

Abstract
The purpose of this study was to combine the advantages of self-nanoemulsifying drug delivery systems and tablets as a conventional dosage form emphasizing the excipients’ effect on the development of a new dosage form. Systems composed of HCO-40, Transcutol® HP, and medium-chain triglyceride were prepared. Essential properties of the prepared systems regarding carvedilol solubility, a model drug, and self-emulsification time were determined. In order to optimize self-nanoemulsifying drug delivery systems (SNEDDS), formulation dispersion–drug precipitation test was performed in the absence and presence of cellulosic polymers. Furthermore, SNEDDS was loaded onto liquisolid powders. P-glycoprotein (P-gp) activity of the selected SNEDDS was tested using HCT-116 cells. Carvedilol showed acceptable solubility in the selected excipients. It also demonstrated improvement in the stability upon dilution with aqueous media in the presence of cellulosic polymers. Use of granulated silicon dioxide improved the physical properties of liquisolid powders containing SNEDDS. It improved the compressibility of the selected powders and the tested SNEDDS showed marked P-gp inhibition activity. Prepared self-nanoemulsifying tablet produced acceptable properties of immediate-release dosage forms and expected to increase the bioavailability of carvedilol.
Key words: carvedilol, dry nanoemulsion, granulated silicon dioxide, liquisolid tablets, SEDDS, SNEDDS, SNET
INTRODUCTION
Oral drug delivery remains the most popular route of administration. However, to achieve successful therapeutic outcomes, several approaches were developed to increase the dissolution rate and thereby oral absorption and bioavailability of poorly water-soluble drugs. Some of these techniques were solid dispersion, anti-solvent, complexation with cyclodextrin, and lipid-based formulations (1–4). Self-emulsifying drug delivery systems (SEDDS) are one of the lipid formulations which represent an attractive alternative to orally administered emulsions since they are physically stable lipid solutions or dispersions (5). SEDDS are isotropic mixtures of natural or synthetic oils, solid or liquid surfactants, and, alternatively, one or more hydrophilic solvents and co-solvents/surfactants (6). Drug substances with adequate solubility in lipid/surfactants/co-solvent (or co-surfactant) blends are candidates for this formulation concept. SEDDS spread readily in the gastrointestinal tract, while the digestive motility of the stomach and intestine provides the agitation necessary for self-emulsification–dispersion process (5,7,8). Benita et al. described SEDDS as systems that produce emulsions with a droplet size between 100 and 300 nm while self-microemulsifying drug delivery systems (SMEDDS) form transparent microemulsions with a droplet size of less than 50 nm (6). However, SEDDS generally refers to all types of self-emulsifying systems unless otherwise described, while self-nanoemulsifying drug delivery systems (SNEDDS) describe systems which form nanoemulsions upon dispersion in aqueous media.
Generally, SEDDS are either administered as liquid dosage forms or incorporated in a soft gelatin capsules. However, it is a fact that solid dosage forms are preferred more than liquid preparations for many reasons including: facility of manufacturing process, convenience to the patient, accuracy, and stability. Incorporation of lipid formulations into solid dosage forms combines the advantages of lipid-based drug delivery systems with those of solid dosage forms thus overcoming the drawbacks of liquid formulations. Some trials were made to formulate liquid SEDDS into solid dosage forms (9–14). One of the most known techniques, liquisolid compacts, is used to transfer liquid medication into acceptably flowing and compressible powders.
Carvedilol, an inherently long-acting beta-blocker, was classified according to the Biopharmaceutical Classification System as a drug with low solubility and it is presented as an immediate-release dosage form in the World Health Organization essential drug list (15,16). Carvedilol has been studied in patients with heart failure, hypertension, and ischemic heart diseases being available in the market in 3.125-, 6.25-, 12.5-, and 25-mg tablets (17). Its serum concentration is not only affected by its low solubility but also by P-glycoprotein (P-gp) activity and first pass metabolism (18,19).
This study was based on subsequent steps. First step included preparation and evaluation of self-emulsifying drug delivery systems. Carvedilol was incorporated into SEDDS composed of different ratios of polyoxyl-40 hydrogenated castor oil, medium-chain triglycerides, and diethylene glycol monoethyl ether. These ingredients were chosen as:
Polyoxyl-40 hydrogenated castor oil. HCO-40 is a nonionic surfactant which has P-gp inhibition activity and an absorption enhancement effect and possesses better emulsification efficiency when compared to Tween 80 (20–22). Presence of HCO-40 in the microemulsion structure, being dispersed in the gastric content, will allow a portion of the added surfactants to be located at the O/W interface. Therefore, the concentration of the free surfactant in the emulsion water phase is probably much lower than its nominal concentration in the entire emulsion, thus decreasing the toxic effect which is attributed to the free surfactant (23). In addition, polyoxyl-40 hydrogenated castor oil is widely used in different oral preparations (24).
Medium-chain triglyceride (MCT), Migliol® 812, has P-gp inhibition activity, good fluidity, and proper self-emulsification properties and it is efficiently digested (25–27).
