

Abstract
The release of verapamil hydrochloride from tablets with Eudragit RLPO or Kollidon®SR with different drug-to-polymer ratios were investigated with a view to develop twice-daily sustained-release dosage form by solid dispersion (SD) technique. The SDs containing Eudragit RLPO or Kollidon®SR at drug-polymer ratios of 1:1, 1:2, and 1:3 with verapamil hydrochloride were developed using solvent evaporation technique. The physical mixtures of drug and both polymers were prepared by using simple mixing technique at the same ratio as solid dispersion. The physicochemical properties of solid dispersion were evaluated by using Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), and differential scanning calorimetry (DSC). The study of DSC, XRD, and FTIR could not show significant interaction between verapamil HCl and Kollidon®SR or Eudragit RLPO. The solid dispersions or physical mixtures were compressed to tablets. The tablets were prepared with solid dispersions containing Eudragit RLPO or Kollidon®SR, with all the official requirements of tablet dosage forms fulfilled. Tablets prepared were evaluated for the release of verapamil hydrochloride over a period of 12 h in pH 6.8 phosphate buffer using US Pharmacopoeia type II dissolution apparatus. The in vitro drug release study revealed that the tablet containing Eudragit has extended the release rate for 12 h whereas the tablet containing Kollidon®SR at the same concentration has extended the release rate up to 8 h. The in vitro release profile and the mathematical models indicate that release of verapamil hydrochloride can be effectively controlled from a tablet containing solid dispersions of Eudragit RLPO. The reduction of size fraction of the SD system from 200–250 to 75–125 μm had a great effect on the drug release.
Key words: Eudragit RLPO, Kollidon®SR, matrix, solid dispersion, sustained release
INTRODUCTION
Many problems are associated with conventional multiple-dosing regimen of long-acting therapy, such as systemic accumulation of the drug leading to side effects or toxicities, flip-flop profile of the plasma drug level, and poor patient compliance. Controlled-release drug delivery systems have the potential of solving these problems. The disease symptoms such as hypertension, ischemic heart disease, asthma, and rheumatoid arthritis exhibit circadian rhythms. For example, blood pressure tends to be lower while asleep and elevated in the early morning. Similarly, the maximum morning occurrence of death from heart attack is around 9 a.m., which coincides with peaks in platelet aggregation, plasma catecholamine levels, and blood pressure. Development of controlled drug delivery system is a better alternative for use of multiple-dose regimen and for treatment of the above-stated diseases (1,2).
Several approaches have been used in an attempt to sustain the drug release from dosage forms. The solid dispersion techniques can be used to enhance the dissolution rate of poorly water-soluble drugs as well as to sustain the drug release by choosing appropriate polymers (3–7,8,9). It has been shown that preparation of matrices with insoluble polymers using solid dispersion (SD) technique is a valuable method in the production of sustained-release products. Many studies have been conducted in order to investigate the effect of formulation and process variables on drug release from tablets of physical mixtures of drug and polymer. It has been shown that polymer particle size, compaction force, and the presence of soluble or insoluble additives are among the factors affecting drug release from tablets of physical mixtures (10,11). However, there are a few studies investigating the effect of these factors on drug release from tablets of SD systems. Khan et al. reported that the presence of diluents and compression pressure considerably affected the drug release from coprecipitates of ibuprofen and Eudragit RSl00 (12). Among the polymers, polymethacrylate resins, like Eudragit RS100 and RL100, have been used as film coatings or inert carriers to formulate oral controlled-release delivery systems of nonsteroidal anti-inflammatory drugs (13,14). Eudragit RLPO is a polymer synthesized from acrylic and methacrylic acid esters, containing low-level quaternary ammonium groups and insoluble at physiological pH values and capable of swelling. Kollidon®SR is a directly compressible polymeric blend composed primarily of polyvinyl acetate (PVAc) and povidone (PVP) (15). The amorphous nature of PVAc coupled with its unusually low glass temperature of 28–31°C imparts certain unique characteristics to this binary matrix (16). Upon compression, this material forms a matrix block releasing the host drug through channels created by the gradual leaching of water-soluble PVP component.
