 390c | 445 | 483 | 439 | 483 |
| toluene | 0.0134 | 349d | 416d | 451 | 507 | 447 | 507 |
| chloroform | 0.1482 | 357 | 440 | 459 | 525 | 458 | 536 |
| THF | 0.2096 | 348c | 431c | 455 | 533 | 450 | 529 |
| acetonitrile | 0.3053 | 350 | 462 | 460 | 551 | 455 | 557 |
| DMF | 0.2745 | 355 | 461 | 464 | 557 | 458 | 557 |
| DMSO | 0.2640 | 358d | 464d | 467 | 566 | 460 | 569 |
| 1-propanol | 0.2493 | 360d | 480d | 456 | 537 | 456 | 585 |
| ethanol | 0.2886 | 360 | 496 | 454 | 545 | 452 | 601 |
| methanol | 0.3084 | 362 | 505 | 457 | 546 | 456 | 604 |
| 1,2-propanediol | 0.2708 | 370 | 510 | 462 | 546 | 462 | 598 |
| ethylene glycol | 0.2745 | 375 | 515 | 464 | 554 | 464 | 617 |
Figure 2.
Normalized absorption of 12 in hexane (1), toluene (2), chloroform (3), DMSO (4), methanol (5), and ethylene glycol (6).
Figure 3.
Normalized absorption of 13 in hexane (1), toluene (2), chloroform (3), DMSO (4), methanol (5), and ethylene glycol (6).
The fluorescence quantum yields of 12 and 13 in toluene, ethanol, and acetone are tabulated in Table 2. These solvents were selected to represent both a range of polarity and the ability to form hydrogen bonds. The quantum yields in these three solvents are comparable and show no obvious trends.
Table 2.
Fluorescence Quantum Yields (QY), λabs,max, and λem,max of 12 and 13 in Selected Solvents
The influence of selected solvents on the emission of 12 and 13 is shown in Figures 4 and 5, respectively. In contrast to the absorption, the solvent polarity has a dramatic effect on the emission wavelength. The emission maximum of 12 ranges from 483 (hexane) to 566 nm (DMSO), and the emission maximum of 13 ranges from 483 (hexane) to 617 nm (ethylene glycol). Examination of Table 1 shows that the emission wavelengths for both 12 and 13 are quite comparable to each other in nonpolar and polar aprotic solvents. One of the distinct differences between these two fluorophores is their response to polar protic solvents. In polar protic solvents, emission of 13 has moved to a longer wavelength relative to 12. The Stokes shift of 13 in a series of alcohols (1-propanol, methanol, 1,2-propanediol, and ethylene glycol) is approximately 40 nm greater than that of 12.
Figure 4.
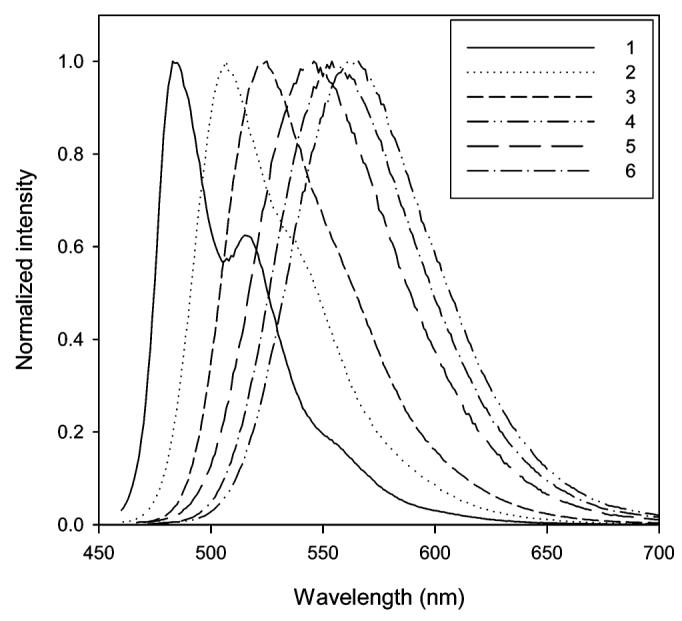
Normalized emissions of 12 in hexane (1), toluene (2), chloroform (3), DMSO (4), methanol (5), and ethylene glycol (6).
