

Abstract
Microparticles of naproxen with Eudragit L100 and Aerosil were prepared by the emulsion solvent diffusion method in order to avoid local gastrointestinal irritation, one of the major side effects of nonsteroidal anti-inflammatory drugs after oral ingestion. The process of preparation involved the use of ethanol as good solvent, dichloromethane as a bridging liquid, water as poor solvent, Aerosil as anti-adhesion agent, and sodium dodecyl sulfate to aid in the dispersion of the drug and excipients into the poor solvent. The obtained microparticles were evaluated for micromeritic properties, yield, encapsulation efficiency, drug physical state, and drug release properties. The influence of formulation factors and preparation condition (polymer/naproxen ratio, Aerosil/polymer ratio, and the initial difference of temperature between the solvent and nonsolvent) on the properties of the microparticles were also examined. The resultant microparticles were finely spherical and uniform with high incorporation efficiency (>79%) and yield (>71%). The incorporation efficiency was enhanced with increasing the ratio of excipients to drug and the initial difference of temperature between the solvent and nonsolvent. The mean diameter of the microparticles was influenced by all of the manufacturing parameters. Studies carried out to characterize the micromeritic properties of formulations, such as flowability and packability, showed that microparticles were suitable for further pharmaceutical manipulation (e.g., capsule filling). Drug release studies of the microparticles confirmed the gastroresistance, and mathematical studies showed that the drug released followed a Hixon and Crowell kinetic. These microparticles represent a simple method for the preparation of drug-loaded enteric microparticles with desired micromeritic properties and gastroresistance release.
Key words: emulsion solvent diffusion method, Eudragit L100, naproxen
INTRODUCTION
Naproxen is a potent nonsteroidal anti-inflammatory drug (NSAID) used for its analgesic, antipyretic, and anti-inflammatory properties in humans (1). Irritation of the gastrointestinal tract (GIT) is, as with most NSAIDs, one of the major side effects reported after oral administration of naproxen (2). Since its gastrointestinal intolerance is not only related to the inhibition of the prostaglandin synthesis, but also to acute local contact of the drug with the gastric mucosa (3), the development of an enteric multiparticulate drug delivery system might reduce or even avoid the mucosal irritation. This objective could be achieved by formulating naproxen-loaded enteric microparticles which offer several additional advantages including good reproducibility of transport through the GIT and minimization of local damage to the GIT mucosal membrane because of their wide distribution over a large area, compared with larger monolithic dosage forms (4,5).
Nowadays, a considerable range of industrial applications rely on the microencapsulation of solids or liquids by polymer coating and entrapment into polymer matrices. Several microencapsulation methods are available for the preparation of the drug delivery system, which depend primarily on the drug solubility in the polymeric material. They are essentially based on solvent evaporation methods (6,7) and phase separation (8,9).
In the general microencapsulation technique using an o/w emulsion system (e.g., solvent evaporation methods), the drug is dissolved, dispersed, or emulsified in an organic polymer solution, which is then emulsified in an external aqueous or oil phase. As the organic solvent is removed by evaporation, the drug and polymer are precipitated in the droplets, thus forming the microspheres. In the phase separation method, the polymer is precipitated around a dispersed drug phase through the addition of a nonsolvent, an incompatible polymer, or a temperature change. This method results in the formation of microcapsules (core shell structure) versus microspheres (matrix structure) formed by the solvent evaporation method.
More recently, an emulsion solvent diffusion method was proposed by Kawashima et al. (10,11) and Kawashima (12) combining the agglomeration and microencapsulation of a drug during crystallization and without any binder. In this process, the drug and the polymer were dissolved in a mixture of good solvent and bridging liquid. The final solution was dispersed into an aqueous medium with constant agitation, forming o/w emulsion droplets. The good solvent diffused out of the dispersed emulsion droplets into the aqueous medium and the precipitated crystals were agglomerated simultaneously with a suitable amount of bridging liquid. The bridging liquid was immiscible with the crystallization medium and preferentially wetted the suspended crystals to form agglomerates. Compared with the solvent evaporation method for the microsphere preparation, the solidification of the liquid droplets in this process was much faster. Therefore, it could reduce the amount of emulsifier employed in the evaporation method to keep a stable o/w or w/o/w emulsion during the evaporation of the solvent. Furthermore, the use of a harmful solvent could be avoided and the reduced pressure and/or the heating to evaporate the solvent would be unnecessary in this method.
The objective of this work was to produce microparticles by the quasi-emulsion solvent diffusion method using Eudragit L100 and Aerosil and incorporating naproxen. The parameters influencing the formation of enteric microparticles such as polymer/drug ratio, Aerosil/polymer ratio, and the initial difference of temperature between the solvent and nonsolvent were investigated.
