

Abstract
The purpose of this study was to investigate physicochemical characteristics and in vitro release of zidovudine from monolithic film of Eudragit RL 100 and ethyl cellulose. Films included 2.5% or 5% (w/w) zidovudine of the dry polymer weight were prepared in various ratios of polymers by solvent evaporation method from methanol/acetone solvent mixture. The release studies were carried out by vertical Franz cells (2.2 cm2 area, 20 ml receptor fluid). Ex vivo studies were done on Wistar rat skin within the films F6 (Eudragit RL100) and F7 (Eudragit RL100/Ethylcellulose, 1:1) consisting 5% (w/w) zidovudine in comparison with the same amount of free drug. Either iontophoresis (0.1 and 0.5 mA/cm2 direct currents, Ag/AgCl electrodes) or dimethyl sulfoxide (pretreatment of 1% and 5%, w/w, solutions) were used as enhancers. Films consisting of ethyl cellulose under the ratio of 50% (w/w) gave similar release profiles, and the highest in vitro cumulative released amount was achieved with F6 film which gave the closest results with the free drug. This result could be due to the high swelling capacity and re-crystallization inhibition effect of RL 100 polymer which also influenced the film homogenization. All the films were fitted to Higuchi release kinetics. It was also observed that both 0.5-mA/cm2 current and 5% (w/w) dimethyl sulfoxide applications significantly increased the cumulative permeated amount of zidovudine after 8 h; however, the flux enhancement ratio was higher for 0.5-mA/cm2 current application, especially within F6 film. Thus, it was concluded that Eudragit RL100 film (F6) could be further evaluated for the transdermal application of zidovudine.
Key words: dimethyl sulfoxide, ethyl cellulose, Eudragit RL100, iontophoresis, transdermal delivery, zidovudine
INTRODUCTION
Transdermal patches are innovative drug delivery systems and can be used for achieving efficient systemic effect bypassing hepatic first-pass metabolism and increasing the fraction absorbed. The monolithic transdermal systems are interesting due to their ease of fabrication and lack of dose dumping (1). Transdermal route shows certain benefits; however, the barrier function of stratum corneum necessitates enhancing the transdermal delivery of therapeutic agents (2). Various drugs can be delivered via skin by physical or chemical enhancement methods. The latter involves the use of chemical penetration enhancers which can decrease the integrity of the skin barrier, while the former can involve the use of ultrasonography or electrically assisted methods such as iontophoresis, electroosmosis, and electroporation (3,4). Iontophoresis is an alternative strategy to facilitate transport which can enhance and/or control the delivery of drug molecules through the skin from annexial pathway by applying a low-density (from 0.1 to 0.5 mA/cm2) electrical current (2,4). Iontophoretic transport promotes penetration of small-molecular-weight cationic drugs into skin by both electrorepulsion, electroosmotic flow, and by increased skin permeability (3–5).
Zidovudine (AZT), the first anti-HIV compound approved for clinical use is one of the most widely used anti-AIDS drug. However, dose-dependent hematological toxicity, low therapeutic index, short biological half-life, and relatively poor bioavailability limits the therapeutic effectiveness of AZT (6). After oral administration, it is rapidly absorbed from the gastrointestinal tract with a peak serum concentration occurring in about 1 h. However, oral bioavailability of AZT is not very high, with a range of 52% to 75%, due to the first-pass metabolism, and the mean half-life is approximately 1 h (7). In order to maintain therapeutic levels, large doses such as 200 mg/4 h should be given frequently in oral route. This dosage often causes toxic levels in blood, and severe adverse effects such as granulocytopenia or anemia occurs (7). The side effects of AZT are usually associated with excessive plasma level of AZT immediately after intravenous or oral administration (6). Hence, delivery from a non-oral pathway such as transdermal route may be helpful in maintaining suitable plasma concentration and could be useful in improving bioavailability and patient compliance and in avoiding side effects (7,8). The benefits of transdermal delivery of AZT are well appreciated in the literatures (7,9–12). AZT is a polar molecule; diffusion of AZT across highly lipophilic stratum corneum is poor and below the level to achieve effective therapeutic plasma concentration (9). Hence, using iontophoresis could be an effective enhancer in achieving therapeutic plasma levels for AZT.
The use of polymethacrylate kind of polymers (Eudragit) in matrix formulations for monolithic transdermal systems has been reported by several authors (13–17). Drug containing films of water-insoluble polymers were generally prepared by casting and drying organic drug–polymer solutions or suspensions, and the reasons for choosing Eudragit polymers were their high capacity for incorporating drugs and skin toleration (18,19). Eudragit RL 100 (ERL) and ethyl cellulose (EC) were often used in the preparation of transdermal delivery systems as well as other dosage forms for modifying the release of drugs. ERL is freely permeable to water, whereas EC forms films which are insoluble in water. Varying the ratio of Eudragit or EC polymers in the composition of the films can provide a control for drug release characteristics (14,17). Also, both Eudragit and EC polymers are nontoxic, non-absorbable, and they do not lose their film-forming properties when formulated with the drug and the excipients (14).
The purpose of this study was to prepare the monolithic film of AZT from ERL and EC polymers; to investigate the physicochemical characteristics, in vitro release profiles, transdermal permeation of AZT from these films; and to modify the transdermal permeation of AZT by using enhancing mechanisms like iontophoresis or dimethyl sulfoxide. Monolithic films were prepared with various combinations of ERL and EC polymers by solvent casting method from methanol/acetone solvent system and evaluated for their variability in thickness, drug amount, swelling properties, and in vitro release kinetics, and the films giving the appropriate properties were further investigated for the AZT penetration from Wistar rat skin in comparison with the AZT solution.
