

Abstract
Poly(propylene succinate) (PPSu) polymers of average molecular weights from 2,800 to 13,100 g/mol were synthesized and characterized with regard to crystallinity, thermal properties, and cytocompatibility. Higher molecular weight samples exhibited lower degree of crystallinity and melted at lower temperatures. Melting of the polymer appeared to begin at 38°C. PPSu cytocompatibility was investigated based on human umbilical vein endothelial cells viability in the presence of increasing concentrations of polymer, and it was found that PPSu exhibited comparable cytocompatibility with poly(dl-lactide). The feasibility of applying PPSu as a drug carrier was shown for the first time, as solid dispersions and nanoparticles of sodium fluvastatin based in PPSu were prepared. Drug release rates decreased with increasing the molecular weight of PPSu in both solid dispersions and nanoparticles. For dispersions prepared from PPSu of the same molecular weight, drug release rates increased with drug loading. It appears that PPSu applicability as a drug carrier warrants further consideration.
Key words: fluvastatin, melt mixing, nanoparticles, poly(propylene succinate), solid dispersions
INTRODUCTION
Aliphatic polyesters like poly(l-lactic acid), poly(ɛ-caprolactone), poly(3-hydroxy butyrare), poly(3-hydroxy valerate), etc., due to their favorable features of biodegradability and biocompatibility, constitute a very important class of biodegradable polymers. They are nowadays commercially available in a variety of types used for several applications. Among them, aliphatic polyesters have found applications in medicine and pharmaceutical technology, for example, in drug delivery systems, surgical sutures, implants, and tissue engineering (1–4).
Poly(propylene succinate) (PPSu) is a novel biodegradable aliphatic polyester, produced using monomers from renewable resources (5–10). PPSu has gained an increasing interest, since it has higher biodegradation rate compared to poly(ethylene succinate) (PESu) and poly(butylene succinate) (PBSu). Due to its low crystallinity, it also degrades faster than most of polyesters used as pharmaceutical excipients, including polycaprolactone (PCL) (11,12). During enzymatic degradation studies, it was found that practically PPSu degrades within 36 h (11). Furthermore, its melting point is just above the human body temperature (43–45°C).
The aims of this work were to synthesize samples of PPSu of different molecular weights and to investigate the feasibility of applying them as drug carriers. To this end, solid dispersions and nanoparticles of sodium fluvastatin based on PPSu were prepared and their basic physicochemical and drug release properties were examined. Fluvastatin is a highly water-soluble drug (Fig. 1). The solubility of its sodium salt is more than 50 g/L and is commercially available as an immediate-release (IR) or a sustained-release (SR) formulation. The release properties of fluvastatin preparations are of particular interest as slow delivery of relatively high doses of fluvastatin using once-daily (13), sustained release formulations can improve compliance of hypercholesterolemic patients with greater risk for cardiovascular heart disease which require more aggressive lowering of low-density lipoprotein levels (14). At high doses, the pharmacokinetics of fluvastatin immediate release are nonlinear, possibly due to saturation of hepatic uptake. Fluvastatin delivery in a slower but sustained fashion would be expected to avoid hepatic saturation, to minimize systemic drug toxicity (15).
Fig. 1.

Chemical structures of a PPSu and b fluvastatin
EXPERIMENTAL
Materials and Equipment
Succinic acid (SA; purum 99+%) was purchased from Aldrich Chemical Co. 1,3-Propanediol (1,3-PD) (CAS number: 504-63-2, purity >99.7%) was kindly supplied by Du Pont de Nemours Co. Tetrabutyl titanate (TBT), used as catalyst, was of analytical grade and it was purchased also from Aldrich Chemical Co. Poly(dl-lactide) (PLA, molecular weight Mw = 109 × 103) was synthesized as described previously (16). All culture media and supplements were from Biochrom AG (Berlin, Germany). Amorphous fluvastatin salt with an assay of 99.9% was supplied from Biocom. All other materials and solvents used for the analytical methods were of analytical grade.
Methods
Synthesis of PPSu Polyesters
Synthesis of aliphatic polyester PPSu was performed following the two-stage melt polycondensation method (esterification and polycondensation) in a glass batch reactor as described previously (6,17). In brief, succinic acid and 1,3-PD at a molar ratio of 1:1.1 were charged into the reaction tube of the polycondensation apparatus. The catalyst (3 × 10−4 mol TBT/mol SA) was added and the apparatus was evacuated several times and filled with argon in order to completely remove the oxygen. The reaction mixture was heated at 180°C under argon atmosphere while stirred at a constant speed (500 rpm). This stage lasted 1 h.