Propranolol hydrochloride, diclofenac sodium, ciprofloxacin hydrochloride, and theophylline matrix tablets were prepared previously using various polymers (17,18). Verapamil hydrochloride sustained-release tablets by matrix-embedded techniques using Kollidon®SR or hydroxy propyl methyl cellulose as retardant were prepared earlier (19,20). Verapamil hydrochloride is a calcium channel blocker used as a peripheral vasodilator. Verapamil hydrochloride has short biological half-life (4 h) and thus frequent administration makes it a potential candidate for the design of sustained-release dosage forms.
The purpose of the present investigation was to design and compare the release characteristics of sustained-release formulations of verapamil hydrochloride by solid dispersion techniques using Eudragit RLPO or Kollidon®SR. The physicochemical characteristics of verapamil hydrochloride in physical mixtures (PMs) and SDs were studied by Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction analysis (XRD), and differential scanning calorimetry (DSC).
MATERIALS AND METHODS
Materials
A gift sample of verapamil hydrochloride was received from Torrent Pharmaceuticals Ltd., (Mumbai, India). Kollidon®SR was received from BASF (Germany). Eudragit RLPO was received from Colorcon Asia Ltd. (Goa). Double-distilled water was used throughout the study and all the other chemicals used were of analytical grade.
Methods
Preparation of Solid Dispersions
The SDs of verapamil HCl with Eudragit RLPO or Kollidon®SR containing three different weight ratios (1:1, 1:2, 1:3; verapamil: Eudragit RLPO or Kollidon®SR) and denoted as formulations F1 to F6, respectively, were prepared by solvent evaporation method. The formulation details are given in Table I. In solvent evaporation method, to a solution of verapamil hydrochloride (1 g in 10 ml acetone) in acetone, an appropriate amount of polymer in solution (1 g in 5 ml acetone) was added and mixed thoroughly by using a mechanical stirrer (Remi Pvt Ltd., Mumbai) at 500 rpm for 1 h at room temperature. From the resulting solutions, the solvent was removed under vacuum in a rotary evaporator at a maximum temperature of 50°C. The solid residue was dried in a desiccator for 24 h at room temperature, pulverized, and passed through sieve number 40. Two size fractions of the SD system with Eudragit RLPO were obtained by passing half of the residue through sieve number 40 and half through sieve number 22.
Table I.
Composition of Mixtures for Preparation of Solid Dispersion
| Formulation Code | Drug: polymer | Verapamil HCl solution | Kollidon SR solution | Eudragit RLPO solution | |||
|---|---|---|---|---|---|---|---|
| Verapamil (g) | Acetone (ml) | Kollidon SR (g) | Acetone (ml) | Eudragit RLPO (g) | Acetone (ml) | ||
| F1 | 1:1 | 2 | 20 | 2 | 10 | ||
| F2 | 1:1 | 2 | 20 | 2 | 10 | ||
| F3 | 1:2 | 2 | 20 | 4 | 20 | ||
| F4 | 1:2 | 2 | 20 | 4 | 20 | ||
| F5 | 1:3 | 2 | 20 | 6 | 30 | ||
| F6 | 1:3 | 2 | 20 | 6 | 30 | ||
The PMs of verapamil with Eudragit RLPO or Kollidon®SR containing three different weight ratios (1:1, 1:2, 1:3; verapamil: Eudragit RLPO or Kollidon®SR) and denoted as formulations f1 to f6, respectively, were prepared by mixing in a mortar and pestle for 10 min.
Characterization of Solid Dispersion
Fourier Transform Infrared Spectroscopy
FTIR spectra were obtained by using an FTIR spectrometer-430 (Jasco, Japan). The samples were previously ground and mixed thoroughly with potassium bromide, an infrared-transparent matrix, at 1:5 (sample to KBr) ratio, respectively. The KBr discs were prepared by compressing the powders at a pressure of 5 tons for 5 min in a hydraulic press. Forty scans were obtained at a resolution of 4 cm−1, from 4,000 to 300 cm−1.
X-ray Diffraction Study
In order to determine the drug carrier interaction and physical state of the drug whether amorphous or crystalline before and after formulations, XRD study was conducted for the pure drug, the polymers, and different formulations. XRD patterns were obtained by using an X-ray diffractometer-PW 1710 (Philips, Holland). The X-ray powder diffraction patterns were obtained at room temperature using Cu as anode material and graphite monochromatic, operated at a voltage of 35 kV, current 20 mA. The samples were analyzed in the 2θ angle range of 10–99°and the process parameters were set as: scan step size of 0.02° (2θ) and scan step time of 0.5 s.