MATERIALS AND METHODS
AZT (Cipla, India), ERL (Röhm Pharma, Darmstadt, Germany), EC, 14 cP (BDH), dibutyl sebacate (DBS; Sigma), HEPES (Fluka), dimethyl sulfoxide (DMSO; Sigma), cellulose acetate membrane (0.22 μm, Sartorius), and dorsal skin from Wistar rats (250–275 g, male) were used in the studies. Animal studies were done according to our Research Ethics Committee of Medical Faculty, Ankara University (protocol number 103-2693).
Preparation of the Films
Polymer films were prepared by the solvent casting method with methanol/acetone (20:80, v/v) mixture in stainless steel rings which give an area of 15.89 cm2. The rings were attached to a glass plate covered with aluminum foil. The ingredients consisted of AZT (2.5%, w/w, or 5.0%, w/w, of dry polymer weight), polymers (ERL, EC), and plasticizer DBS (25%, w/w, of dry polymer weight) and dissolved in 10 mL of solvent mixture by stirring for 60 min at 500 rpm (WiseStir, Japan). Then, 3 mL of this solution dropped onto the aluminum foil in the steel ring with an injector. The solution was evaporated at room temperature overnight and kept in 50% relative humidity, 25°C conditions for 4 days to form films (20). The amounts of the ingredients were calculated according to the area of the films, and the compositions of the films were given in Table I.
Table I.
Composition and Characterization of the Films
| Film code | Ratio of ERL/EC (%, w/w) | AZT (%, w/w) | DBS (%, w/w) | Thickness (μm) | AZT amount (μg/cm2) |
|---|---|---|---|---|---|
| F1 | 100:0 | 2.5 | 25 | 293 ± 1.5 | 632.5 ± 6.7 |
| F2 | 80:20 | 2.5 | 25 | 307 ± 2.0 | 628.6 ± 20.1 |
| F3 | 60:40 | 2.5 | 25 | 313 ± 2.0 | 656.1 ± 13.7 |
| F4 | 50:50 | 2.5 | 25 | 317 ± 2.6 | 686.5 ± 9.9 |
| F5 | 0:100 | 2.5 | 25 | 327 ± 3.4 | 473.0 ± 43.9 |
| F6 | 100:0 | 5.0 | 25 | 297 ± 1.9 | 1475.8 ± 28.5 |
| F7 | 50:50 | 5.0 | 25 | 322 ± 1.2 | 1401.8 ± 21.9 |
All values are expressed as means ± SD (n = 3)
EC ethyl cellulose, ERL Eudragit RL 100, AZT zidovudine, DBS dibuthyl sebacate
Evaluation of the Film Properties
Thicknesses and Drug Amounts of the Films
Preparation method was validated by standardization of thickness and amount of drug in films. Each film was accepted as a single batch in the studies. Film thickness and drug amount studies were done on three batches for each formulation and repeated on three discs cut from each batch. Films were formulated to give 300-μm thicknesses theoretically according to the method given in Röhm Pharma Catalogue (21), and AZT contents of the films were calculated from the area of the films as 682 μg/cm2 (F1–F5) and 1533.9 μg/cm2 (F6–F7) theoretically.
Thickness of the films were determined using micrometer (NSK, Japan) from the 1-cm2 discs cut from different sides of the films with a circular metallic die. In order to show the uniformity of drug distribution in the films, the contents of these 1-cm2 discs were dissolved in ethanol with magnetically stirring at 500 rpm for 1 h. AZT contents were determined by spectrophotometer at 265 nm (Shimadzu 1404, Japan) against a blank solution of ethanol. Film thicknesses (μm ±SD) and AZT amounts (μg/cm2 ±SD) for 1-cm2 discs were given in Table I.
Analytical validation of the spectrophotometric method was done in the HEPES buffer and ethanol media by performing linearity and range, precision, accuracy, and specificity according to ICH Guidelines Q2R1(22). Correlation coefficient (r) values were 0.9995 and 0.9999 for HEPES buffer and ethanol, respectively, and the results showed linearity of the method. Accuracy was found higher than 93% for the concentration levels between 1.0 and 30 μg/mL, and the paired t test results for the data of two consecutive days showed intermediate precision (P > 0.05). LOQ values were 0.215 and 0.0712 μg/mL for HEPES buffer and ethanol, respectively. The results given established validation for the analytical method used in the study.
Swelling Experiments
Swelling properties of polymers (ERL 100%, w/w, EC 100%, w/w, and ERL/EC 1:1 combination) were evaluated. A piece of 1-cm2 film was dried in an oven at 50°C for 24 h. Then, dried film was accurately weighed and immersed in a flask containing dissolution media at 37°C. The swollen sample was withdrawn from the medium at the end of 1, 8, and 24 h. Sample was weighed after the removal of excess surface water with a filter paper. The percentage swelling, Is (%), was calculated as follows:
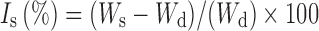 |
1 |
where Wd is the weight of dried polymer film, and Ws denotes the weight after swelling (23). Also, the change in thickness was evaluated in the same manner in order to explain swelling. The results were given in Table II.
Table II.