In the second step of polycondensation, vacuum (5.0 Pa) was applied slowly over a time period of about 30 min, to avoid excessive foaming and to minimize oligomer sublimation. The temperature was slowly increased to 230°C, while stirring speed was also increased to 720 rpm. The polycondensation continued for about 60 min. After the end of the polycondensation reaction, the polymer obtained was milled and washed with methanol. PPSu samples with different molecular weight were obtained by applying different polycondensation times (20, 40, and 90 min).
Intrinsic Viscosity
Intrinsic viscosity [η] measurements were performed using an Ubbelohde viscometer at 25°C in chloroform. All polyesters were dissolved at room temperature in order to prepare solutions of 1 wt.% and filtered through a disposable membrane filter 0.2 μm (Teflon). Intrinsic viscosity was calculated using the Solomon–Ciuta equation (18):
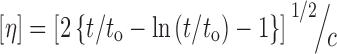 |
where c is the concentration of the solution; t, the flow time of solution, and to the flow time of pure solvent.
Gel Permeation Chromatography
For the estimation of molecular weight gel permeation chromatography (GPC) analysis was performed using a Waters 150C GPC equipped with differential refractometer as detector and three ultrastyragel (103, 104, and 105 Å) columns in series. CHCl3 was used as the eluent (1 ml/min) and the measurements were performed at 35°C. Calibration was performed using polystyrene standards with a narrow molecular weight distribution.
End Group Analysis
Carboxyl end-group content (C.C.) of the resins was determined as follows. About 0.1 g of polyesters was dissolved in chloroform at room temperature, and the solution was titrated using a standard NaOH solution in methanol (N/10) and phenol red as indicator.
Thermal Analysis
A Perkin-Elmer, Pyris 1 differential scanning calorimeter (DSC), calibrated with indium and Zinc standards, was used. The instrument was connected with an Intracooler 2P cooling device for operation to sub-ambient conditions. A sample of about 10 mg was studied in each test, placed in an aluminum seal and heated to 35°C above the melting point at a heating rate 20°C/min. The sample remained at that temperature for 5 min in order to erase any thermal history. After that, it was quenched into liquid nitrogen and scanned again using the same heating rate as before. The glass transition temperature (Tg), the melting temperature (Tm), and the heat of fusion (ΔΗm) were measured.
Nuclear Magnetic Resonance
1H-NMR spectra of polyesters were obtained with a Bruker AMX 400 spectrometer operating at a frequency of 400 MHz for protons. Deuterated chloroform was used as solvent in order to prepare solutions of 5% w/v. The number of scans was 10 and the sweep width was 6 kHz.
Preparation of Solid Dispersions by Melt Mixing
Four samples of PPSu, with [η] values of 0.10, 0.18, 0.28 and 0.50 dl/g, respectively, were used for the preparation of solid dispersions of fluvastatin following the melt mixing method. Physical mixtures of fluvastatin and PPSu were transferred to a reaction tube immersed in an oil bath and heated to 60°C while stirred under an argon atmosphere. The mixture was held to this temperature for 15 min. Τwo series of solid dispersions were prepared: one series with constant drug loading (30 wt.%) using PPSu polymers with different molecular weights and one series using PPSu of constant molecular weight (PPSu with [η] = 0.28 dl/g) and varying the drug loading (10, 20, 30, and 50 wt.%).
Preparation of PPSu Nanoparticles Loaded with Fluvastatin
The nanoparticles were prepared by w/o/w emulsification-solvent evaporation method (19). PPSu (100 mg) dissolved in 2 ml dichloromethane and drug fluvastatin (5 mg) dissolved in 0.5 ml water were mixed, and the mixture was probe-sonicated at 15 W for 2 min using a Bioblock Scientific, model 75038, sonicator. The prepared solution was then transferred to an aqueous solution of sodium cholate (V = 6 ml, C = 12 mM), and it was probe sonicated for 2 min at 15 W. The w/o/w emulsion was gently stirred for 6 h under a fume hood. The nanoparticles formed were purified by centrifugation (20,000 rpm for 20 min) and reconstitution in deionized water. The nanoparticles suspension was filtered using a microfilter with pore size of 1.2 μm (Millex AP, Millipore) in order to remove polymer aggregates. The size of the prepared nanoparticles was determined using photon correlation spectroscopy in a Malvern Z-sizer 5000 instrument (five runs per sample).
Wide Angle X-Ray Diffractometry
Wide angle X-Ray diffractometry (WAXD) was used for the identification of the crystal properties of the pure materials and dispersions. WAXD study was performed over the range 2θ of 5°C to 50°C, using a Philips PW 1710 diffractometer with Bragg–Brentano geometry (θ, 2θ) and Ni-filtered CuKa radiation.