Differential Scanning Calorimetry
The DSC measurements were performed on a DSC-6100 (Seiko Instruments, Japan) differential scanning calorimeter with a thermal analyzer. All accurately weighed samples (about 1.675 mg of verapamil hydrochloride or its equivalent) were placed in sealed aluminum pans, before heating under nitrogen flow (20 ml/min) at a scanning rate of 10°C min−1 from 25°C to 200°C. An empty aluminum pan was used as reference.
Preparation of Tablets
Two hundred tablets were prepared for each formulation. The solid dispersions of each formulation were mixed thoroughly with 1% Aerosil and 1% of talc in a mixing polybag for 10 min. The tablets of desired weight were prepared from the solid dispersions by using hydraulic pellet press (type KP) without using diluents. The equipment was adjusted to produce tablets with consistent weight using a constant compaction force of 30 kg/cm2. Six different formulae, having different ratio of Eudragit RLPO or Kollidon®SR, were developed to evaluate the drug release and to study the effect of polymer concentration on drug release. Three batches of tablets were prepared for each formulation. The tablet characteristics are given in Table II.
Table II.
Physical Properties of Compressed Tablets
| Formulation series | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Drug content (milligram per tablet) | 81.2 ± 1.3 | 81.3 ± 1.5 | 82.8 ± 1.2 | 81.2 ± 0.9 | 82.1 ± 0.8 | 81.3 ± 0.8 |
| Weight variation | ±5.8 | ±4.9 | ±4.2 | ±5.4 | ±5.2 | ±4.3 |
| Hardness (kg/cm2) | 8.2 ± 0.3 | 8.1 ± 0.8 | 8.4 ± 0.9 | 8.3 ± 0.6 | 8.4 ± 0.4 | 8.0 ± 0.5 |
| Friability (%) | 0.72 | 0.53 | 0.52 | 0.79 | 0.91 | 0.65 |
Evaluation of Tablets
Physical Properties of Tablets
The prepared tablets were evaluated for weight variation (n = 20), hardness, friability (n = 6), and drug content as per IP 1996. Hardness of tablet was determined by using a Monsanto tablet hardness tester (Campbell Electronics, Mumbai, India). Friability test (n = 20) was conducted using Roche friabilator (F. Hoffmann-La Roche Ltd., Basel, Switzerland). The drug content of the manufactured tablets was estimated using UV spectrophotometric method. From each batch, 20 tablets were taken, weighed, and finely ground. An adequate amount of this powder equivalent to 10 mg of the drug was accurately weighed, dissolved, and suitably diluted in double-distilled water and analyzed by UV spectrophotometric method at 278 nm (Shimadzu, model no-1700).
Surface Morphology of Tablet
The surface morphology of verapamil hydrochloride tablet was visualized using scanning electron microscope (SEM 515, PHILIPS, Holland). The tablet was sputtered with gold (E5200 Autosputter coater, BioRad, UK) before microscopy.
Dissolution Methodology
Dissolution studies of formulations were performed by using the US Pharmacopoeia model digital tablet dissolution test apparatus-2 (Lab India Ltd., Mumbai, India) at a paddle rotation speed of 50 rpm in 900-ml phosphate buffer (pH 6.8) at 37 ± 0.5°C. Six tablets were used in each run. At the specified times (every 1 h for 12 h), 5-ml samples were withdrawn using a syringe filter (0.45 μm; Sepyrane, Mumbai, India) and then assayed for verapamil hydrochloride content by measuring the absorbance at 278 nm on a UV-Visible spectrophotometer (Shimadzu UV-1700, Pharm Spec). Fresh medium (5 ml), which was prewarmed at 37°C, was added to the dissolution medium after each sampling to maintain a constant volume throughout the test.
In Vitro Dissolution Data
To compare dissolution profiles, several approaches can be used such as analysis of variance, model-independent and model-dependent approaches (21). In this dissolution study, model-dependent approaches are used for comparison of dissolution profiles.
In model-dependent approaches, release data were fitted to five kinetic models including the zero-order (Eq. 1), first-order (Eq. 2), Higuchi matrix (Eq. 3), Peppas–Korsmeyer (Eq. 4), and Hixson–Crowell (Eq. 5) release equations to find the equation with the best fit using PCP Disso v3 software (Pune, India) (22).