Swelling Properties of the Free Polymers
| Ratio of ERL/EC (%, w/w) | Change in thickness (%) | Percentage swelling, I s (%) |
|---|---|---|
| 100:0 | 37.7 ± 6.35 | 129 ± 14.0 |
| 50:50 | 26.6 ± 1.56 | 73.7 ± 21.8 |
| 0:100 | 7.03 ± 3.46 | 6.12 ± 5.47 |
All values are expressed as means ± SD (n = 3)
EC ethyl cellulose, ERL Eudragit RL 100, AZT zidovudine, DBS dibuthyl sebacate
In Vitro Release Studies
The in vitro drug release studies were carried out by jacketed vertical Franz diffusion cells with a surface area of 2.2 cm2 and a receptor compartment capacity of 20 mL. Discs containing 1 mg AZT (F1–F5) or 2 mg AZT (F6, F7) were cut from the formulated films. These discs were applied between the chambers on a 0.22-μm cellulose acetate membrane as a donor phase and wetted with 0.1 mL of 100 mM, pH 7.4, HEPES buffer in order to ensure the humidity (24). Also, the stock solutions of AZT in HEPES buffer (1 and 2 mg/mL) were applied to the donor compartment to show the effect of polymers on the release of AZT. The whole assembly was continuously stirred at 500 rpm and the temperature maintained at 37 ± 1°C. The amount of AZT released to the receptor compartment was determined by collecting 1-mL samples up to 8 h, and the receptor phase was replenished by adding 1 mL of buffer and then analyzed spectrophotometrically at 267 nm. The results were given in Figs. 1 and 2.
Fig. 1.

In vitro release profiles of AZT from the films (F1–F7)
Fig. 2.

Comparison of in vitro release profiles of AZT from the films with AZT in solution. a Comparison of 1 mg/mL AZT with F1–F5. b Comparison of 2 mg/mL AZT with F6–F7
Preparation of the Skin
Full thickness dorsal skins (~200 μm) were obtained from male Wistar rats weighing 250–275 g. The rats were anaesthetized by ketamine HCl (75 mg/kg, i.p.) before the experiments and were shaved using electric and hand razors. The dorsal skin was surgically removed from the animal, and adhering subcutaneous fat was carefully cleaned. The skin pieces were washed with serum physiologic covered with aluminum foil and stored in −20°C for 2 weeks (25,26).
Ex Vivo Release Studies
Skin penetration of drug was modified by either iontophoresis or DMSO in the ex vivo studies.
Ex vivo skin permeation studies using ~200-μm full thickness dorsal skin of Wistar rats were carried out using jacketed vertical Franz diffusion cells with a diffusional surface area of 2.2 cm2 and 20 mL of receptor cell volume. The receptor compartments were filled with 100 mM HEPES buffer, pH 7.4, containing 0.02% (w/w) of sodium azide in order to prevent microbial contamination (27), and receptor phase was stirred at 500 rpm with small magnetic beads to mix the contents uniformly. In order to attain ~32°C in the skin surface, the receptor phase was maintained at 37 ± 1°C. The skin pieces were mounted over the diffusion cells with the dermal side in contact with the receptor phase, equilibrated for 1 h, and then the air bubbles were removed. Subsequently, the film which contains 2 mg AZT was applied to the stratum corneum side in the donor compartment and wetted with 0.1 mL of 100 mM, pH 7.4, HEPES buffer in order to ensure the humidity (17,24). The amount of AZT penetrated from dorsal skin was determined by collecting 1-mL samples up to 8 h and the receptor phase replenished by adding 1 mL of buffer.
In order to show the effect of iontophoresis on the release rate of AZT, 0.1- and 0.5-mA/cm2 direct currents were applied both on the films and with AZT solution (2 mg/mL) in donor compartment continuously for 3 h comparing with the application without any enhancement method. Ag/AgCl electrodes attached to a constant current source were placed to vertical Franz-type cells in order to ensure anodal iontophoresis. HEPES buffer (100 mM, pH 7.4) was used as medium and extraneous ions were not used with the buffer. The effect of chemical enhancer (DMSO) was compared with the effect of iontophoresis. The alcoholic solution of the DMSO (1%, w/w, and 5%, w/w, 600 μL) was pretreated for 2 h onto the dorsal skin pieces after the membranes were fixed between the compartments. Then the treatment region was washed five times with the HEPES buffer and the films placed onto the skin (28). Ex vivo experiments were carried out with the same conditions given before. The skin penetration of AZT released from F6 and F7 films, with or without using enhancement methods, were compared with 2 mg/mL AZT solution and the results were given in Fig. 3.
Fig. 3.

Cumulative amount of AZT penetrated from rat skin at the end of 8 h
The ex vivo studies were analyzed with high-performance liquid chromatography (HPLC; Agilent 1100 Series) method under the conditions given as follows (29): G1379A Degasser, G1311A Quat pump, a G1313A ALS, G1316A COLCOM auto-injector, Luna C18 column, 5 μm, 150 × 4.6 mm with CTO-10A column oven, DAD detector (G1315B), acetonitrile/methanol/water (10:70:20, v/v/v) as mobile phase, temperature was set at 37°C, λ, 265 nm as detector wavelength, 0.45-mL/min flow rate, 20-μL injection volume, and the retention time was found as 3.8 min.