Figure 5.
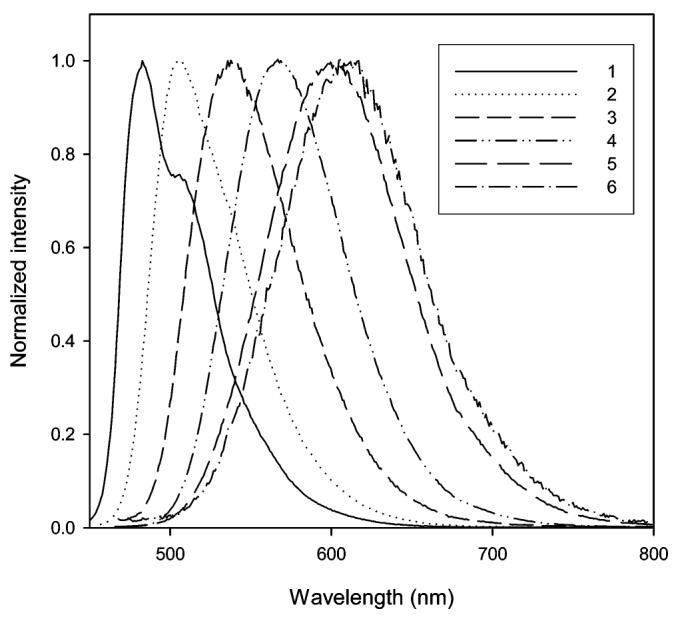
Normalized emissions of 13 in hexane (1), toluene (2), chloroform (3), DMSO (4), methanol (5), and ethylene glycol (6).
To better explore the solvatochromic behavior of these fluorophores, we used a standard dipolar model for solvent stabilization. When a fluorophore is excited from the ground to excited state, the solvent dipole can reorient around the excited-state dipole moment. This solvent relaxation can lower the energy of the excited state. This effect becomes larger as the solvent polarity is increased, thus the emission will be red shifted. More precisely, this solvent effect is determined by the index of refraction and the relative dielectric constant of the solvent, described by the Lippert equation in which the fluorophore is considered to be a dipole located in a cavity with a radius of a in a continuous solvent—dipole environment (eq 1).1b
| (1) |
In the equation, and are the wavenumbers of the absorption and emission, n is the refractive index, ∊ is the relative low-frequency dielectric constant of the solvent, h is Planck’s constant, c is the speed of light, and Δf is called the orientation polarizability. The linear correlation between the Stokes shifts and Δf predicted by the Lippert equation does not hold for a specific interaction between the fluorophore and solvent molecules such as hydrogen bonding.
Plots of the Stokes shift as a function of the solvent orientational polarizability of 12 and 13 are shown in Figure 6A and B, respectively. The linear correlation between the Stokes shift and orientational polarizability shown in Figure 6A reveals only a dipolar solvent effect here. On the other hand, in Figure 6B, all the protic solvents are located on the top and right side of the plot indicative of a specific solvent effect most likely due to hydrogen bonding.
Figure 6.
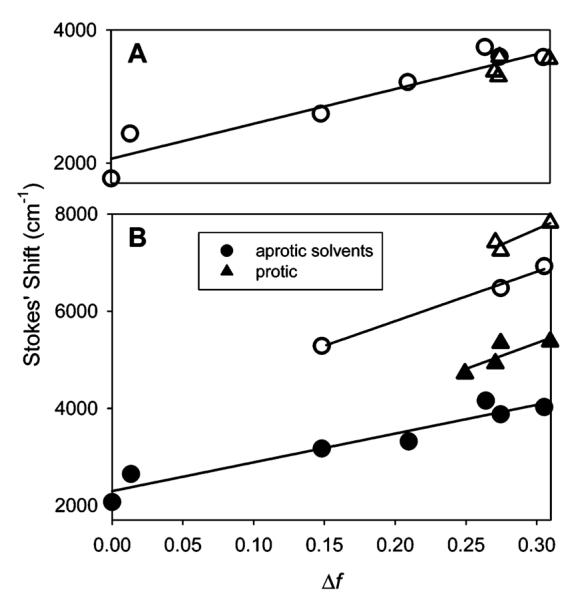
Lippert plots. Circles denote aprotic solvents, and triangles denote protic solvents. (A) Lippert plot for fluorophore 12. The slope of the fit to all solvents is 5223 cm-1 (R2 = 0.9015). (B) Lippert plots for Anthradan 13 (closed symbols) and PRODAN (open symbols). For Anthradan, the slope for the fit to aprotic solvents is 5921 cm-1 (R2 = 0.9233) and for protic solvents is 10 656 cm-1 (R2 = 0.6845). The Lippert plot for PRODAN yields a slope for the fit to aprotic solvents of 10 162 cm-1 (R2 = 0.9933) and that for protic solvents of 12 803 cm-1 (R2 = 0.5627).