MATERIALS AND METHODS
Materials
Naproxen (Shasun Chemicals, India), Eudragit L100, Eu L100 (Rohm, Germany), Aerosil (Mingtai Chemical, Taiwan), sodium dodecyl sulfate (SDS; Sigma-Aldrich, Deisenhofen, Germany), and ethanol and dichloromethane (Merck, Germany) were used.
Preparation of the Microparticles
Naproxen (0.5 g) was dissolved with Eudragit L100 (1–2 g) in a mixed solution of ethanol (good solvent, 5 mL), and dichloromethane (bridging liquid, 5 mL) at 25°C. Then, Aerosil (0.3 and 0.6 g) was suspended uniformly in the drug/polymer solution under vigorous agitation. The resultant drug/polymer/Aerosil suspension was poured into 100 mL distilled water containing 0.4% of SDS (poor solvent) under agitation (800 rpm) and thermally controlled at 0°C and 25°C. After agitating the system for 60 min, the solidified microparticles were recovered by filtration. The resultant products were dried in an oven at 50°C for 6 h. The microparticle formulations are shown in Table I.
Table I.
Formulation and Process Conditions for the Microparticles Preparation
| Aerosil/drug | Nonsolvent temperature (°C) | Batch |
|---|---|---|
| 0.6 | 25 | 1 |
| 0.3 | 25 | 2 |
| 0.6 | 0 | 3 |
Determination of Drug Loading and Incorporation Efficiency of Microparticles
The weighed amount of microparticle powder was dissolved in ethanol and the drug content was measured spectrophotometrically at 330 nm for naproxen. The drug loading and incorporation efficiency (in percent) were calculated by using Eqs. 1 and 2, respectively:
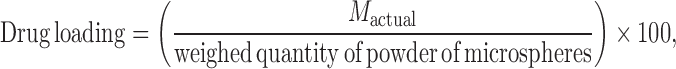 |
1 |
 |
2 |
where Mactual is the actual naproxen content in weighed quantity of powder of microparticles and Mtheoretical is the theoretical amount of naproxen in microparticles calculated from the quantity added in the fabrication process. The means of three assays were reported.
In Vitro Release Studies
The in vitro dissolution/release of naproxen as pure drug and microparticles was determined with a USP rotating paddle method (900 mL 0.1 N HCl or pH 7.4 phosphate buffer; 100 rpm; 37°C; n = 3). At preset time intervals, aliquots were withdrawn and replaced by an equal volume of dissolution medium to maintain constant volume. After suitable dilution, the samples were analyzed spectrophotometrically at 330 nm. The kinetics data obtained from release rates were also evaluated.
X-Ray Powder Diffraction
In order to investigate polymorphism, X-ray was used. The cavity of the metal sample holder of the X-ray diffractometer was filled with the ground sample powder and then smoothed with a spatula. The X-ray diffraction pattern of naproxen samples was obtained using the X-ray diffractometer (Siemens, Model D5000, Germany) at 40 kV, 30 mA and a scanning rate of 0.06°/min over the range 2θ = 5–40, using CuKα1 radiation of wavelength 1.5405 Å.
Differential Scanning Calorimetry
The thermal characteristics of the microparticles and pure drug were determined using a differential scanning calorimeter (DSC60, Shimadzu, Japan). After calibration with indium and lead standards, samples of the crystals (3–5 mg) were heated (range 25–300°C) at 10°C/min in crimped aluminum pans under a nitrogen atmosphere.
Scanning Electron Microscopy
The morphology of crystals was examined using a scanning electron microscope (SEM; LEO 440i, Cambridge, UK) operating at 15 kV. The samples were sputter-coated with gold before examination.
Powder Flow Measurement
Flowability of the crystals was assessed by determining the compressibility index (Carr index) and the angle of repose. The Carr index (13) is a measure of the propensity of a powder to consolidate. Changes occurring in packing arrangement during the tapping procedure are expressed as the Carr index (Eq. 3):
 |
3 |
where ρt and ρb are the tap and bulk densities of the powder bed, respectively.
The Carr index reflects the compressibility of the powders, and there is a correlation between the compressibility index and the flowability of the crystals. The angle of repose was measured by a fixed funnel method (14). The results presented are the mean value of six determinations.
Size and Roundness Determination of Particles
A small amount of powder (about 20 mg) was suspended in mineral oil (Sigma Chemical, St. Louis, USA) and the suspension was spread onto a microscope slide. A cover slip was applied, allowing the suspension to settle homogeneously between the two glass surfaces. Particle size and shape were assessed in tandem using image analysis software (designed in-house at King’s College London) installed on an Archimedes computer, which was attached to an optical microscope (NikonLabophot, Tokyo, Japan) via a miniature video camera.