Analytical validation of the HPLC method was done in the mobile phase by performing linearity and range, precision, accuracy, and specificity according to ICH Guidelines Q2R1(22). Correlation coefficient (r) value of 0.9998 showed linearity of the method. Accuracy was found higher than 93% for the concentration levels between 0.4 and 40 μg/mL, and the paired t test results for the data of two consecutive showed intermediate precision (P > 0.05). LOQ value was 0.0103 μg/mL. The results given established validation for the analytical method used in the study.
Data Analysis
Multivariate one-way analysis of variance (ANOVA) with Tukey–Kramer multiple comparison post test (GraphPad, InStat 3.0) was applied after the in vitro and ex vivo studies in order to interpret the similarity or difference between the release profiles of AZT from the films. Paired t test with a two-tailed p value was also used to show that the differences were significant or not between the applications. Also, regression analysis of the ex vivo permeation curves was carried out. The slope of the straight line obtained after plotting the mean cumulative amount released per area of the film versus time (hour) was taken as the experimental flux for AZT for the ex vivo studies.
Interaction Studies
The interaction studies were conducted on the films containing AZT which were used in the ex vivo studies by comparing the pure drug on the basis of UV analysis and infrared (IR) spectrums (30).
UV Analysis
The alcoholic solutions of the pure drug and films containing AZT were filtered with cellulose acetate membrane (0.45 μm) and scanned for UV absorption between 200 and 400 nm using Shimadzu 1204 spectrophotometer.
IR Analysis
The IR absorption spectra of the pure drug and films containing AZT were taken in the range of 400–4000 cm−1 using potassium bromide disc method by Jasco 420, FTIR.
RESULTS AND DISCUSSION
Film Characterization
Each individual film formulation was evaluated separately for their thickness and drug amount characteristics and the results were given with Table I. The uniformity of polymeric films containing various ratios of ERL and EC was shown with the low SD values in thickness measurements. The highest SD value for thickness measurement was observed with F5 formulation containing 100% (w/w) EC. Also, increasing ratio of EC polymer affected the increase in the thickness of the films (F1–F5, consisting 2.5%, w/w, AZT). When drug amounts in the films were evaluated, the lowest drug amount value with the highest SD was observed with F5 formulation. These results indicated that the repeatability of film preparation was poor when EC was used alone (F5) and best results were achieved when using only ERL polymer as film former (F1). However, the combination of ERL/EC films did not significantly differ from each other for their thickness and drug amount characteristics (F2–F4). The results observed using EC polymer could be attributed to the swelling property of this polymer in organic solvents such as acetone and methanol, which was explained in the technical literature EC.
In the preparation method, polymer and AZT were both dissolved in methanol/acetone solvent mixture, and when the solvent was evaporated, homogenous dispersion was handled using ERL polymer in contrast to EC. AZT crystal structure was also evaluated with the visible microscopy, and the same crystal structure could not be observed in the films even if excess amount of AZT was used (5%, w/w). This give rise to thought that ERL polymer could have some inhibitor effect on the re-crystallization of AZT in the matrix system, as the quaternary amine groups consisting of ERL polymer could increase the solubility of drugs incorporated into the polymer blends (31).
According to these reasons, films prepared by 100% (w/w) ERL were thought to give the best results for their preparation characteristics with the used method. However, in order to see the effect of EC on the release of AZT, EC was not preferred to be used alone but incorporated with F1 and F6 films in a ratio of 50% (w/w).
In Vitro release studies
The cumulative amount of AZT released from the ERL (100%, w/w) films were higher (414 μg/cm2 for F1 and 1091 μg/cm2 for F6) than the other films studied (Fig. 1). The reason of the high release from ERL polymer could be explained by the hydrophilic nature of this polymer and the existence of the quaternary ammonium groups which could affect the release of AZT from the films because of the hydration and swelling of the films (23). ERL polymer was reported to give a water-permeable and swelling film rather than other types of Eudragits and also EC which is hydrophobic (14). ERL swells in the presence of buffer medium based on ion exchange and mutual repulsion of cationic groups which create some large pores and expand the polymeric chains which results in more hydration and also permeability to drug molecules. After swelling experiments (Table II), films containing ERL had been expanded in size, whereas formulation prepared with EC did not show so much expansion (23). Another reason for this high release could be explained by the choice of plasticizer, DBS. This plasticizer was demonstrated as an appropriate one for a more rapid in vitro release (32).
The film coded F5 (EC 100%, w/w) showed the slowest AZT release (64 μg/cm2). This could be attributed to the hydrophobic nature of this polymer which helps to retain the drug in the matrix system by reducing the penetration of solvent molecules into the film, in contrast to ERL polymer (6,16). However, the release profile of AZT from the films prepared with different combinations of ERL/EC (F2–F4) seemed to be similar (318.9, 334.8, and 326.8 μg/cm2, respectively) with each other (Fig. 1). Thus, multivariate one-way ANOVA with Tukey–Kramer multiple comparison post test (GraphPad, InStat 3.0) was applied to the release profiles, and significant difference was not found (p > 0.05) between the release profiles of these films with the exception of the first 2 h. When the release in the first 2 h was evaluated with post tests, the release profile of F4 significantly differed from F2 and F3 formulations (33). The reason for this difference could be attributed to the EC polymer rather than the ERL.
In order to show the effect of polymer type on the in vitro release of drug, AZT solutions (1 and 2 mg/mL) in HEPES buffer were also studied with the same release conditions. As it can be seen from Fig. 2a, b, the initial release of AZT from solution was very fast and the in vitro release was finished in approximately 2 h. Even if the initial fast release was decreased when AZT was formulated in ERL film, cumulative amount of AZT released from F6 formulation at the end of 8 h was found closer to free AZT (2-mg/mL solution).