Fluorophore 13 is structurally related to PRODAN, and comparisons of the Stokes shift vs Δf in various solvents are shown in Figure 6B. Although the slope for PRODAN is slightly higher, it is clear that the sensitivity to solvent polarity is quite similar for PRODAN and Anthradan (13) which both contain the propionyl acceptor group. Fluorophore 12 with a nitrile acceptor group has no specific solvent effect and is slightly less sensitive to the solvent polarity compared with both PRODAN and Anthradan (13).
Single-Molecule Properties
A strong test of the quality of a fluorophore is its ability to be imaged at the single-molecule level, which requires bright fluorescence, weak coupling with dark states, and photostability. Our single-molecule analysis reveals that single copies of Anthradan molecules in poly(methyl methacrylate) (PMMA) are visible in an epifluorescence image of a typical sample, as shown in Figure 7. However, the photostability of these molecules needs further development. To characterize a single-molecule emitter using one simple parameter, we recorded the distribution of the number of photons emitted from single fluorophores before photobleaching. After recording single-molecule distributions, which were exponential in shape as expected, a fit to the exponential yields an average number of detected photons, Ntot,d ∼ 20 000 per molecule. The photon collection efficiency21 of our setup is D = 0.10. Correcting the Ntot,d value for the collection efficiency yields a value for the total number of photons emitted, Ntot,e ∼ 2.0 × 105 photons emitted per molecule. This value is an order of magnitude lower than that for DCDHF-6 or R6G (2.4 × 106 and 1.9 × 106 photons emitted per molecule, respectively),22 both of which are good single-molecule fluorophores. Nevertheless, we demonstrated that single copies of Anthradan are observable, which gives us hope in being able to further synthetically develop this fluorophore for enhanced photophysical properties required for future single-biomolecule imaging.
Figure 7.

Epifluorescence images of single copies of Anthradan in a PMMA film imaged with 488 nm excitation.
Conclusions
New 2,6-dipolar anthracene fluorophores have been synthesized which have bathochromically shifted absorption and emission relative to their well-known naphthalene analogues. These new fluorophores retain the same photophysical properties that have made the naphthalene compounds so useful, namely a pronounced environmental sensitivity for the emission. The anthracene compound Anthradan has been successfully imaged at the single-molecule level, but the number of photons emitted and detected is low relative to other high-performance single-molecule chromophores, perhaps reflecting instability problems intrinsic to chromophores containing larger acene substructures.
Experimental Section
Sample Preparation
Solutions of fluorophores were prepared in toluene, and bulk absorption and emission spectra were taken on conventional spectrometers. Quantum yields were measured against DCDHF-6 in toluene and were corrected for differences in optical density and solvent refractive index.1b
Single-molecule samples were prepared by spin-casting 1% (by mass) solutions of PMMA in toluene, doped with nanomolar fluorophore concentrations, onto clean glass coverslips. After drying, these samples were studied using an inverted microscope in an epifluorescence configuration.21 Samples were illuminated using a continuous-wave 488 nm laser; the intensity at the sample was approximately 0.5 kW/cm2. The emission was collected through a 100×, 1.4 N.A. microscope objective, filtered to remove scattered excitation light, and imaged onto a back-illuminated frame-transfer Si CCD camera with an integration time of 100 ms/frame.