Hundreds of particles were measured in each sample and the surface volume mean diameter and roundness were recorded. Roundness was calculated using the following equation:
 |
4 |
Packability Determination
Twenty-five grams of the sample was poured slowly and gently into a 25-mL measuring cylinder and tapped up to 1,200 times. The packability was evaluated by the tapped density according to the Kawakita and Ludde (15) equation as follows:
 |
5 |
where n is the tap number, C denotes the volume reduction which can be calculated according to the Eq. 6, 1/a defines the degree of volume reduction at the limit of tapping, termed compactibility, and 1/b is a constant related to cohesion, termed cohesiveness (16):
 |
6 |
where V0 and Vn are the powder bed volumes at the initial and nth tapped state, respectively.
The plot of n/C versus n is linear and the compactibility 1/a and cohesiveness 1/b are obtained from the slope (1/a) and the intercept (1/ab) of the plot of the modified Kawakita and Ludde equation (15). The correlation coefficients of Eq. 5 for each sample were listed in Table II.
Table II.
Mean Diameter, Angle of Repose, and Carr’s Index of Microparticles at Various Polymer/Drug Ratios (±SD, n = 3)
| Samples | Untreated | Treated without additives | Microparticles | |||
|---|---|---|---|---|---|---|
| Batch | Polymer/drug ratio | |||||
| 2:1 | 3:1 | 4:1 | ||||
| Arithmetic mean diameter±σ (mm) | 0.01 ± 0.02 | 0.55 ± 0.05 | 1 | 1.15 ± 0.11 | 1.32 ± 0.10 | 1.40 ± 0.10 |
| 2 | 1.35 ± 0.12 | 1.42 ± 0.12 | 1.42 ± 0.12 | |||
| 3 | 1.40 ± 0.13 | 1.40 ± 0.10 | 1.42 ± 0.10 | |||
| Bulk density (g/cm3) | 0.34 ± 0.01 | 0.23 ± 0.01 | 1 | 0.22 ± 0.01 | 0.29 ± 0.02 | 0.30 ± 0.01 |
| 2 | 0.18 ± 0.02 | 0.22 ± 0.01 | 0.22 ± 0.02 | |||
| 3 | 0.23 ± 0.02 | 0.31 ± 0.02 | 0.26 ± 0.02 | |||
| Angle of repose (°) (n = 6) | 58.7 ± 0.6 | 36.5 ± 0.5 | 1 | 33.3 ± 1.3 | 27.2 ± 0.6 | 23.9 ± 0.5 |
| 2 | 34.7 ± 1.1 | 30.3 ± 0.5 | 26.1 ± 0.5 | |||
| 3 | 25.1 ± 1.5 | 21.6 ± 0.6 | 29.2 ± 0.6 | |||
| Carr’s index (%) | 51.7 ± 0.1 | 26.8 ± 0.6 | 1 | 9.1 ± 0.7 | 10.8 ± 0.7 | 6.9 ± 0.6 |
| 2 | 13.3 ± 0.7 | 8.3 ± 0.5 | 12.6 ± 0.7 | |||
| 3 | 10.2 ± 0.6 | 7.9 ± 0.6 | 8.5 ± 1.1 | |||
| a | 0.53 (r = 0.998) | 0.298 (r = 0.998) | 1 | 0.295(r = 0.998) | 0.126(r = 0.998) | 0.087(r = 0.998) |
| 2 | 0.158(r = 0.998) | 0.082(r = 0.998) | 0.149(r = 0.998) | |||
| 3 | 0.197(r = 0.998) | 0.092(r = 0.998) | 0.096(r = 0.998) | |||
| b | 0.061 | 0.02 | 1 | 0.002 | 0.019 | 0.028 |
| 2 | 0.014 | 0.026 | 0.037 | |||
| 3 | 0.004 | 0.032 | 0.042 | |||
RESULTS AND DISCUSSION
Preparation Process of the Microparticles
When the resultant mixed solution of good solvent and bridging liquid containing drug and additives was poured into the poor solvent under stirring, finely dispersed droplets were formed immediately and semitransparent emulsion were observed visually. With the diffusion of the good solvent out of the droplets into the poor solvent, the drug and the polymer coprecipitated in the droplets. The dichloromethane commixed into the good solvent, bridged the drug, Aerosil, and polymer in the droplets, and the droplets solidified gradually into the microparticles. The formation of the microparticles could be described in the following processes: the formation of quasi-emulsion droplets, the diffusion of the organic solvent, and the solidification of the droplets. In the previous researches (17,18) for the preparation of microparticles with methacrylic copolymer (Eudragit L100, S100, RS, and RL) using the quasi-emulsion solvent diffusion method, a large ratio of drug to Eudragit (>4/1) and also a large amount of organic solvent had to be used, since the polymer revealed a high viscosity during the formation of droplets and resulted in droplets often agglomerating into an irregular mass and adhering to the propeller or the vessel wall. In this study, however, these problems could be avoided effectively due to the good anti-adhesion property of Aerosil. During the preparation process of microparticles, Aerosil commixed with the polymer uniformly. A decrease in viscosity prevented conglutination between the emulsified droplets. It is a good anti-adhesion agent against the viscous characteristic of polymers. At the same time, Aerosil could accelerate the solidification of droplets and be packed in the microparticles well. It was also found that introducing proper amount of additives into the distilled water was a favorable method to prevent coalescence of the droplets. In this study, the distilled water containing SDS (0.4% w/v) was selected as a poor solvent and spherical microparticles of naproxen were prepared successfully as seen in Fig. 1, the microparticles were invariably spherical, though the plasticizer was not added in this formulation. It was indicated that Eu L100 was one of the suitable polymers for the preparation of microparticles using this method due to its good plastic deformation property.