When evaluated for release kinetics, the in vitro release of AZT from the films was best fitted to the Higuchi’s equation, which indicates that the transport of the drug from the films was governed by a diffusion mechanism. Also, the n values calculated from the Korsmayer–Peppas kinetic indicated that the amount of drug released by Fickian diffusion (n < 0.5) predominated from the formulations (34). The exception was the EC film (F5) which was n > 1 could be interpreted as the transport of AZT governed not only by diffusion but also erosion mechanism, and the release shows a zero-order release kinetic. Also, the combination of ERL/EC (1:1) showed an anomalous release (F4); however, by increasing the amount of AZT in this film, the n value was decreased to show diffusion (Table III).
Table III.
Drug Release Kinetics of AZT from the Films
| Code | Zero-order | First-order | Higuchi | Korsmeyer–Peppas | ||||
|---|---|---|---|---|---|---|---|---|
| r 2 | k 0 | r 2 | k 1 | r 2 | k H | r 2 | n | |
| F1 | 0.724 | 0.115 | 0.817 | 0.0020 | 0.861 | 2.41 | 0.984 | 0.476 |
| F2 | 0.727 | 0.088 | 0.798 | 0.0013 | 0.897 | 1.79 | 0.946 | 0.350 |
| F3 | 0.763 | 0.095 | 0.818 | 0.0014 | 0.816 | 2.19 | 0.905 | 0.473 |
| F4 | 0.857 | 0.096 | 0.901 | 0.0014 | 0.920 | 2.52 | 0.922 | 0.687 |
| F5 | 0.950 | 0.033 | 0.950 | 0.0004 | 0.975 | 0.98 | 0.974 | 1.017 |
| F6 | 0.706 | 0.068 | 0.733 | 0.0012 | 0.819 | 2.22 | 0.889 | 0.422 |
| F7 | 0.735 | 0.049 | 0.758 | 0.0007 | 0.846 | 1.58 | 0.886 | 0.398 |
r 2 indicates determination coefficient; k 0, k 1, k H are kinetic constants, and n is diffusional exponent indicative of the mechanism of drug release
Ex Vivo Studies
The ex vivo studies were done on F6 and F7 films in comparison with the same amount of AZT in solution (2 mg/mL). The studies examined on dorsal skin of Wistar rats (approximately 200 μm in thickness) showed that application of 0.5-mA/cm2 direct current significantly increased the skin transport of AZT in contrast to passive diffusion for the studied formulations. Highest cumulative amounts of AZT permeated from skin were observed by the application 0.5-mA/cm2 current (72.5 μg/cm2 for AZT solution, 60.35 μg/cm2 for F6, and 39.75 μg/cm2 for F7). When any enhancement method was not applied, these values were found as 24.7, 16.73, and 12.78 μg/cm2 for AZT solution, F6, and F7 films, and the cumulative amounts permeated from skin with the application of 0.1 mA/cm2 were found as 59.9, 44.97, and 17.93 μg/cm2 for AZT solution, F6, and F7, respectively (Fig. 3). When the effects of 0.1- and 0.5-mA/cm2 direct currents were compared, significant differences were not found between the cumulative amounts of AZT permeated from skin for the films F6 and F7. However, by calculating the enhancement ratio from the flux values, highest enhancement ratio values were observed with the application of 0.5-mA/cm2 current.
Also, pretreatment of DMSO affected the increase in the transport of AZT from dorsal skin of rats. However, the increase in the concentration of DMSO did not have significant effect on the cumulative amounts of AZT permeated from skin for F6 (37.19 μg/cm2 for 1%, w/w, DMSO and 41.93 μg/cm2 for 5%, w/w, DMSO) and F7 (33.64 μg/cm2 for 1%, w/w, DMSO and 35.24 μg/cm2 for 5%, w/w, DMSO). Cumulative amounts of drug permeated from AZT solution into skin was 38.5 μg/cm2 for 1% (w/w) DMSO pretreatment and 64.3 μg/cm2 for 5% (w/w0 DMSO pretreatment. Also, the effects of 0.5-mA/cm2 current and 5% (w/w) DMSO was not found significant for the studied films. However, when the enhancement ratios from the flux values were calculated, it was observed that the values were lower than the application of 0.1- and 0.5-mA/cm2 current.
While AZT released from the films could be quantitated from receptor compartment of Franz cells at the end of 15 min, this duration increased up to 60 min for the other applications evaluated on the films. This result could indicate that AZT transport from the skin via iontophoresis had a decrease in the lag time of transport.
Also, regression analysis of the ex vivo permeation curves was carried out. The slope of the straight line obtained after plotting the mean cumulative amount released per area of the film versus time (hour) was taken as the experimental flux for AZT for the ex vivo studies. The results were supported by the data evaluated by ANOVA. Experimental fluxes (μg cm−2 h−1) for free AZT were higher than films as expected; however, the calculated flux values for F6 were close to the values of AZT in solution. The experimental fluxes were highest for the 0.5-mA/cm2 current application, and the flux value for the permeation of AZT from F6 film was very close to the flux value for AZT solutions; 7.07 ± 0.026 and 7.56 ± 0.03 μg cm−2 h−1, respectively. The results were given in Table IV.
Table IV.