Fourier Transformed-Infrared Spectroscopy
Fourier transformed-infrared spectroscopy (FTIR) spectra were obtained using a Perkin-Elmer FTIR spectrometer, model Spectrum 1000. A small amount of each material was mixed with KBr (1 wt.% content) and compressed to tablets. The IR spectra of these tablets were obtained in absorbance mode and in the spectral region of 450 to 4,000 cm−1 using a resolution of 4 cm−1 and 64 co-added scans.
Scanning Electron Microscopy
The morphology of the prepared nanoparticles was examined by a scanning electron microscopy (SEM) system, Jeol (JMS-840). The samples were covered with carbon coating in order to increase conductivity of the electron beam. Operating conditions were: accelerating voltage of 20 kV, probe current of 45 nA, and counting time of 60 s.
Nanoparticles Yield, Drug Loading Content, and Entrapment Efficiency
Freeze-dried nanoparticles samples were weighed, dissolved in 0.2 N NaOH, and assayed spectrophotometrically for fluvastatin at 310.5 nm. The nanoparticle yield, percent drug loading, and percent drug entrapment efficiency were calculated by the following Eqs. 1–3, respectively:
 |
1 |
 |
2 |
 |
3 |
Size Measurements of Nanoparticles
The particle size distribution of PPSu/fluvastatin nanoparticles was determined by dynamic light scattering using a Zetasizer Nano Instrument (Malvern Instruments, Nano ZS, ZEN3600, UK) operating with a 532 nm laser. A suitable amount of nanoparticles was dispersed in distilled water creating a total concentration 1‰ and was kept at 37°C under agitation at 100 rpm. For particle size measurements, three different batches were used.
Cell Culture
The human umbilical vein endothelial cells (HUVEC) were grown routinely in RPMI-1640 medium supplemented with 15% fetal bovine serum, 15 mg ECGS, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamycin, and 2.5 μg/ml amphotericin B. Cultures were maintained at 37°C, 5% CO2, and 100% humidity.
In Vitro Cytocompatibility Study
The cytocompatibility of PPSu was evaluated by measuring the viability of HUVEC cells in the presence of different concentrations of the polymer. The same procedure was followed for determining cytocompatibility of PLA which is a well-known biocompatible polymer and was used in this study for comparison. Cell viability was determined by the MTT assay. HUVEC cells were seeded in 24-well plates at a density of 30,000 cells per well in 500 μl cell culture medium. Twenty-four hours after plating, with different amounts of PPSu, nanoparticles (suspended in water) were added in the wells. After 24 h of incubation at 37°C, 50 μl of MTT solution (5 mg/ml in PBS pH 7.4) was added into each well, and plates were incubated at 37°C for 2 h. The medium was withdrawn and 200 μl of acidified isopropanol (0.33 ml HCl in 100 ml isopropanol) was added in each well and agitated thoroughly to dissolve the formazan crystals. The solution was transferred to 96-well plates and immediately read on a microplate reader (Biorad, Hercules, CA, USA), at a wavelength of 490 nm. The experiments were performed in triplicate. Cytocompatibility of polymers was expressed as percent cell viability, which was calculated from the ratio between the number of cells treated with the nanoparticles and that of nontreated cells (control).
Study of In Vitro Drug Release from the Nanoparticles
Nanoparticle samples (10 mg) were suspended in 20 ml phosphate buffer (pH = 7.0) inside glass vials. The vials were transferred to a mildly shaken water bath (37°C). At predetermined time intervals, 500-μl samples were withdrawn from the vials and centrifuged at 28,000 rpm for 30 min in a Heraeus Biofuge 28RS centrifuge. The supernatants were assayed for drug at 303 nm using a Shimadzu UV 1205 spectrophotometer. After sampling, 500 μl of fresh medium was added in the incubation medium. For drug release measurements, three different batches were used.
RESULTS AND DISCUSSION
Preparation and Characterization of PPSu Samples
The PPSu samples were synthesized by a two-step melt polycondensation method. In the first stage, succinic acid was esterified by 1,3-propanediol forming oligomers. The second stage involved polycondensation of these oligomers giving the PPSu polymer as shown in Fig. 2. Varying the reaction parameters, i.e., temperature or time of the second stage, samples of different intrinsic viscosities, and thus of different molecular weights, were obtained. As it can be seen in Table I, the molecular weight increased with increasing the polycondensation time. Polymers with molecular weights ranging between 2,000 and 4,000 g/mol can be obtained after short polycondensation times (till 20 min). At longer polycondensation times, effective removal of polycondensation by-products (ethylene glycol and water) can be achieved, allowing for the preparation of polyesters of higher molecular weight. Thus molecular weight values up to 13,130 g/mol could be achieved after 90 min of polycondensation.