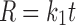 |
1 |
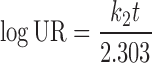 |
2 |
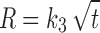 |
3 |
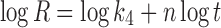 |
4 |
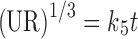 |
5 |
Where R and UR are the released and unreleased percentages, respectively, at time (t); k1, k2, k3, k4, and k5 are the rate constants of zero-order, first-order, Higuchi matrix, Peppas–Korsmeyer, and Hixon–Crowell model, respectively.
RESULTS AND DISCUSSION
Characterization of Solid Dispersion
Fourier Transform Infrared Spectroscopy
The IR spectra of solid dispersions of verapamil hydrochloride with Eudragit and verapamil hydrochloride with Kollidon®SR were compared with the standard spectrum of verapamil hydrochloride (Fig. 1 A). IR spectrum of verapamil hydrochloride is characterized by the absorption of NH group at 3,467 cm−1(Fig. 1 A). In spectra of solid dispersions of verapamil hydrochloride with Eudragit and verapamil hydrochloride with Kollidon®SR, this band was shifted towards lower frequencies at 3,282 and 3,280 cm−1, respectively (23). The shift in this band could be due to some sort of interactions between the drug and polymer. These interactions might be due to the intermolecular hydrogen bonding or complexation (23).
Fig. 1.

FTIR spectrograms of pure verapamil hydrochloride (a), Pure Kollidon®SR (b), Pure Eudragit RLPO (c), Verapamil hydrochloride–Kollidon®SR SD (d), Verapamil hydrochloride–Eudragit RLPO SD (e)
X-ray Diffraction Study
The diffraction spectrum of pure verapamil HCl showed that the drug was crystalline as demonstrated by numerous peaks observed at 2θ of 10.59°, 14.45°, 17.07°, 18.1°, 18.84°, 20.29°, 21.32°, 23.06°, 23.75°, and 26.29° (fingerprint region), etc. (Fig. 2 A) (23). Pure Kollidon®SR showed two peaks with intensity at 2θ and d-spacings of 18.11 and 4.893 and 31.74 and 2.816 Å. Some changes in peak position of verapamil HCl were observed in SDs. The prominent peaks from pure verapamil HCl at 2θ of 10.59°, 14.45°, 17.07°, 18.1°, 18.84°, 20.29°, 21.32°, 23.06°, 23.75°, and 26.29°, etc. were clearly seen at the same position in the SDs but the peak intensities were decreased to some extent. From the stated observations, we can conclude that the crystalline nature of the drug was still maintained, but the relative reduction of diffraction intensity of verapamil HCl in Kollidon®SR SDs suggests reduction in quality of the crystals and/or change in crystal size and/or presence of high-concentration polymer. The positions of Kollidon®SR peak patterns in the SDs were the same and super imposable, which again ruled out the possibility of chemical interaction and new compound formation between the two components. Results of this study indicate that verapamil HCl is still present in crystalline form with reduced crystal size in the SDs.
Fig. 2.

X-ray diffractograms of pure verapamil hydrochloride (a), Pure Kollidon®SR (b), Pure Eudragit RLPO (c), Verapamil hydrochloride–Kollidon®SR SD (d), Verapamil hydrochloride–Eudragit RLPO SD (e)
Pure Eudragit showed one peak with intensity of four counts at 2θ and d-spacings of 82.12 and 1.172 Å. No significant changes in peak positions of verapamil HCl were observed in SDs with Eudragit. The prominent peaks from pure verapamil HCl at 2θ of 10.59°, 14.45°, 17.07°, 18.1°, 18.84°, 20.29°, 21.32°, 23.06°, 23.75°, and 26.29°, etc. were clearly seen at the same position in the SDs but the peak intensities were decreased to a lesser extent. From the stated observations, we can conclude that the crystalline nature of the drug was still maintained, but the small reduction of diffraction intensity of verapamil HCl in Eudragit SDs suggests that the quality of the crystals was reduced and/or presence of high-concentration polymer.
Differential Scanning Calorimetry
The DSC curve of pure verapamil HCl exhibited a single endothermic response corresponding to the melting of drug. Onset of melting was observed at 148.1°C (Fig. 3 A). Eudragit shows an endothermic peak at 45.3°C where as DSC thermogram of Kollidon®SR shows a broad peak probably due to presence of water. In SD systems, the drug endothermic peak was observed at 148.3°C with Kollidon®SR and at 149.2°C with Eudragit RLPO, respectively. These observations of DSC study indicate absence of significant interactions between drug and polymer in solid dispersions.