2-Bromo-6-dihexylaminoanthraquinone (8)
A mixture of 2,6-dibromo-9,10-anthraquinone (3.66 g, 10 mmol), anhydrous CsF (1.62 g, 11 mmol), and 200 mL of anhydrous DMSO was heated under nitrogen at 140–150 °C for 5 h with magnetic stirring. The black reaction mixture was then cooled to 70 °C. Then, dihexyl-amine (3.71 g, 20 mmol) and potassium carbonate (2.76 g, 20 mmol) were added to the flask, and the mixture was stirred at 70 °C for 36 h. The resulting red—brown mixture was poured into 1000 mL of water and extracted with dichloromethane (200 mL × 3). The combined organic layers were washed with water and brine and dried over anhydrous MgSO4. After removal of the dichloromethane, the residue was separated by flash chromatography with hexane and a hexane/dichloromethane (4:1) mixture as eluent. The 2-bromo-6-dihexylaminoanthraquinone was obtained as a red solid (0.84 g, 18% yield). Mp 114.2–115.4 °C (CH2Cl2/hexane). 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 2.0 Hz, 1H), 8.12 (d, J = 9.0 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 7.81 (dd, J = 8.2, 2.0 Hz, 1H), 7.39 (d, J = 2.7 Hz, 1H), 6.88 (dd, J = 9.0, 2.7 Hz, 1H), 3.43 (t, J = 7.7 Hz, 4H), 1.70–1.61(m, 4H), 1.42–1.31 (m, 12H), 0.93 (t, J = 7.0 Hz, 6H). 13C NMR (100 Hz, CDCl3) δ 183.6, 179.8, 152.3, 135.8, 135.7, 134.9, 132.3, 130.1, 129.9, 129.5, 128.6, 120.9, 115.6, 108.3, 51.2, 31.6, 27.2, 26.7, 22.6, 14.0. Anal. Calcd for C26H32BrNO2: C, 66.38; H, 6.86; N, 2.98. Found: C, 66.42; H, 7.09; N, 3.27. HRMS (m/z): [M + Na] calcd for C26H32BrNO2Na, 492.1514; found, 492.1517.
2-Bromo-6-dihexylaminoanthracene (11)
A suspension of 2-bromo-6-dihexylaminoanthraquinone (0.84 g, 1.79 mmol) and NaBH4 (3.0 g, 79.3 mmol) in i-PrOH (100 mL) was stirred for 12 h at room temperature and then heated under reflux for 10 h. After cooling to room temperature, the solvent was removed by rotary evaporation and the residue was treated with water. The mixture was neutralized with 6 M HCl and heated under reflux for 4 h. After cooling to room temperature, the mixture was extracted with dichloromethane and the organic layers were dried over MgSO4. After the solvent was removed, the residue was added to i-PrOH (50 mL) and treated with another portion of NaBH4 (2.0 g, 52.2 mmol) with subsequent refluxing for 12 h. After cooling, i-PrOH was removed and aqueous 3 M HCl (10 mL) was added carefully until bubbling ceased. The mixture was refluxed for 2 h under nitrogen protection. The mixture was cooled down to room temperature and extracted with dichloromethane. The combined organic layers were washed with water and brine and dried over anhydrous MgSO4. The solvent was removed, and the residue was purified by flash chromatography with hexane as eluent to afford 2-bromo-6-dihexylaminoanthracene as a glassy yellow solid (420 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 8.04 (s, 1H), 8.02 (d, J = 2.0 Hz, 1H), 7.81 (d, J = 9.4 Hz, 1H), 7.70 (d, J = 9.1 Hz, 1H), 7.37 (dd, J = 9.1, 2.0 Hz, 1H), 7.18 (dd, J = 9.4, 2.4 Hz, 1H), 6.83 (d, J = 2.4 Hz), 3.38 (t, J = 7.7 Hz, 4H), 1.69–1.61 (m, 4H), 1.40–1.31 (m, 12H), 0.92 (t, J = 7.0 Hz, 6H). 13C NMR (100 Hz, CDCl3) δ 145.7, 134.1, 130.6, 129.91, 129.87, 129.14, 129.11, 128.5, 126.8, 124.9, 122.4, 118.5, 116.7, 102.5, 51.2, 31.8, 29.7, 27.5, 26.9, 22.7, 14.1. Anal. Calcd for C26H34BrN: C, 70.90; H, 7.78; N, 3.18. Found: C, 70.71; H, 7.84; N, 3.47. HRMS (m/z): [M + H] calcd for C26H35BrN, 440.1953; found, 440.1955.