Fig. 2.

Synthesis of PPSu following the two-stage polycondensation method in the melt phase
Table I.
Properties of PPSu Samples with Different Molecular Weights
| a/a | IV (dl/g) | M n (g/mol) | M w (g/mol) | T m (°C) | T g (°C) | X c (%) | [−COOH] (eq/10−6g) |
|---|---|---|---|---|---|---|---|
| 1 | 0.10 | 2,800 | 7,630 | 50.8 | −31.8 | 37.6 | 762 |
| 2 | 0.18 | 3,740 | 9,910 | 48.2 | −32.6 | 36.1 | 203 |
| 3 | 0.28 | 6,480 | 15,320 | 47.8 | −33.7 | 35.1 | 95 |
| 4 | 0.50 | 13,130 | 32,210 | 45.4 | −40.6 | 33.9 | 23 |
X c degree of crystallinity measured from WAXD patterns, T g glass transition temperature, T m melting temperature, M w molecular weight, M n number molecular weight
The samples with low molecular weights, lower than 3,740 g/mol, are soft, while those with high molecular weight present progressive hardening. The carboxyl end groups content gradually decreased from 762 eq/10−6 g, for the sample with IV = 0.1 dl/g, to 23 eq/10−6 g for the sample with intrinsic viscosity of 0.50 dl/g (Table I), revealing the progress of polycondensation reaction. The DSC traces of the synthesized PPSu samples (Fig. 3) showed that the polyester melting point was about 45.5°C. The melting point, determined as the peak temperature, increased from 45°C to about 50°C with decreasing the molecular weight of the samples. It is important here to note that independent of the molecular weight, melting of the PPSu polyesters begins at about 38°C and is completed a few degrees above temperature of the human body (45–50°C). As it can be seen in Fig. 3a, the lower molecular weight samples show higher crystallinity since these samples have higher melting peak area (percent crystallinity values are presented in Table I). This is because of the higher chain mobility in the case of low molecular weight polymers, which results in easier and faster crystallization. In general, PPSu is slowly crystallizing. Crystallization behavior of polymers is determining the final physical structure that specimens will show, and the latter is crucial for solid dispersions or other pharmacotechnical systems since the behavior of polymeric carriers affects drastically the solid state of the active pharmaceutical ingredient and other properties after long-term storage. Crystallization behavior of polymers in the simplest case is evaluated from their behavior on cooling from the melt at 10°C/min and on heating amorphous samples from the glass. PBSu crystallizes rapidly on cooling from the melt. In fact, it is rather difficult to obtain amorphous PBSu samples. PESu on the other hand does not crystallize on cooling from the melt at 10°C/min, but it shows cold crystallization when amorphous samples are heated from the glass even at rates as high as 20°C/min. Finally, PPSu shows slower crystallization among the tested biodegradable succinate polyesters. PPSu samples do not crystallize on cooling from the melt not only on cooling at 10°C/min, but even at very slow cooling rates such as 1°C/min. Also, in general, glassy PPSu samples prepared by quenching in liquid nitrogen show only limited crystallization on heating. As it is expected, crystallization rates of PPSu samples decrease with molecular weight, the PPSu 0.28 and 0.50 samples showing the slower rates. At this point, however, it is important to note that PPSu 0.50 corresponds to a moderate molecular weight for polymers, the others corresponding to low molecular weights. Thus, apart to what might be important for the specific study, in general, the behavior of PPSu 0.50 should be considered to be indicative for the polyester. As one can see in Fig. 3b, PPSu 0.28 showed cold crystallization on heating from the glassy state at 2.5°C/min. PPSu 0.50 showed cold crystallization only at heating at even slower rates, such as 0.5°C/min (Fig. 3b). Slower crystallization behavior of PPSu compared to those of the other two succinates (PBSu and PESu) is attributed to the reduced symmetry of the repeating unit of the polymer and especially to the presence of the propylene segment having an odd number of carbon atoms. Although to explore cold crystallization very slow heating tests were performed, always in the literature values for glass transition temperature (Tg) of polymers correspond to amorphous samples, prepared by quenching in liquid nitrogen and are obtained on heating at a rate of 20°C/min. Thus, and for heating at a rate of 20°C/min and using samples of about the same molecular weight, the Tg is about −34°C for PPSu 0.28, compared to −11°C and −41°C for PESu 0.28 and PBSu 0.28 samples, respectively. Finally, comparison among different PPSu samples showed that the Tg of PPSu increases with increasing molecular weight for constant heating rate of 20°C/min. It is well known that the glass transition is associated with segmental motions of the macromolecular chains which are characterized by specific relaxation times.