Fig. 3.

DSC thermograms of pure verapamil hydrochloride (a), Pure Kollidon®SR (b), Pure Eudragit RLPO (c), Verapamil hydrochloride–Kollidon®SR SD (d), Verapamil hydrochloride–Eudragit RLPO SD (e)
Evaluation of Tablets
Physical Characterization of Designed Tablets
Physical appearance, tablet hardness, friability, weight variation, and drug content uniformity of different formulations were found to be satisfactory as can be observed from Table II. Tablet hardness varied between 8.0 and 8.5 kg/cm2. The friability was less than 0.8% (w/w). The matrix tablet showed low weight variation and a high degree of content uniformity indicating that the solid dispersion method is suitable for manufacture of good-quality verapamil hydrochloride matrix tablets.
Scanning electron microphotographs of tablets have shown that the surface of the tablet is smooth along with small pores ranging from 18.85 to 30.13 μm (Fig. 4).
Fig. 4.

Scanning electron microscopy of tablet surface
Dissolution Rate Study
In vitro dissolution studies were conducted on three tablets of each of the formulations (formulations F1 to F6). The mean cumulative percent of verapamil hydrochloride released at different time intervals is shown in Fig. 5. It was observed that the initial rate of release for F1 and F2 was high and above 90% of the drug was released within 4 h. In formulation F3, around 98% of the drug was released within 10 h; however, in case of formulation F4, the release was faster as 95% of drug was released within 5 h. The formulations F1 to F 4 were unable to sustain the drug release for 12 h, whereas formulation F5 sustained the release for 12 h. Formulation F6 released around 95% of the drug within 8 h. So the formulation F5 contains the optimum quantity of polymer to sustain the drug release for 12 h. Formulation f 5 showed 97.35% drug release within 5 h, whereas the F5 showed only 66.35% drug release during this time interval, followed by continuous release until 100% release was achieved within 12 h. This indicates that the SD system is more efficient than the physical mixture in retarding drug release from tablets. F5 sustained the drug release for about 12 h and F6 controlled the release for 8 h. The release rate of verapamil hydrochloride decreased by increase in concentration of Eudragit in SD systems, such as F5 sustained the drug release for 12 h but F1 sustained the release for 3–4 h. Stated evidence confirms that Eudragit SD system provides better control of release of drug compared to Kollidon®SR SD system. But Kollidon®SR and verapamil hydrochloride physical mixture provides better control of drug release compared to the physical mixture of verapamil with Eudragit (Fig. 4). Similar results were reported by other researchers for other drugs and polymer systems. Tablets of the SD system retained their shape until the end of the dissolution test, and only a small amount of erosion of the edges of the tablets was observed.
Fig. 5.

In vitro dissolution profile of formulations from F1to F6 and f1 to f6
Table III shows the regression parameters obtained after fitting various release kinetic models to the in vitro dissolution data. The goodness of fit for various models was investigated for F1, F2, F3, F4, F5, and F6, but in case of F5 it was ranked in order of Hixson–Crowell cube root law > Korsmeyer–Peppas > zero order > first order > Higuchi. In vitro release data of F5 best fitted to Hixson–Crowell cube root law model and n value for Korsmeyer–Peppas is 0.9985, hence exhibits non-Fickian diffusion.
Table III.