2-Cyano-2-(dihexylamino)anthracene (12)
A mixture of 2-bromo-6-dihexylaminoanthracene (220 mg, 0.5 mmol) and cuprous cyanide (269 mg, 3 mmol) in 6 mL of anhydrous DMF was stirred and refluxed under a nitrogen atmosphere for 12 h. The reaction mixture was then cooled and treated with 1 mL of ethylenediamine and 10 mL of water at 50 °C for a half hour. Water (50 mL) was added, and the resulting mixture was extracted with dichloromethane (60 mL × 2). The combined organic layer was washed with water (50 mL × 2), dried (MgSO4), concentrated, and loaded on silica gel. Purification by silica gel flash chromatography with hexane/dichloromethane elution afforded the product as a yellow solid (160 mg, 83% yield). Mp 81.9–82.5 °C (hexane). 1H NMR (CDCl3) δ 8.29 (d, J = 1.5 Hz, 1H), 8.27 (s, 1H), 8.09 (s, 1H), 7.91–7.87 (m, 2H), 7.4 (dd, J = 8.8, 1.5 Hz, 1H), 7.27 (dd, J = 9.5, 2.5 Hz, 1H), 6.87 (d, J = 2.5 Hz, 1H), 3.45 (t, J = 7.7 Hz, 4H), 1.75–1.65 (m, 4H), 1.47–1.33 (m, 12H), 0.95 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3) δ 146.7, 136.0, 135.8, 132.4, 129.7, 128.5, 127.5, 127.1, 126.9, 124.3, 122.2, 120.2, 118.6, 105.8, 102.0, 51.2, 31.7, 27.5, 26.9, 22.7, 14.1. UV—vis (THF) λ1 = 253 nm, ∊1 = 5.20 104 L cm-1 mol-1, λ2 = 318 nm, ∊2 = 6.38 × 104 L cm-1 mol-1, λ3 = 456 nm, ∊3 = 1.03 × 104 L cm-1 mol-1. IR (cm-1) 2926, 2215, 1626. Anal. Calcd for C27H34N2: C, 83.89; H, 8.87; N, 7.25. Found: C, 83.85; H, 9.15; N, 7.61. HRMS (m/z): [M + H] calcd for C27H35BrN2, 387.2800; found, 387.2797.
6-Propionyl-2-(dihexylamino)anthracene (Anthradan, 13)
A mixture of 2-cyano-6-dihexylaminoanthracene (157 mg, 0.406 mmol) was dissolved in anhydrous THF (10 mL). Ethyl magnesium chloride (0.18 mL, 25% weight in THF, 0.49 mmol) and a trace of cuprous chloride were added, and the mixture was stirred and heated to reflux under a nitrogen atmosphere for 24 h. The reaction flask was covered by aluminum foil to avoid possible photoreactions. The reaction mixture was then cooled, and hydrochloric acid (5 mL, 1:1) was added. The mixture was refluxed for another 12 h. Then the reaction was cooled, and the solution was neutralized with NaHCO3. Water (100 mL) was added, and the resulting mixture was extracted with dichloromethane (60 mL × 3). The combined organic layers were washed with water (50 mL × 2), dried (MgSO4), concentrated, and loaded on silica gel. Purification by silica gel flash chromatography with hexane/dichloromethane (up to 1:2) as eluent afforded the product as yellow solid (130 mg, 77% yield). Mp 66.2–66.9 °C (hexane). 1H NMR (CDCl3) δ 8.57 (s, 1H), 8.36 (s, 1H), 8.09 (s, 1H), 7.94 (dd, J = 9.0, 1.6 Hz, 1H), 7.91–7.86 (m, 2H), 7.27 (dd, J = 9.4, 2.5 Hz, 1H), 6.89 (d, J = 2.5 Hz, 1H), 3.44 (t, J = 7.7 Hz, 4H), 3.16 (q, J = 7.3 Hz, 2H), 1.75–1.65 (m, 4H), 1.46–1.36 (m, 12H), 1.33 (t, J = 7.3 Hz, 3H), 0.95 (t, J = 6.9 Hz, 3H). 13C NMR (CDCl3) δ 200.6, 146.4, 135.8, 133.7, 131.7, 131.4, 129.6, 128.7, 127.7, 126.6, 122.7, 121.9, 118.0, 102.3, 51.2, 31.8, 31.5, 27.5, 26.9, 22.7, 14.1, 8.7. UV—vis (THF) λ1 = 257 nm, ∊1 = 4.46 × 104 L cm-1 mol-1, λ2 = 323 nm, ∊2 = 5.31 × 104 L cm-1 mol-1, λ3 = 449 nm, ∊3 = 1.21 × 104 L cm-1 mol-1. IR (cm-1) 2929, 1682, 1622. Anal. Calcd for C29H39NO: C, 83.40; H, 9.41; N, 3.35. Found: C, 83.11; H, 9.67; N, 3.59. HRMS (m/z): [M + H] calcd for C29H40NO, 418.3110; found, 418.3110.