Fig. 3.

DSC thermograms of a as received at a heating rate of 20°C/min and b quenched PPSu samples on heating
The WAXD patterns and the 1H-NMR spectra of the synthesized PPSu samples were examined to further characterize the material. The WAXD patterns of all samples showed characteristic peaks at 2θ = 18.25°, 19.45°, 20.62°, 22.67°, 24.16°, 26.05°, 29.98°, and 34.39° and a little increased crystallinity for the lower molecular weight polymers (data not shown) which is in agreement with the DSC results. Apart from such differences in the crystalline and amorphous background contributions, the WAXD patterns were found to be essentially the same for all PPSu samples. The 1H-NMR spectra confirmed the identity and purity of PPSu polyesters synthesized here. The chemical shifts are assigned to different protons in the repeating unit of the polymer:
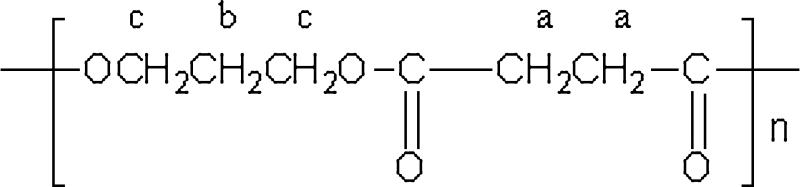
Thus, PPSu samples exhibited a single peak at 2.63 ppm attributed to methylene protons of succinic acid, a; a triple peak at 4.09–4.21 ppm attributed to b protons; and a multiple peak between 1.9 and 2.02 ppm corresponding to c protons (Fig. 4).
Fig. 4.

1H-NMR spectra of the PPSu 0.28
Solid Dispersions Prepared by Melt Mixing
PPSu has never been used before in pharmaceutical technology applications. In this work, solid dispersions of fluvastatin using PPSu matrix were prepared, and the effect of the molecular weight of PPSu and percent drug loading on the properties of the solid dispersions was studied. The solid dispersion samples were prepared by slow heating to above the melting temperature of the polymer. Then, complete melting of the polymer allowed dissolution of the drug fluvastatin in the polymer melt, under continuous stirring. The melt mixing method was applied in solids dispersion preparation because it offers many advantages compared with the solvent evaporation method as it is a simpler, faster, and solvent-free method (20–23).
WAXD patterns of the solid dispersions revealed only peaks corresponding to the reflections of the polymer crystals, irrespective of the percent drug loading (Fig. 5a) or the molecular weight of the polymer (Fig. 5b), indicating that the drug dispersed in the polymers was amorphous. This was expected since the used fluvastatin is amorphous and only a broad peak is recorded in its WAXD patterns. The intensity of the crystalline reflections of PPSu decreased with increasing percent drug loading (Fig. 5a). This could have been expected because of the decrease of the polymer content, but also because of a drop in the crystallinity of the polymer as a result of the mixing since the drug acts as inhibitor to the crystal growth reducing the mobility of macromolecular chains.
Fig. 5.

WAXD patterns of the fluvastatin/PPSu solid dispersions a using PPSu 0.28 and various drug loads and b 30% w/w drug in different PPSu matrices
The nature of possible interactions between PPSu and fluvastatin was investigated with FTIR spectroscopy since any kind of physicochemical interactions that may take place, such as the formation of hydrogen bonds between the carrier and the drug, should lead to frequency shifts or splitting in absorption peaks. PPSu with low molecular weights has a lot of carboxyl end groups, which could form hydrogen bonds with hydroxyl groups of fluvastatin. Two characteristic peaks are important in the fingerprint of fluvastatin. The first appears at 1,573 cm−1 and corresponds to the carboxyl anion group of the drug. The second appears at 3,300–3,650 cm−1 and is associated with the hydroxyl groups of fluvastatin. In the spectra of the solid dispersions of fluvastatin in PPSu, there were no shifts or other changes in the characteristic peaks of fluvastatin or PPSu (Fig. 6). Thus, it is unlikely that fluvastatin and PPSu interact with each other.
Fig. 6.

FT-IR spectra of PPSu, fluvastatin, and fluvastatin/PPSu solid dispersions
Thermal analysis of the prepared solid dispersions was carried out using differential scanning calorimetry. The thermograms of fluva/PPSu solid dispersions containing different fluvastatin amounts are shown in Fig. 7. All samples exhibited multiple melting endothermic peaks. More specifically, the 10/90 sample showed triple melting peak whereas the 20/80 and 30/70 w/w samples showed double melting peaks. The lower temperature melting peak appearing in the case of the 10/90 sample is due to melting of imperfect PPSu crystals. As this peak in the particular case of low drug load is located at about 15°C, which is below the storage temperature, it seems that these polymer crystals most probably were nucleated on the already existing crystals and grown on cooling to sub-ambient temperature, prior to recording the DSC heating scan from −60°C to 60°C. Also, it should be pointed out that the middle peak (appearing in all cases) is located close to the human body temperature and the ultimate one is close to the melting point of the used PPSu matrix (47.8°C; Table I). Although the ultimate melting peak always appeared at the same temperature, the middle one progressively shifted to higher temperatures with increasing proportion of fluvastatin in the dispersion.