Statistical Parameters of Various Formulations After Fitting Drug Release Data to Various Release Kinetics Models
| Zero-order model | First-order model | H-M model | P-K model | H-C model | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Formulations | R | K 1 | R | K 2 | R | K 3 | R | K 4 | R | K 5 |
| F1 | 0.9212 | 22.589 | 0.9827 | −0.6036 | 0.9973 | 43.9834 | 0.9943 | 42.5253 | 0.9967 | −0.1354 |
| F2 | 0.8505 | 29.088 | 0.9987 | −0.8051 | 0.9895 | 51.9070 | 0.9928 | 59.3105 | 0.9797 | −0.1785 |
| F3 | 0.9899 | 12.867 | 0.9631 | −0.1893 | 0.9249 | 24.1354 | 0.9580 | 14.9449 | 0.9758 | −0.0550 |
| F4 | 0.8878 | 18.835 | 0.9746 | −0.4772 | 0.9993 | 40.0509 | 0.9989 | 40.8484 | 0.9920 | −0.1095 |
| F5 | 0.9817 | 10.283 | 0.9693 | −0.2204 | 0.9419 | 26.9247 | 0.9850 | 10.4969 | 0.9895 | −0.0548 |
| F6 | 0.9411 | 12.752 | 0.9707 | −0.3345 | 0.9824 | 32.3677 | 0.9889 | 22.4618 | 0.9979 | −0.0755 |
H-M Higuchi matrix, P-K Peppas–Korsmeyer, H-C, Hixon–Crowell, R correlation coefficient, K 1–K 5 constants of release kinetics, PM physical mixture, F1–F6 solid dispersion of prepared by solvent evaporation method
Effect of Size Fraction on Drug Release
The release profiles for tablets prepared from two size fractions of the SD system with Eudragit RLPO are shown in Fig. 6. The 75–125-μm size fraction exhibited low percent drug release and release rate when compared to size fraction 200–250 μm. Tablets prepared from the coarse size fraction disintegrated rapidly and released their drug content rapidly. However, tablets prepared from smaller-sized fractions remained nearly intact during dissolution testing. In a study on release of drug from different-sized fractions of the SD system with either Eudragit RS or RL, it was shown that the release rate of indomethacin was reduced by increasing the particle size of the SD system in the untableted system (3). It has also been reported that polymer particle size could affect drug release rate as well as the hardness of inert insoluble matrices prepared from physical mixtures. The finer the particle size of the polymer, the more retardation in the drug release rate (5,6,24).
Fig. 6.

Dissolution profile of different-sized fractions of solid dispersion
CONCLUSIONS
The SD system provides better control of drug release rate than the physical mixture at the same ratio of drug and polymer. The SD system of verapamil with Eudragit RLPO provides better control of drug release (up to 12 h) than the one with Kollidon®SR. However, physical mixture of drug with Kollidon®SR provides better control of drug release than the physical mixture with Eudragit RLPO. Hence, the SD system is more efficient than physical mixture for preparation of verapamil HCl sustained-release tablets. The reduction of size fraction of the SD system from 200–250 to 75–125 μm had a significant effect on the drug release. The study of DSC, XRD, and FTIR could not show any significant interaction between verapamil HCl and Kollidon®SR or Eudragit RLPO.
References
- 1.Elliott W. J. Circadian variation in the timing of stroke onset. A metaanalysis. Stroke. 1998;29:992–996. doi: 10.1161/01.str.29.5.992. [DOI] [PubMed] [Google Scholar]
- 2.Gallerani M., Manfredini R., Ricci L., Grandi E., Cappato R., Calò G., L Pareschi P., Fersini C. Sudden death from pulmonary thromboembolism: chronobiological aspects. Eur. Heart J. 1992;6:305–323. doi: 10.1093/oxfordjournals.eurheartj.a060232. [DOI] [PubMed] [Google Scholar]
- 3.Lovrecich M., Nobile F., Rubessa F., Zingone G. Effect of ageing on the release of indomethacin from solid dispersion with eudragits. Int. J. Pharm. 1996;131:247–255. doi: 10.1016/0378-5173(95)04354-3. [DOI] [Google Scholar]
- 4.Karnachi A. A., De Hon R. A., Khan M. A. Compression of indomethacin coprecipitates with polymer mixtures: effect of preparation methodology. Drug Dev. Ind. Pharm. 1995;21(12):1473–14483. doi: 10.3109/03639049509063034. [DOI] [Google Scholar]
- 5.Goldberg A. H., Gibaldi M., Kanig J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures. I. Theoretical considerations and discussion of literature. J. Pharm. Sci. 1965;54:1145–1148. doi: 10.1002/jps.2600540810. [DOI] [PubMed] [Google Scholar]