Acknowledgment
This work was supported in part by the Department of Energy, Grant No. DE FG02-04ER63777, and by the National Institutes of Health, Grant No. 5P20-HG00363802.
Footnotes
Supporting Information Available: Hard copies of H1 and C13 NMR spectra for compounds 8 and 11–13 are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Valeur B. Molecular Fluorescence, Principles and Applications. Wiley-VCH; New York: 2002. For review: [Google Scholar]; (b) Lakowicz JR. Principles of fluorescence spectroscopy. 2nd ed Kluwer Academic/Plenum; New York: 1999. [Google Scholar]; (c) Willets KA, Nishimura SY, Schuck PJ, Twieg RJ, Moerner WE. Acc. Chem. Res. 2005;38:549–556. doi: 10.1021/ar0401294. [DOI] [PubMed] [Google Scholar]; (d) de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem. ReV. 1997;97:1515–1566. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- (2).Weber G, Farris FJ. Biochemistry. 1979;18:3075–3078. doi: 10.1021/bi00581a025. [DOI] [PubMed] [Google Scholar]
- (3) (a).Samanta A, Fessenden RW. J. Phys. Chem. A. 2000;104:8972–8975. [Google Scholar]; (b) Davis BN, Abelt CJ. J. Phys. Chem. A. 2005;109:1295–1298. doi: 10.1021/jp046050y. [DOI] [PubMed] [Google Scholar]; (c) Vazquez ME, Blanco JB, Imperiali B. J. Am. Chem. Soc. 2005;127:1300–1306. doi: 10.1021/ja0449168. [DOI] [PubMed] [Google Scholar]; (d) Lobo BC, Abelt CJ. J. Phys. Chem. A. 2003;107:10938–10943. [Google Scholar]; (e) Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY. Science. 2002;296:1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]; (f) Xiao Y, Liu FY, Qian XH, Cui JN. Chem. Commun. 2005:239–241. doi: 10.1039/b413537g. [DOI] [PubMed] [Google Scholar]; (g) Petric A, Jacobson AF, Barrio JR. Bioorg. Med. Chem. Lett. 1998;8:1455–1460. doi: 10.1016/s0960-894x(98)00241-8. [DOI] [PubMed] [Google Scholar]; (h) Agdeppa ED, Kepe V, Petric A, Satyamurthy N, Liu J, Huang SC, Small GW, Cole GM, Barrio JR. Neuroscience. 2003;117:723–730. doi: 10.1016/s0306-4522(02)00907-7. [DOI] [PubMed] [Google Scholar]; (i) Pierce DW, Boxer SG. J. Phys. Chem. 1992;96:5560–5566. [Google Scholar]
- (4) (a).Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baird GS, Zacharias DA, Tsien RY. Proc. Natl. Acad. Sci. U.S.A. 2000;97:11984–11989. doi: 10.1073/pnas.97.22.11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5) (a).Willets KA, Ostroverkhova O, He M, Twieg RJ, Moerner WE. J. Am. Chem. Soc. 2003;125:1174–1175. doi: 10.1021/ja029100q. [DOI] [PubMed] [Google Scholar]; (b) Nishimura SY, Lord SJ, Klein LO, Willets KA, He M, Lu Z, Twieg RJ, Moerner WE. J. Phys. Chem. B. 2006;110:8151–8157. doi: 10.1021/jp0574145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Lee SK, Yang WJ, Choi JJ, Kim CH, Jeon SJ, Cho BR. Org. Lett. 2005;7:323–326. doi: 10.1021/ol047658s. [DOI] [PubMed] [Google Scholar]; (b) Quesada E, Ardhammar M, Norden B, Miesch M, Duportail G, Bonzi-Coulibaly Y, Nakatani Y, Ourisson G. Helv. Chim. Acta. 2000;83:2464–2476. [Google Scholar]; (c) Mathauer K, Vahlenkamp T, Frank CW, Wegner G. Langmuir. 1993;9:1582–1586. [Google Scholar]