Fig. 1.

Scanning electron micrographs of samples: untreated naproxen (I, II) and from batch 1 (III, IV) using polymer/drug ratio 2:1
Micromeritic Properties of the Microparticles
The scanning microphotographs of the original crystals of naproxen and naproxen/Eu L100/Aerosil microparticles are shown in Fig. 1. It can be observed that the original particles having an irregular shape (Fig. 1, I) can be spherically agglomerated and simultaneously microencapsulated by the quasi-emulsion solvent diffusion process. Sphericity of the microparticles is confirmed by comparing the roundness of the microparticles, which is close to 1, with the roundness of the untreated sample (1.50).
The surface topography of the microparticles is shown in Fig. 1, IV. It was observed that the Aerosil particles and polymer (amorphous) were commixed uniformly in dense texture and naproxen crystals were not observed visually.
Table II shows that the mean diameters of the microparticles were approximately 115–140 times higher than those of the untreated crystals and the mean diameter of the microparticles was influenced by the manufacturing parameters. According to the results, larger particles were obtained at higher polymer/drug ratios. Increasing the polymer load leads to a more viscous solution and, when the viscous polymeric solution is poured into the aqueous phase, larger droplets and thus larger microparticles are formed (17,19,20).
Table II also shows that the mean diameters from batch 3 at all polymer/drug ratios were significantly greater than those produced from batch 1 (p < 0.05).
The lower temperature of the nonsolvent at batch 3 could result in an increase in the rate of polymer and drug solidification. An increased solidification rate would also tend to reduce the time available for subdivision of larger globules into smaller ones during stirring. As a result, the globules of the internal phase were larger in diameter than batch 1 on solidification (21). Therefore, the temperature of the system was an important factor determining the properties of the resultant microparticles, and it should be controlled properly in the preparation process (22).
In the case of batch 2 where the mean microparticle diameters were significantly higher than batch 1 at all polymer/drug ratios, the decreased Aerosil/polymer ratio possibly increased the viscosity of the polymeric solution and, consequently, the microparticles’ diameter as discussed above.
The bulk density of all the spherical microparticles is lower than those of the untreated naproxen (Table II), although their particle size and sphericity, in general, are higher. Therefore, these low densities should be related to the intraparticle porosity or particle density. A reduction in bulk densities of the treated samples indicates the greater porosity within the microparticles.
Table II also shows that the flowability that represented in terms of the angle of repose and Carr’s index in the microparticles was much improved compared to those of the original powders (untreated naproxen).
The microparticles were easily packed by tapping, the process which was described by Kawakita and Ludde’s equation (15). As shown in Table II, the smaller values of parameter a in Kawakita and Ludde’s equation for the microparticles indicated their higher packability than the untreated naproxen. The apparent packing velocity by tapping, represented by parameter b, for microparticles was faster than that for conventional crystals, since the microparticles were packed closely even without tapping due to their excellent flowability and packability.
On the basis of these findings, it could be concluded that good flowability and packability for microparticles may be attributed to the spherical shape and the bigger particle size. These results suggest that the microparticles might be suitable for capsule filling and the capsules formed from them would attain uniformity in weight because of their very good flowability and packability of the microparticles.