Fig. 7.

DSC thermograms of fluvastatin/PPSu 0.28 solid dispersions containing different amounts of fluvastatin.
Drug release profiles of fluvastatin from the solid dispersions in PPSu are presented in Fig. 8. For dispersions of the same percent drug loading, an increase in the molecular weight of the polymer tended to decrease the rate of drug release (Fig. 8a). This may be attributed to the decreased hydrophilicity of the higher molecular weight polymers due to the lower proportion of polar end groups in these polymers. For dispersions prepared from PPSu of the same molecular weight, an increase in percent drug loading tended to increase the rate of drug release (Fig. 8b), probably due to the increased hydrophilicity of the dispersion when the proportion of the hydrophilic, water-soluble drug in the dispersion is increased (it should be pointed here that fluvastatin used was in the form of the water-soluble sodium salt). The released results obtained (Fig. 8) suggest that extended drug release profiles from solid dispersions in PPSu could only be obtained when a high molecular weight PPSu is used and the drug/polymer weight proportion is kept relatively low. The particular dosage forms are appropriate for oral administration.
Fig. 8.

Release profiles of fluvastatin from solid dispersions: a fluvastatin/PPSu 30/70 (w/w) using PPSu matrices of different molecular weights and b fluvastatin/PPSu solid dispersions containing different drug proportions (a PPSu 0.28 matrix was used)
PPSu Nanoparticles Loaded with Fluvastatin
Even though PPSu has a low melting point, it was found that it can form fluvastatin-loaded nanoparticles under the experimental conditions applied here. Αn increase in PPSu molecular weight tended to increase the particle size of the nanoparticles (Table II), a fact that has been previously reported for microparticles with the increase of the molecular weight of PCL (24). This may be associated with the increased macromolecular chain length when the molecular weight of the polymer is increased. The yield of nanoparticles tended to decrease with increasing PPSu molecular weights, possibly due to the formation of increased proportions of big nanoparticles and/or aggregates when PPSu with a relatively high molecular weight is used for the preparation of the nanoparticles. These big nanoparticles and aggregates are removed through filtration and, thus, relatively low nanoparticles yield is obtained.
Table II.
Nanoparticle Characteristics
| Sample | DL (%) | EE (%) | Yield (%) | Mean diameter (nm) | PdI |
|---|---|---|---|---|---|
| PPSu 0.18 | 1.19 | 5.55 | 39.90 ± 3.27 | 157.67 ± 9.29 | 0.138 ± 0.022 |
| PPSu 0.28 | 1.21 | 5.63 | 37.09 ± 2.51 | 158.67 ± 11.15 | 0.169 ± 0.039 |
| PPSu 0.50 | 1.18 | 4.67 | 17.97 ± 2.41 | 181.67 ± 0.57 | 0.226 ± 0.009 |
DL drug loading, EE encapsulation efficiency, PdI polydispersity index
The SEM study showed that the drug-loaded nanoparticles had a discrete spherical shape with a diameter about 180 nm or less, a fact that is in agreement with the measurements of dynamic light scattering (Table II).
Figure 9 shows fluvastatin release from PPSu nanoparticles prepared using PPSu with different molecular weights. Irrespective of the molecular weight of the polymer, the release profiles are characterized by a significant burst release which is followed by a period of slower rates. During the burst release phase, 67–78% of the drug was released, indicating that a high proportion of the drug was located at or close to the surface of the nanoparticles. As in the case of solid dispersions of fluvastatin in PPSu (Fig. 8a), and probably for similar reasons, an increase of the molecular weight of the polymer tended to slow down the release of fluvastatin from the nanoparticles (Fig. 9). However, the rates in case of nanoparticles were rather high especially in the PPSu 0.50.
Fig. 9.