- (7).Perochon E, Lopez A, Tocanne JF. Chem. Phys. Lipids. 1991;59:17–28. doi: 10.1016/0009-3084(91)90059-k. [DOI] [PubMed] [Google Scholar]
- (8).Coleman RS, Mortensen MA. Tetrahedron Lett. 2003;44:1215–1219. [Google Scholar]
- (9).Ishikawa J, Sakamoto H, Nakao S, Wada H. J. Org. Chem. 1999;64:1913–1921. doi: 10.1021/jo9819584. [DOI] [PubMed] [Google Scholar]
- (10) (a).Ihmels H, Meiswinkel A, Mohrschladt CJ, Otto D, Waidelich M, Towler M, White R, Albrecht M, Schnurpfeil A. J. Org. Chem. 2005;70:3929–3938. doi: 10.1021/jo047841z. [DOI] [PubMed] [Google Scholar]; (b) Ihmels H. Eur. J. Org. Chem. 1999:1595–1600. [Google Scholar]; (c) Ihmels H, Meiswinkel A, Mohrschladt CJ. Org. Lett. 2000;2:2865–2867. doi: 10.1021/ol006291y. [DOI] [PubMed] [Google Scholar]; (d) Bohne C, Ihmels H, Waidelich M, Yihwa C. J. Am. Chem. Soc. 2005;127:17158–17159. doi: 10.1021/ja052262c. [DOI] [PubMed] [Google Scholar]
- (11).Gundermann KD, Klockenbring G, Roker C, Brinkmeyer H. Liebigs Ann. Chem. 1976:1873–1888. [Google Scholar]
- (12).Sun S, Desper J. Tetrahedron. 1998;54:411–422. [Google Scholar]
- (13).Criswell TR, Klanderman BH. J. Org. Chem. 1974;39:770–774. [Google Scholar]
- (14).Yanagimoto T, Takimiya K, Otsubo T, Ogura F. J. Chem. Soc., Chem. Commun. 1993:519–520. [Google Scholar]
- (15).Hodge P, Power GA, Rabjohns MA. Chem. Commun. 1997:73–74. [Google Scholar]
- (16).Quick AJ. J. Am. Chem. Soc. 1920;42:1033–1042. [Google Scholar]
- (17).Mallory FB, Mallory CW, Butler KE, Lewis MB, Xia AQ, Luzik ED, Fredenburgh LE, Ramanjulu MM, Van QN, Francl MM, Freed DA, Wray CC, Hann C, Nerz-Stormes M, Carroll PJ, Chirlian LE. J. Am. Chem. Soc. 2000;122:4108–4016. [Google Scholar]
- (18).Friedman L, Shechter H. J. Org. Chem. 1961;26:2522–2524. [Google Scholar]
- (19).Tamaki T. Bull. Chem. Soc. Jpn. 1978;51:2817–2821. [Google Scholar]
- (20).Rotkiewicz K, Grellmann KH, Grabowski ZR. Chem. Phys. Lett. 1973;19:315–318. [Google Scholar]; Rettig W. In: Top. Curr. Chem. Mattay J, editor. Vol. 169. Springer-Verlag; New York: 1994. pp. 253–299. For a review, see. [Google Scholar]
- (21).Moerner WE, Fromm DP. ReV. Sci. Instrum. 2003;74:3597–3619. [Google Scholar]
- (22).Soper SA, Nutter HL, Keller RA, Davis LM, Shera EB. Photochem. Photobiol. 1993;57:972–977. [Google Scholar]
- (23).Bunker CE, Bowen TL, Sun YP. Photochem. Photobiol. 1993;58:499–505. [Google Scholar]
- (24).Davis BN, Abelt CJ. J. Phys. Chem. A. 2005;109:1295–1298. doi: 10.1021/jp046050y. [DOI] [PubMed] [Google Scholar]