Yield, Drug Loading, and Encapsulation Efficiency
The results of the determination of microparticles’ yield, drug loading, and encapsulation efficiency for various polymer/drug ratios are shown in Table III. The yield of microparticles was over 71% and the encapsulation efficiency over 79% (w/w) irrespective of the drug loading. The yield and average drug loading was in rank order of batch 3>batch 1>batch 2.
Table III.
Microparticle Yield, Drug Loading, and Encapsulation Efficiency (%) at Various Polymer/Drug Ratios (±SD, n = 3)
| Samples | Batch | Polymer/drug ratio | ||
|---|---|---|---|---|
| 2:1 | 3:1 | 4:1 | ||
| Yield (%) | 1 | 73.2 ± 1.2 | 76.9 ± 0.89 | 80.6 ± 1.9 |
| 2 | 71.2 ± 1.5 | 74.6 ± 2.1 | 79.7 ± 2.2 | |
| 3 | 77.3 ± 0.94 | 80.5 ± 1.1 | 83.4 ± 1.4 | |
| Drug loading (%) | 1 | 23.9 ± 0.2 | 19.1 ± 0.1 | 16.3 ± 0.1 |
| 2 | 23.0 ± 0.1 | 18.9 ± 0.3 | 16.0 ± 0.2 | |
| 3 | 24.3 ± 0.3 | 19.9 ± 0.1 | 16.7 ± 0.2 | |
| Encapsulation efficiency (%) | 1 | 82.8 ± 0.5 | 87.8 ± 0.6 | 89.5 ± 0.4 |
| 2 | 79.1 ± 0.3 | 85.9 ± 0.3 | 88.5 ± 0.6 | |
| 3 | 87.4 ± 0.2 | 89.6 ± 0.2 | 91.1 ± 0.4 | |
This may have been due to their faster rates of solidification resulting from the use of a higher ratio of Aerosil/drug (in batch 1 compared to batch 2) and a lower nonsolvent temperature (in batch 3 compared to batch 1). Moreover, a fast solidification rate may reduce the partitioning of drug and additives into the external phase of the emulsion. Consistent with the higher yields for batch 3, higher encapsulation efficiencies were also produced from this batch. Losses for the relatively insoluble naproxen may be due to sedimentation of the drug prior to drop formation and some dissolution during the hardening process (23). According to this fact, we suppose that any factor which could modify the time needed to reach the physical state transition boundaries of the polymeric solution (solution–gel–glass) during the hardening process will be effective in the polymer precipitation and, consequently, in encapsulation efficiency results (17). The polymer/drug ratio can be a critical factor during microparticle formation.
The significant enhancement in the drug entrapment was observed when the polymer concentration increased. For instance, the naproxen encapsulation efficiency was increased from 79% to 88.5% as the polymer/dug ratio was increased from 2:1 to 4:1 at batch 2. This improved encapsulation efficiencies may be simply due to the greater proportion of polymer with respect to the amount of drug.
On the other hand, the increase in polymer load leads to a shorter time for the composition of the polymer solution to reach the viscous (gelation) boundary, resulting in a rapid film-like membrane formation on the droplets periphery. If the film-like polymeric membrane is quickly solidified, the microparticle structure is more fixed and thus solvent and nonsolvent counterdiffusion is delayed. As a consequence, less water may be allowed to diffuse into the dispersed phase and less drug will be carried by the solvent into the aqueous phase (17).
According to Table III, a reduction in encapsulation efficiency was observed by reduction of the ratio of Aerosil to polymer (batch 2 compared to batch 1).
Aerosil, used in this study, is known to accelerate the solidification of droplets (24). Therefore, the slower solidification of the polymeric shell at the droplet periphery due to decreasing Aerosil/polymer ratio delays the creation of diffusional barrier to mass transfer, increasing ethanol and water counter diffusion, and consequently, the amount of dissolved drug carried by the solvent into the aqueous phase.
It has been shown that decreasing the nonsolvent phase temperature enhances polymer precipitation and the further crystallization of the drug dissolved (17). Based on this fact, it could be concluded that decreasing the temperature of the nonsolvent phase in batch 3 increases the drug entrapment compared to batch 1 by helping to promote solidification of the dispersed droplets. Moreover, decreasing nonsolvent temperature would reduce the solubility of naproxen possibly causing a reduction in the rate of drug partitioning into the aqueous phase.
In Vitro Release of Naproxen
To evaluate the pH-dependent release profiles of naproxen from the samples, in vitro release tests were performed under sink condition in 0.1 N HCl and pH 7.4 phosphate buffer. In the dissolution medium at pH 1.2, nearly 22% of the drug was released in 2 h (Fig. 2) and after 10 min at pH 7.4 nearly 90% of the drug was released from untreated sample (Fig. 3). As clearly shown in Fig. 2, a slow drug release behavior was observed for microparticles at pH 1.2. Since the polymer is insoluble in the release media with pH 1.2, the microparticles were only slightly swollen and remained intact in this case.