Experimental Flux Values (μg cm−2 h−1) for the Skin Transport of AZT from F6, F7 Films and 2 mg/mL AZT Solution
| Passive | 0.1-mA/cm2 current | 0.5-mA/cm2 current | 1% (w/w) DMSO | 5% (w/w) DMSO | |
|---|---|---|---|---|---|
| F6 | 2.08 ± 0.04 | 5.66 ± 0.55 | 7.07 ± 0.03 | 4.64 ± 0.37 | 5.83 ± 0.36 |
| F7 | 1.89 ± 0.1 | 3.79 ± 0.2 | 4.99 ± 0.45 | 3.78 ± 0.36 | 3.94 ± 0.17 |
| Free AZT | 2.49 ± 0.09 | 6.05 ± 0.20 | 7.56 ± 0.03 | 4.83 ± 0.69 | 5.17 ± 0.69 |
All values are expressed as means ± SE (n = 3)
DMSO dimethyl sulfoxide
Iontophoresis was reported to cause the formation of transient aqueous pores by electroporating the skin, which constituted a significant transport route during the application. On the other hand, sulfoxides such as DMSO are aprotic solvents that can denature proteins and change the intercellular keratin confirmation. DMSO can also interact with the intercellular lipid domains of human stratum corneum. Further, DMSO within skin membranes may facilitate drug partitioning from the formulation to the tissues (35,36). Thus, even if the results were found as not significant for the two applications, this could be interpreted as an advantage of iontophoresis as an enhancement mechanism for the transdermal application of AZT.
Interaction Studies
The maximum UV absorption for the pure drug and films containing AZT were found to be at 265 nm. The accuracy of the UV spectrophotometric method was between 94% (±0.27) and 98% (±0.29) for the concentration range of 1–30 μg/mL in the presence of polymers. Also, the drug recovery from the films was higher than 90%. The results indicate that UV spectrophotometric method used in the analysis was selective and AZT remained intact in the films without any chemical interaction (30).
On analysis of IR spectra of the pure drug and films containing AZT and the major IR peaks of AZT could be determined from the pattern (Fig. 4). The IR spectrum of AZT (Fig. 4a) showed –OH and –NH stretching bands in the 3500–3200 cm−1, C=N=N=N (azide group) at 2082 cm−1, a band at 1684 cm−1 for amide C=O group, 1379 cm−1 for –CH2 group, 1282 cm−1 for C–O–C group, and a band for NH bending at 1513 cm−1. The characteristic IR peaks of AZT could also be determined from the films (Fig. 4b–d). However, some of the peaks in the spectra of the films got merged. This might be due to a physical interaction (not chemical) between the drug and the polymers. Especially, the quaternary ammonium groups of ERL give peaks near 1684 cm−1. However, the increase in the intensity of this peak could be considered as the existence of AZT. Anyway, the presence of characteristic azide band at 2082 cm−1 and amide C=O bands at 1684 cm−1 showed that AZT was stable in the films (37).
Fig. 4.

IR spectra of AZT. a Pure drug, b AZT with ERL, c AZT with EC, d AZT with ERL/EC, 1:1 mixture
CONCLUSION
In this study, monolithic films of AZT were prepared by various combinations of ERL and EC polymers. The reasons for choosing these polymers were their ease of preparation with solvent evaporation method and the release modifying characteristics of these polymers.
All the films prepared had various degree of sustained release effect on the in vitro release of AZT. However, by using only ERL polymer in the film, cumulative released amounts of drug were highest (F1, F6). This could be attributed to the hydrophilic nature and swelling of ERL polymer. AZT released rapidly from ERL film than the EC or ERL/EC films because pores of the polymer should expand by swelling effect. ERL polymer could also prevent AZT from crystallization in the film. Even if excess amount of AZT was loaded, there was not any crystal deposits of AZT in the films prepared with ERL. It has been demonstrated that this polymer has some inhibitor effect on the crystallization of active agents dispersed in films (31). So, higher drug loading should be possible using ERL polymer in the films to achieve higher flux values. Thus, F6 film gave the highest flux value amongst the films prepared and this value was close with free AZT. Contrarily, drug release from hydrophobic EC film was slow, as the polymer was not swellable in the buffer. AZT could not diffuse from the film in the absence of the pores. The slow release of AZT from EC film could be attributed to this situation.
Stratum corneum is a barrier to maintain therapeutic levels via transdermal route. Thus, a penetration enhancement mechanism must be used to overcome this barrier function. Iontophoresis is a good alternative method to enhance the skin permeability of drugs. Though AZT is uncharged, it has been shown that skin permeation of AZT can be increased by iontophoresis due to the electroosmotic flow and alteration in skin permeability (7). In this study, pretreatment of DMSO was used as an alternative enhancement mechanism to iontophoresis, and it was observed that iontophoresis was more effective on the permeation of AZT; also, it more effectively increased the flux values rather than DMSO. However, iontophoresis was not found so efficient on the ERL/EC film (F7). Contrarily, flux values with ERL film (F6) were close to free AZT, especially with the effect of 0.5-mA/cm2 current. Thus, it was thought that the films prepared with 100% (w/w) ERL polymer had better film and release properties in contrast to films consisting of EC polymer, and F6 film consisting ERL polymer with a higher drug loading should be improved in further studies for transdermal delivery either by using permeation enhancers or combining this polymer with suitable alternative polymer which could be prepared with solvent evaporation method.
Acknowledgment
The authors wish to thank Drogsan Pharmaceuticals, Ankara, Turkey/Cipla Inc., India for their support in providing zidovudine.