- 6.Chiou W. L., Riegelman S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971;60:1281–1302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 7.Sadeghi F., Afrasiabi Garekani H., Sadr A. Influence of polymer viscosity and plasticizer addition on ethylcellulose matrix characteristics prepared from solid dispersion system. STP Pharma. Sci. 2003;13:105–110. [Google Scholar]
- 8.Hernandez J. I., Ghalj E. S., Malave A., Marti A. Controlled release matrix of acetaminophen ethylcellulose solid dispersion. Drug Dev. Ind. Pharm. 1994;20:1253–1265. doi: 10.3109/03639049409038365. [DOI] [Google Scholar]
- 9.Goracinova K., Klisarova L. J., Simov A. Physical characterization and dissolution properties of verapamil HCl coprecipitates. Drug Dev. Ind. Pharm. 1995;21:383–391. doi: 10.3109/03639049509048119. [DOI] [PubMed] [Google Scholar]
- 10.Dabbagh M. A., Ford J. L., Rubinstein M. H., Hogan J. E. Effect of polymer particle size, compaction pressure and hydrophilic polymers on drug release from matrices containing ethylcellulose. Int. J. Pharm. 1996;140:85–95. doi: 10.1016/0378-5173(96)04599-1. [DOI] [Google Scholar]
- 11.Katikaneni P. R., Upadrashta S. M., Neau S. H., Mitra A. K. Ethylcellulose matrix controlled release tablets of a water-soluble drug. Int. J. Pharm. 1995;123:119–125. doi: 10.1016/0378-5173(95)00060-V. [DOI] [Google Scholar]
- 12.Khan M. A., Karnachi A. A., Singh S. K., Sastry S. V., Kislalioglu S.M., Bolton S. Controlled release coprecipitates: formulation considerations. J. Cont. Rel. 1995;37:131–141. doi: 10.1016/0168-3659(95)00073-H. [DOI] [Google Scholar]
- 13.Benita S., Hoffman A., Donbrow M. Microencapsulation of paracetamol using polyacrylate resins (eudragit retard), Kinetics of drug release and evaluation of kinetic model. J. Pharm. Pharmcol. 1985;37:391–395. doi: 10.1111/j.2042-7158.1985.tb03021.x. [DOI] [PubMed] [Google Scholar]
- 14.Ho C., Hwang G. C. C. Development of extended release solid dispersion of non-steroidal anti-inflammatory drugs with aqueous polymeric dispersions: optimization of drug release via a curve-fitting technique. Pharm. Res. 1992;9:206–210. doi: 10.1023/A:1018933306162. [DOI] [PubMed] [Google Scholar]
- 15.Technical Information, Kollidon®SR; BASF Aktiengesellschaft, Germany. 1–10 (1999).
- 16.W. Daniels. Vinyl ester polymers, In transitions and relaxations to Zwitterionic Polymerisation, 2nd Ed., Encyclopedia of polymer science and Engineering, John Wiley and sons, New York. 17:402–425 (1989).
- 17.Sahoo J., Murthy P. N., Biswal S., Mahapatra A. K., Sahoo S. K. Comparative study of propranolol hydrochloride release from matrix tablets with Kollidon®SR or hydroxy propyl methyl cellulose. AAPS Pharma. SciTech. 2008;9(2):577–582. doi: 10.1208/s12249-008-9092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saha R. N., Sanjeev C., Sahoo J. A comparative study of controlled release matrix tablets of diclofenac sodium, ciprofloxacin hydrochloride, and theophylline. Drug Delivery. 2001;8:149–154. doi: 10.1080/107175401316906919. [DOI] [PubMed] [Google Scholar]
- 19.J. Sahoo, P. N. Murthy, S. Biswal, A. K. Mahapatra, and S. K. Sahoo. Preparation and release rate study of controlled release matrix tablets of verapamil hydrochloride using hydroxy propyl methyl cellulose. Int. J. Pharma. Ex. In press.
- 20.Sahoo J., Murthy P. N., Biswal S., Mahapatra A. K., Sahoo S. K. Preparation and release rate study of controlled release matrix tablets of verapamil hydrochloride using Kollidon®SR. Pharm. BIT. 2007;XVI(2):119–124. [Google Scholar]
- 21.Biswal S., Sahoo J., Murthy P. N., Giradkar P. R., Avari J. G. Enhancement of dissolution rate of gliclazide using solid dispersions with polyethylene glycol 6000. AAPS Pharma. SciTech. 2008;9(2):563–570. doi: 10.1208/s12249-008-9079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa P., Lobo J. M. S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001;13:123–133. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 23.Rustichelli C., Gamberini M. C., Ferioli V., Gamberini G. Properties of the racemic species of verapamil hydrochloride and gallopamil hydrochloride. Int. J. Pharm. 1999;178:111–120. doi: 10.1016/S0378-5173(98)00355-X. [DOI] [PubMed] [Google Scholar]
- 24.Oth M. P., Moes A. J. Sustained release solid dispersions of indomethacin with Eudragit RS and RL. Int. J. Pharm. 1989;55:157–164. doi: 10.1016/0378-5173(89)90037-9. [DOI] [Google Scholar]