Fig. 2.

Release of naproxen from microparticles into 0.1 N HCl from batch 1 using polymer/drug ratios 2:1, 3:1, and 4:1 (±SD, n = 3)
Fig. 3.

Release of naproxen from microparticles into pH 7.4 phosphate buffer from batch 1 using polymer/drug ratios 2:1, 3:1, and 4:1 (±SD, n = 3)
At a pH of 7.4, the polymer dissolved rapidly and the microparticles’ disintegration resulted in a faster drug release rate compared with pH 1.2. It can be seen that incorporation of naproxen into the anionic polymer Eu L100 led to a retardation of the dissolution rate of the drug (Fig. 3). The retarding effect on drug release is frequently attributed to the creation, by the polymer, of a diffusional barrier between the drug and the dissolution medium (10,25). This behavior can explain the slower release of naproxen from microparticles compared to that obtained from agglomerated crystals without the polymer.
The results showed that the release rate of naproxen from the microparticles could be modulated with adjusting the ratios of polymer/drug in the formulation. As seen in Fig. 3, decreasing the ratio of Eu/drug from 4:1 to 3:1 resulted in a marked increase in drug release rate. In addition to the retarding effect of the polymer, this result could be attributed to the hardening sequence of microparticles.
During the formation of microparticles, the low viscosity of the dispersed phase delayed polymer precipitation and the complete solidification of the dispersed droplets. Due to this fact, more water could diffuse into the droplets before their solidification, thus forming more water pockets (pores). A higher number of pores will allow for easier penetration of dissolution medium and consequent dissolution of naproxen from the microparticles into the dissolution medium (17). Moreover, the size of the microparticles decreased significantly with decreasing polymer/drug ratio, which also contributed to the faster release of drug at lower polymer/drug ratios.
The microparticles at polymer/drug ratio of 2:1 showed rapid burst release followed by the more constant release thereafter, whereas burst release was not noticeable at 3:1 and 4:1 ratios. The unexpected biphasic release profile of naproxen observed at polymer/drug ratio of 2:1 may be explained by the heterogeneous distribution of the drug within the matrix. Whereas the higher proportions of polymer at the 3:1 and 4:1 polymer/drug ratios may have been sufficient to entrap the drug completely and distribute it evenly.
As seen in Fig. 4, the rate of drug release from the microparticles of batch 2 and batch 3 at 2:1 polymer/drug ratio was significantly slower than that produced from batch 1. The inhibitory effect on the drug release rate observed with decreasing nonsolvent temperature (batch 3) may be explained by the influence of temperature on the microparticle hardening sequence. Decreasing nonsolvent temperature enhances polymer precipitation, favoring a rapid solid crust formation at the droplet surface. The faster solidification of the dispersed droplets causes forming fewer pores and, consequently, reducing the dissolution rate of naproxen from the microparticles, as discussed above.
Fig. 4.

Release of naproxen from microparticles into pH 7.4 phosphate buffer from batches 1, 2, and 3 using polymer/drug ratio 2:1 (±SD, n = 3)
Aerosil was considered to be helpful to promote the dispersibility of the drug in the microparticles and, consequently, accelerate the drug release rate (26). This could account for the slightly faster drug release rate obtained in batch 1 compared with batch 2. Although Aerosil had large specific surface area for the drug dispersion, there was slight increase in the drug release rate by increasing the proportion of Aerosil which may be due to its property of insolubility (22) and its accelerating effect on the hardening sequences of microparticles (as discussed previously).
No noticeable differences could be observed in the drug release behavior of various batches at 3:1 or 4:1 polymer/drug ratios. This may be due to fast solidification of the microparticles at these ratios because of high polymer/drug ratio which possibly make the effect of Aerosil/polymer and nonsolvent temperature on dissolution profile negligible.
To characterize the release mechanism of naproxen from the microparticles, three kinetic models were applied, i.e., Wagner, Hixon and Crowell, and Higuchi (27). According to the data obtained from release in phosphate buffer, the best fit was obtained with the Hixon and Crowell model. The fact that the release obeys the cube root law suggests that the release of naproxen is governed by the dissolution of the microparticles.