Release profiles of fluvastatin from nanoparticles prepared using PPSu of different molecular weights (the number following the word PPSu denotes the inherent viscosity of the polymer). The drug loading and size characteristics of the nanoparticles are given in Table II
In Vitro Cytocompatibility
In order to be suitable for drug delivery applications, PPSu should exhibit satisfactory biocompatibility. The PPSu polymer exhibited low toxicity against HUVEC cells, with appreciable cytotoxicity (more than 20% reduction of cell viability) being observed only after exposing the cells at high nanoparticle concentrations, i.e., higher than 800 μg/ml (Fig. 10). Based on the viability of HUVEC cells in the presence of the polymer (Fig. 10), the cytocompatibility of PPSu is comparable to the cytocompatibility of PLA, which is a polymer of high biocompatibility and widely used in biomedical applications (25).
Fig. 10.

HUVEC cells viability after incubation for 24 h with different concentrations of PPSu and PLA
CONCLUSIONS
PPSu polymers of different molecular weights were synthesized and their thermal properties, crystallinity, and cytocompatibility were investigated. Higher molecular weight samples exhibited lower degree of crystallinity but melted at lower temperatures. Based on HUVEC cells viability in the presence of increasing concentrations of polymer, PPSu exhibited comparable cytocompatibility with poly(dl-lactide). The feasibility of applying PPSu as drug carrier was shown, as solid dispersions and nanoparticles of sodium fluvastatin based in PPSu were readily prepared using established methods. Drug release properties were affected by drug loading and polymer molecular weight.
Acknowledgment
This work was funded by the General Secretariat of Research and Technology of Greece in the framework of the project PABET NE.
References
- 1.Rizzarelli P., Puiglisi C., Montaudo G. Soil burial and enzymatic degradation in solution of aliphatic co-polyesters. Polym. Degrad. Stab. 2004;85:855–863. doi: 10.1016/j.polymdegradstab.2004.03.022. [DOI] [Google Scholar]
- 2.Avella M., Martuscelli E., Raimo M. Review properties of blends and composites based on poly(3-hydroxy)butyrate (PHB) and poly(3-hydroxybutyrate-hydroxyvalerate) (PHBV) copolymers. J. Mater. Sci. 2000;35:523–545. doi: 10.1023/A:1004740522751. [DOI] [Google Scholar]
- 3.Kumagai Y., Doi Y. Enzymatic degradation and morphologies of binary blends of microbial poly(3-hydroxybutyrate) with poly(e-caprolactone), poly(1,4-butylene adipate and poly(vinyl acetate) Polym. Degrad. Stab. 1992;36:241–248. doi: 10.1016/0141-3910(92)90062-A. [DOI] [Google Scholar]
- 4.Mainardes R. M., Evangelista R. C. Praziquantel-loaded PLGA nanoparticles : preparation and characterization. J. Microencapsul. 2005;22(1):13–24. doi: 10.1080/02652040400026285. [DOI] [PubMed] [Google Scholar]
- 5.Papageorgiou G. Z., Bikiaris D. N. Crystallization and melting behavior of three biodegradable poly(alkylene succinates). A comparative study. Polymer. 2005;46:12081–12092. doi: 10.1016/j.polymer.2005.10.073. [DOI] [Google Scholar]
- 6.Bikiaris D. N., Achilias D. S. Synthesis of poly(alkylene succinate) biodegradable polyesters I. Mathematical modelling of the esterification reaction. Polymer. 2006;47:4851–4860. doi: 10.1016/j.polymer.2006.04.044. [DOI] [Google Scholar]
- 7.Ranucci E., Söderqvist Lindblad M., Albertsson A. C. New biodegradable polymers from renewable sources. High molecular weight poly(ester carbonate)s from succinic acid and 1,3-propanediol. Macromol. Rapid Commun. 2000;21:680–684. doi: 10.1002/1521-3927(20000601)21:10<680::AID-MARC680>3.0.CO;2-Y. [DOI] [Google Scholar]
- 8.Liu Y., Ranucci E., Söderqvist Lindblad M., Albertsson A. C. Polyester-carbonates based on 1,3-propylene-co-1,4-cyclohexanedimethylene succinate. J. Polym. Sci. Part A: Polym. Chem. 2001;39:2508–2519. doi: 10.1002/pola.1227. [DOI] [Google Scholar]
- 9.Hartlep H., Hussmann W., Prayitno N., Meynial-Salles I., Zeng A. P. Study of two-stage processes for the microbial production of 1,3-propanediol from glucose. Appl. Microbiol. Biotechnol. 2002;60:60–66. doi: 10.1007/s00253-002-1111-8. [DOI] [PubMed] [Google Scholar]
- 10.Kim D. Y., Yim S. C., Lee P. C., Lee W. G., Lee S. Y., Chang H. N. Batch and continuous fermentation of succinic acid from wood hydrolysate by Mannheimia succiniciproducens MBEL55E. Enzyme Microb. Technol. 2004;35:648–653. doi: 10.1016/j.enzmictec.2004.08.018. [DOI] [Google Scholar]
- 11.Bikiaris D. N., Papageorgiou G. Z., Achilias D. S. Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s. Polym. Degrad. Stab. 2006;91:31–43. doi: 10.1016/j.polymdegradstab.2005.04.030. [DOI] [Google Scholar]
- 12.Umare S. S., Chandure A. S., Padney R. A. Synthesis, characterization and biodegradable studies of 1,3-propanediol based polyesters. Polym. Degrad. Stab. 2007;92:464–479. doi: 10.1016/j.polymdegradstab.2006.10.007. [DOI] [Google Scholar]
- 13.E. Karavas, E. Koutris, E. Ioannidou, E. Stathaki, and D. Bikiaris. Pharmaceutical formulation containing an HMG-CoA reductase inhibitor and method for the preparation thereof. WO 2007/131517 A1 (2007).