References
- 1.Pongjangyakul T., Prakongpan S., Priprem A. Acrylic matrix type nicotine transdermal patches: In vitro evaluations and batch-to-batch uniformity. Drug Dev. Ind. Pharm. 2003;29:843–853. doi: 10.1081/DDC-120024180. [DOI] [PubMed] [Google Scholar]
- 2.Kalia Y. N., Guy R.H. Interaction between penetration enhancers and iontophoresis: Effect on human skin impedance in vivo. J. Control. Release. 1997;44:33–42. doi: 10.1016/S0168-3659(96)01500-3. [DOI] [Google Scholar]
- 3.Banga A. K. Electrically assisted transdermal delivery of drugs. In: Wise D. L., editor. Handbook of Pharmaceutical Controlled Release Technology. New York: Marcel Dekker; 2000. pp. 567–581. [Google Scholar]
- 4.Denet A. R., Ucakar B., Preat V. P. Transdermal delivery of timolol and atenolol using electroporation and iontophoresis in combination: A mechanistic approach. Pharm. Res. 2003;20(12):1946–1951. doi: 10.1023/B:PHAM.0000008041.86042.c0. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch A. C., Upasani R. S., Banga A. K. Factorial design approach to evaluate interactions between electrically assisted enhancement and skin stripping for delivery of tacrine. J. Control. Release. 2005;103(1):113–121. doi: 10.1016/j.jconrel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 6.A. Kuksal, A. K. Tiwary, N. K. Jain, and S. Jain. Formulation and in vitro, in vivo evaluation of extended-release matrix tablet of zidovudine: influence of combination of hydrophilic and hydrophobic matrix formers. AAPS Pharm. Sci. Tech.7(1):Article 1, E1–E9. doi:10.128/pt070101 (2006). [DOI] [PMC free article] [PubMed]
- 7.Oh S. Y., Jeong S. Y., Park T. G., Lee J. H. Enhanced transdermal delivery of AZT (Zidovudine) using iontophoresis and penetration enhancer. J.Controlled Rel. 1998;51:161–168. doi: 10.1016/S0168-3659(97)00162-4. [DOI] [PubMed] [Google Scholar]
- 8.Kim D. D., Chien Y. W. Transdermal delivery of dideoxynucleoside-type anti-HIV drugs. 1. Stability studies for hairless rat skin permeation. J. Pharm. Sci. 1995;84(9):1061–1066. doi: 10.1002/jps.2600840906. [DOI] [PubMed] [Google Scholar]
- 9.Narishetty S., Panchagnula R. Effect of l-menthol and 1-8-cineole on phase behaviour and molecular organisation of SC lipids and skin permeation of zidovudine. J. Control. Release. 2005;102:59–70. doi: 10.1016/j.jconrel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Thomas N. S., Panchagnula R. Combination strategies to enhance transdermal permeation of zidovudine (AZT) Pharmazie. 2003;58:895–898. [PubMed] [Google Scholar]
- 11.Kim D. D., Chien Y. W. Transdermal delivery of dideoxynucleoside-type anti-HIV drugs. 2. The effect of vehicle and enhancer on skin permeation. J. Pharm. Sci. 1996;85(2):214–219. doi: 10.1021/js950141c. [DOI] [PubMed] [Google Scholar]
- 12.Kararli T. T., Kirchhoff C. F., Penzotti Jr S. C. Enhancement of transdermal transport of azidothymidine (AZT) with novel terpene and terpene like enhancers: In vivo–in vitro correlations. J. Control. Release. 1995;34:43–51. doi: 10.1016/0168-3659(94)00128-H. [DOI] [Google Scholar]
- 13.Acartürk F., Şencan A. Investigation of the effect of different adjuvants on felodipine release kinetics from sustained release monolithic films. Int. J. Pharm. 1996;131:183–189. doi: 10.1016/0378-5173(95)04321-7. [DOI] [Google Scholar]
- 14.Kusum Devi V., Saisivam S., Maria G. R., Deepti P. U. Design and evaluation of matrix controlled transdermal patches of verapamil hydrochloride. Drug Dev. Ind. Pharm. 2003;29(5):495–503. doi: 10.1081/DDC-120018638. [DOI] [PubMed] [Google Scholar]
- 15.Gupta S. P., Jain S. K. Development of matrix-membrane transdermal drug delivery system for atenolol. Drug Delivery. 2004;11:281–286. doi: 10.1080/10717540490493943. [DOI] [PubMed] [Google Scholar]
- 16.Mutalik S., Udupa N., Kumar S., Agarwal S., Subramanian G., Ranjith A. K. Glipizide matrix transdermal systems for diabetes mellitus: Preparation, in vitro and preclinical studies. Life Sci. 2006;79:1568–1577. doi: 10.1016/j.lfs.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 17.U. Ubaidulla, M. Reddy, K. Ruckmani, F. J. Ahmad, and R. K. Khar. Transdermal therapeutic system of carvedilol: Effect of hydrophilic and hydrophobic matrix on in vitro and in vivo characteristics. AAPS Pharm. Sci. Tech.8(1):Article 2. doi:10.1208/pt0801002 (2007). [DOI] [PMC free article] [PubMed]
- 18.Bodmeier R., Paeratakul O. Propranolol HCl release from acrylic films prepared from aqueous latexes. Int. J. Pharm. 1990;59:197–204. doi: 10.1016/0378-5173(90)90109-H. [DOI] [Google Scholar]
- 19.Lin S. Y., Lin Y. Y., Cheng C. L. Studies on the compatibility, efficiency and permanence of plasticizers on drug-free or drug-loaded Eudragit-E-films. Pharmazie. 1995;50:801–809. [Google Scholar]
- 20.İnal Ö., Kılıçarslan M., Arı N., Baykara T. In vitro and in vivo transdermal studies of atenolol using iontophoresis. Acta Pol. Pharm. Drug Res. 2008;65(1):29–36. [PubMed] [Google Scholar]
- 21.Röhm Pharma Catalogue. 2000. Röhm Pharma Data Sheet, HTML\files\pharma4_ formulation\4.6.1.pdf.