DSC
The DSC curves and X-ray diffraction pattern of the microparticles are shown in Figs. 5 and 6, respectively. As shown in Fig. 5, the melting peak of naproxen in the microparticles disappeared gradually with increasing the ratio of Eu L100 to drug in the formulation. When the ratio of Eu L100 to drug was 4:1, no melting peak of the drug was found in the microparticles, though the melting peak was observed in the physical mixtures of the drug and excipients with the same formulation. These indicated that naproxen had been disordered in the microparticles at 4:1 polymer/drug ratio, suggesting that naproxen had been highly dispersed at this ratio, so as amorphous state. This was further supported by the X-ray analysis data given in Fig. 6, which shows the representative diffraction patterns for the pure naproxen, the microparticles (polymer/drug ration of 4:1), and physical mixture (polymer/drug ratio of 4/1). No crystalline peaks of naproxen were found in the microparticles at 4:1 polymer/drug ratio, but crystalline peaks were observed in the physical mixtures of the drug and excipients with the same ratio. A decrease in the intensity of the peaks with the physical mixture is only due to a lower loading of the drug per unit weight of the physical mixture compared to pure drug. The scans and X-ray powder diffraction (XRPD) patterns are similar for all batches of microparticles produced at all polymer/drug ratios and hence not all of them are shown. In summary, the DSC and XRPD data indicated no sign of interaction between the drug and the additives and showed only a change in crystallinity of naproxen present in microparticles.
Fig. 5.

DSC scans of untreated naproxen, Aerosil, Eu L100, and microparticles produced using batch 1 at polymer/drug ratios of 2:1, 3:1, and 4:1 and physical mixture of drug/polymer (4:1)
Fig. 6.

The X-ray diffraction patterns of untreated naproxen, Eu L100, and microparticles produced using batch 1 at a polymer/drug ratio of 4:1 and physical mixture of drug/polymer (4:1)
CONCLUSION
Naproxen was successfully encapsulated in the enteric microparticles by the quasi-emulsion solvent diffusion method. Microparticles with high drug encapsulation efficiency, spherical shape, and almost monodispersed particle size distribution could be prepared. The rate of polymer solidification appeared to be the main factor that determined the efficiency of drug encapsulation. The resultant microparticles have the desired micromeritic properties (good flowability and packability) and were suitable to be incorporated into capsules, while the in vitro release profiles confirmed their gastroresistance, thus allowing pH-dependent release of naproxen in the GIT. The release rate of naproxen from the microparticles could be modulated as desired by adjusting the formulation of the microparticles and preparation conditions. Finally, the main advantages of this technique are the one-step production method, since it does not need a coating process, and being inexpensive and simple.
References
- 1.Todd P. A., Clissold S. P. Naproxen : a reappraisal of its pharmacology., and use in rheumatic disease and pain states. Drugs. 1990;40:91–137. doi: 10.2165/00003495-199040010-00006. [DOI] [PubMed] [Google Scholar]
- 2.Sweetman S. C. Martindale. 33. London, Chicago: Pharmaceutical Press; 2002. p. pp 60. [Google Scholar]
- 3.Bjorkman D. J. Nonsteroidal anti-inflammatory drug-induced gastrointestinal injury. Am. J. Med. 1996;101:25S–32S. doi: 10.1016/s0002-9343(96)00135-0. [DOI] [PubMed] [Google Scholar]
- 4.Eskilson C. Controlled release by microencapsulation. Manuf. Chem. 1985;56(3):33–39. [Google Scholar]
- 5.Mesiha M. S., Valles J. A screening study of lubricants in wet powder masses suitable for extrusion–spheronization. Drug Dev Ind Pharm. 1993;19(8):943–959. doi: 10.3109/03639049309062993. [DOI] [Google Scholar]
- 6.M. Donbrow (ed.). Microcapsules and nanoparticles in medicine and pharmacy, chapter 2, CRC, Boca Raton, FL, 1992, pp. 17–46.
- 7.Jeyanthi R., Thanoo B. C., Metha R. C., DeLuca P. P. Effect of solvent removal technique on the matrix characteristics of polylactide/glycolide microspheres for peptide delivery. J. Control. Release. 1996;38:235–244. doi: 10.1016/0168-3659(95)00125-5. [DOI] [Google Scholar]
- 8.Madan P. L. Microencapsulation. 1. Phase separation or coacervation. Drug. Dev. Ind. Pharm. 1978;4:95–116. doi: 10.3109/03639047809055641. [DOI] [Google Scholar]
- 9.Palmieri G. F., Martell S., Lauri D., Wehrle P. Gelatin-acacia complex coacervation as a method for ketoprofen microencapsulation. Drug. Dev. Ind. Pharm. 1996;22(9/10):951–957. doi: 10.3109/03639049609065925. [DOI] [PubMed] [Google Scholar]
- 10.Kawashima Y., Niwa T., Takeuchi H., Hino T., Itoh Y. Hollow microspheres for use as a floating controlled drug delivery system in the stomach. J. Pharm. Sci. 1992;81(2):135–140. doi: 10.1002/jps.2600810207. [DOI] [PubMed] [Google Scholar]
- 11.Kawashima Y., Iwamoto T., Niwa T. Size control of ibuprofen microspheres with an acrylic polymer by changing the pH in an aqueous dis-persion medium and its mechanism. Chem. Pharm. Bull. 1993;41(1):191–195. [Google Scholar]
- 12.Y. Kawashima. In: D. Chulia, M. Deleuil. Y. Pourcelot (Eds.) Powder Technology and Pharmaceutical Processes, Elsevier, Amsterdam, Netherland, 1994, pp. 493–511.