- 14.Ballantyne C. M., McKenney J., Trippe B. S. Efficacy and safety of an extended-release formulation of fluvastatin for once-daily treatment of primary hypercholesterolemia. Am. J. Cardiol. 2000;86:759–763. doi: 10.1016/S0002-9149(00)01076-6. [DOI] [PubMed] [Google Scholar]
- 15.Ballantyne C. M., Pazzucconi F., Pinto X., Reckless J. P., Stein E., McKenney J., Bortolini M., Chiang Y. T. Efficacy and tolerability of fluvastatin extended-release delivery system: a pooled analysis. Clin. Ther. 2001;23:177–192. doi: 10.1016/S0149-2918(01)80001-1. [DOI] [PubMed] [Google Scholar]
- 16.Beletsi A., Leontiadis L., Klepetsanis P., Ithakissios D. S., Avgoustakis K. Effect of preparative variables on the properties of PLGA-mPEG copolymers related to their application in controlled drug delivery. Int. J. Pharm. 1999;182:187–197. doi: 10.1016/S0378-5173(99)00058-7. [DOI] [PubMed] [Google Scholar]
- 17.Papageorgiou G. Z., Bikiaris D. N. Synthesis, cocrystallization and enzymatic degradation of novel poly(butylene-co-propylene succinate) copolymers. Biomacromolecules. 2007;8:2437–2449. doi: 10.1021/bm0703113. [DOI] [PubMed] [Google Scholar]
- 18.Solomon O. F., Ciuta I. Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962;6:683. doi: 10.1002/app.1962.070062414. [DOI] [Google Scholar]
- 19.Avgoustakis K., Beletsi A., Panagi Z., Klepetsanis P., Karydas A. G., Ithakisios D. S. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vitro drug resistance in blood properties. J. Control Release. 2002;79:123–135. doi: 10.1016/S0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- 20.Papageorgiou G. Z., Bikiaris D., Karavas E., Politis S., Docoslis A., Yong P., Stergiou A., Georgarakis E. Effect of physical state and particle size distribution on dissolution enhancement of nimodipine/PEG solid dispersions prepared by melt mixing and solvent evaporation. AAPS J. 2006;8(4):E623–631. doi: 10.1208/aapsj080471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L., Cui F. D., Sunada H. Preparation and evaluation of solid dispersions of nitrendipine prepared with fine silica particles using the melt-mixing method. Chem. Pharm. Bull. 2006;54:37–43. doi: 10.1248/cpb.54.37. [DOI] [PubMed] [Google Scholar]
- 22.Six K., Berghmans H., Leuner C., Dressman J., Van Werde K., Mullens J., Benoist L., Thimon M., Meublat L., Verreck G., Peeters J., Brewster M., Van den Mooter G. Characterization of solid dispersions of itraconazole and hydroxypropylmethylcellulose prepared by melt extrusion, part II. Pharm. Res. 2003;20:1047–1054. doi: 10.1023/A:1024414423779. [DOI] [PubMed] [Google Scholar]
- 23.Brucec L. D., Shahb N. H., Malickb A. W., Infeldb M. H., McGinity J. W. Properties of hot-melt extruded tablet formulations for the colonic delivery of 5-aminosalicylic acid. Eur. J. Pharm. Biopharm. 2005;59:85–97. doi: 10.1016/j.ejpb.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Jeong J. C., Lee J., Cho K. Effect of crystalline microstructure on drug release behavior of poly(ɛ-caprolactone) microspheres. J. Control Release. 2003;92:249–258. doi: 10.1016/S0168-3659(03)00367-5. [DOI] [PubMed] [Google Scholar]
- 25.Athanasiou K. A., Niederauer G. G., Agrawal C. M. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials. 1996;17:93–102. doi: 10.1016/0142-9612(96)85754-1. [DOI] [PubMed] [Google Scholar]