- 22.ICH Guideline. 2005. Validation of Analytical Procedures: Text and Methodology Q2 (R1), http:/www.ich.gov/LOB/media/MEDIA417.pdf.
- 23.Akhgari A., Farahmand F., Afrasiabi Garekani H., Sadeghi F., Vandamme T.F. Permeability and swelling studies on free films containing inulin in combination with different polymethacrylates aimed for colonic drug delivery. Eur. J. Pharm. Sci. 2006;28:307–314. doi: 10.1016/j.ejps.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Padula C., Nicoli S., Colombo P., Santi P. Single-layer transdermal film containing lidocaine: Modulation of drug release. Eur. J. Pharm. Biopharm. 2007;66(3):422–428. doi: 10.1016/j.ejpb.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Santoyo S., Arellano A., Ygartua P., Martin C. Penetration enhancer effects on the in vitro percutaneous absorption of piroxicam trough rat skin. Int. J. Pharm. 1995;105(1):219–224. doi: 10.1016/0378-5173(94)00344-5. [DOI] [Google Scholar]
- 26.Pillai O., Panchangula R. Transdermal delivery of insulin from poloxamer gel: Ex vivo and in vivo permeation studies in rat using iontophoresis and chemical enhancers. J. Control. Release. 2003;89:127–140. doi: 10.1016/S0168-3659(03)00094-4. [DOI] [PubMed] [Google Scholar]
- 27.Narishetty S. T., Panchagnula R. Transdermal delivery system for zidovudine: In vitro, ex vivo, in vivo evaluation. Biopharm. Drug Disp. 2004;25(1):9–20. doi: 10.1002/bdd.381. [DOI] [PubMed] [Google Scholar]
- 28.Femenia-Font A., Balaguer-Fernandez C., Merino V., Rodilla V., Lopez-Castellano A. Effect of chemical enhancers on the in vitro percutaneous absorption of sumatriptan succinate. Eur. J. Pharm. Biopharm. 2005;61:50–55. doi: 10.1016/j.ejpb.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 29.Uslu B., Özkan S. A. Determination of lamivudine and zidovudine in binary mixtures using first derivative spectrophotometric, first derivative of the ratio-spectra and high-performance liquid chromatography–UV methods. Anal. Chim. Acta. 2002;466:175–185. doi: 10.1016/S0003-2670(02)00545-7. [DOI] [Google Scholar]
- 30.Aqil M., Ali A. Monolithic matrix type transdermal drug delivery systems of pinacidil monohydrate: In vitro characterization. Eur. J. Pharm. Biopharm. 2002;54:161–164. doi: 10.1016/S0939-6411(02)00059-0. [DOI] [PubMed] [Google Scholar]
- 31.Kotiyan P. N., Vavia P. R. Eudragits: Role as crystallization inhibitors in drug-in-adhesive transdermal systems of estradiol. Eur. J. Pharm. Biopharm. 2001;52:173–180. doi: 10.1016/S0939-6411(01)00174-6. [DOI] [PubMed] [Google Scholar]
- 32.Siepmann J., Lecomte F., Bodmeier R. Diffusion-controlled drug delivery systems: Calculation of the required composition to achieve desired release profiles. J. Control. Release. 1999;60:379–389. doi: 10.1016/S0168-3659(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 33.Yuksel N., Kanık A.E., Baykara T. Comparison of in vitro dissolution profiles by ANOVA-based, model-dependent and-independent methods. Int. J. Pharm. 2000;209:57–67. doi: 10.1016/S0378-5173(00)00554-8. [DOI] [PubMed] [Google Scholar]
- 34.Costa P., Sausa Lobo J. M. Evaluation of mathematical models describing drug release from estradiol transdermal systems. Drug Dev. Ind. Pharm. 2003;29(1):89–97. doi: 10.1081/DDC-120016687. [DOI] [PubMed] [Google Scholar]
- 35.Williams A. C., Barry B. W. Penetration enhancers. Adv. Drug Del. Rev. 2004;56:603–618. doi: 10.1016/j.addr.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 36.Anigbogu A. N. C., Williams A. C., Barry B. W., Edwards H. G. M. Fourier transform Raman spectroscopy of interactions between the penetration enhancer dimethyl sulfoxide and human stratum corneum. Int. J. Pharm. 1995;125(2):265–282. doi: 10.1016/0378-5173(95)00141-5. [DOI] [Google Scholar]
- 37.Araujo A. A. S., Storpirtis S., Mercuri L. P., Carvalho F. M. S., Filho M. S., Matos J. R. Thermal analysis of the antiretroviral zidovudine (AZT) and evaluation of the compatibility with excipients used in solid dosage forms. Int. J. Pharm. 2003;260(2):303–314. doi: 10.1016/S0378-5173(03)00288-6. [DOI] [PubMed] [Google Scholar]