- 13.Carr R. L. Evaluating flow properties of solids. Chem. Eng. 1965;72:163–168. [Google Scholar]
- 14.Carr R. L. Classifying flow properties of solids. Chem. Eng. 1965;72:69–72. [Google Scholar]
- 15.Kawakita K., Ludde K. H. Some considerations on powder compression equations. Powder Technol. 1970/1971;4:61–68. doi: 10.1016/0032-5910(71)80001-3. [DOI] [Google Scholar]
- 16.H. Vromans. Studies on consolidation and compaction properties of lactose, Ph.D. Thesis, University of Groningen, Groningen, The Netherlands (1987).
- 17.Re M. I., Biscans B. Preparation of microparticles of ketoprofen with acrylic polymers by a quasi-emulsion solvent diffusion method. Powder Technol. 1999;101:120–133. doi: 10.1016/S0032-5910(98)00163-6. [DOI] [Google Scholar]
- 18.Kachrimanis K., Nikolakakis I., Malamataris S. Spherical crystal agglomeration of ibuprofen by the solvent-change technique in presence of methacrylic polymers. J. Pharm. Sci. 2000;89(2):250–258. doi: 10.1002/(SICI)1520-6017(200002)89:2<250::AID-JPS12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 19.Yang J. F., Qiu L. Y., Jin Y., Zhang J. X. Thymosin-loaded enteric microparticles for oral administration: preparation and in vitro release studies. Int. J. Pharm. 2005;301:41–47. doi: 10.1016/j.ijpharm.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Scully D. B. Scale-up in suspension polymerization. J. Appl. Polym. Sci. 1976;20:2299–2302. doi: 10.1002/app.1976.070200826. [DOI] [Google Scholar]
- 21.Thomopson C. J., Hansford D., Higgins S., Rostron C., Hutcheon G. A., Munday D. L. Evaluation of ibuprofen-loaded microspheres prepared from novel copolyesters. Int. J. Pharm. 2007;329:53–61. doi: 10.1016/j.ijpharm.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 22.Cui F., Yang M., Jiang Y., Cun D., Lin W., Fan Y., Kawashima Y. Design of sustained-release nitrendipine microparticles having solid dispersion structure by quasi-emulsion solvent diffusion method. J. Control. Release. 2003;91:375–384. doi: 10.1016/S0168-3659(03)00275-X. [DOI] [PubMed] [Google Scholar]
- 23.Zaniboni H. C., Fell J. T., Collett J. H. Production and characterization of enteric beads. Int. J. Pharm. 1995;125:151–155. doi: 10.1016/0378-5173(95)00146-A. [DOI] [Google Scholar]
- 24.Yang M. S., Cui F., You B., Fan Y., Wang L., Yue P., Yang H. Preparation of sustained-release nitrendipine microparticles with Eudragit RS and Aerosil using quasi-emulsion solvent diffusion method. Int. J. Pharm. 2003;259:103–113. doi: 10.1016/S0378-5173(03)00209-6. [DOI] [PubMed] [Google Scholar]
- 25.Ribardiere A., Tchoreloff P., Couarraze G., Puisieux F. Modification of ketoprofen bead structure produced by the spherical crystallization technique with a two-solvent system. Int. J. Pharm. 1996;144:195–207. doi: 10.1016/S0378-5173(96)04746-1. [DOI] [Google Scholar]
- 26.Gao Y., Cui F., Guan Y., Yang L., Wang Y., Zhang L. Preparation of roxithromycin-polymeric microparticles by the emulsion solvent diffusion method for taste masking. Int. J. Pharm. 2006;318:62–69. doi: 10.1016/j.ijpharm.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Brossard C., Wouessidjewe D. Control de dissolution des forms pharmaceutiques orals solids a liberation ralenite. STP Pharma. 1990;6:728–741. [Google Scholar]