Diethylene glycol monoethyl ether, Transcutol® HP, as a co-solvent, is considered as a component that decreases the fluidity of SEDDS, enhances drug incorporation into the SEDDS, improves self-emulsification properties, and possesses penetration enhancement effect (22,28,29). Cellulosic polymers were added to the SEDDS to study their effect as drug precipitation inhibitors (30).
Additionally, incorporating one of the prepared systems in liquisolid tablets was done using the selected powders to produce self-nanoemulsifying tablets (SNET). The results showed successful incorporation of carvedilol within the SNEDDS, which also improved its stability upon addition of cellulosic polymers. Use of granulated SiO2 was able to reduce the amount of powder needed to transfer SNEDDS into free-flowing compressible powder. The appropriate choice of excipients reduced the weight of the produced tablets and ensured drug stability within the gastrointestinal condition. SNET presented a successful dosage form that incorporated a drug dissolved in a liquid SNEDDS.
MATERIALS AND METHOD
Materials
Polyoxyl-40 hydrogenated castor oil, HCO-40, was provided by CISME (Italy). MCT (Miglyol® 812) was a gift from Sasol (Germany). Diethylene glycol monoethyl ether, Transcutol® HP, was kindly supplied by Gattefosse (France). Carvedilol was provided by Hetero drugs (India). HPMC 5cp (Methocel® E5 LV), HPMC 15cp (Methocel® E15 LV), and methyl cellulose 15cp (Methocel® A15 LV, MC) were kindly provided by Colorcon (UK). Aerosil® 200 pharma (amorphous silicone dioxide) and Aeroperl® 300 pharma (granulated silicon dioxide) were obtained from Degussa. Ac-Di-Sol® (crosscarmellose sodium) was purchased from FCM Corp. (USA). Microcrystalline cellulose (Vivapur® PH 102, MCC) was obtained from JRS (Germany). Human colon carcinoma cells (HCT-116) were purchased from the American Type Culture Collection (VA, USA). All cell culture materials were obtained from Cambrex, BioScience (Copenhagen, Denmark), while all other fine chemicals were from Sigma-Aldrich (CT, USA).
Preparation and Evaluation of SEDDS and SNEDDS
Construction of ternary-phase diagram
Different ratios of HCO-40 (surfactant), Transcutol® HP (co-solvent), and MCT (oil) were mixed in order to prepare different systems. The prepared systems were visually analyzed for any phase separation under storage for 72 h at ambient temperature.
One hundred milligrams of each system were introduced into a beaker containing 20 ml of 0.1 N HCl at 37°C and the contents were stirred using a magnetic stirrer at 125 rpm. The clarity of the formed aqueous dispersion was visually assessed using the following grading to identify the tested systems:
-
A
Denoting the formation of a clear microemulsion
-
B
Denoting the formation of a translucent microemulsion
-
C
Represent the formation of a slightly less clear emulsion which had a bluish white appearance
-
D
Denoting the formation of a bright white emulsion (similar in appearance to milk)
-
E
Denoting the formulations which exhibited either poor emulsification with large oil droplets on the surface or the emulsion was not formed.
Phase diagram was constructed to identify these results and experiments were established in triplicates. Type A and B systems are most likely expected to have particle size less than 50 nm and referred as a SNEDDS (31). Dispersion time required for type A or B systems were recorded while those of type C, D, or E were not easily observed due to aqueous media turbidity.
Determination of carvedilol saturated solubility in different systems
Excess carvedilol was added to the obtained type (A) systems (Table I) at 40°C and mixed using a vortex mixer. Systems were kept further in a shaker with a thermodynamically controlled water bath at 25°C for 48 h (32). One gram of each system was centrifuged at 3,000 rpm. For each system, 10 mg were evaluated for carvedilol content after dilution to 50 ml using 0.1 N HCl.
Table I.
Percent Composition of Type (A) Systems
| System no. | %HCO-40 | %Transcutol® HP | %MCT |
|---|---|---|---|
| 0 | 90 | 0 | 10 |
| 1 | 80 | 0 | 20 |
| 2 | 80 | 10 | 10 |
| 4 | 70 | 10 | 20 |
| 5 | 70 | 20 | 10 |
| 8 | 60 | 20 | 20 |
| 9 | 60 | 30 | 10 |
| 13 | 50 | 30 | 20 |
| 14 | 50 | 40 | 10 |
| 20 | 40 | 50 | 10 |
| 27 | 30 | 60 | 10 |
| 35 | 20 | 70 | 10 |
Preparation and evaluation of carvedilol SEDDS
Carvedilol (12.5% W/W) was added to type (A) systems (Table I), heated to 40°C, and mixed using a vortex mixer until the appearance of a clear system. They were stored at ambient temperature for 1 week and checked for any precipitation. The resulting appearance was visually assessed using the same grading as mentioned before in “Construction of ternary-phase diagram.”
The efficiency of self-emulsification was assessed using a standard US Pharmacopeia (USP) dissolution apparatus II (33). One hundred milligrams of different type (A) systems containing 12.5% W/W carvedilol were loaded onto a glass support and placed in the bottom of a dissolution vessel containing 500 ml 0.1 N HCl at 37°C. Gentle agitation was provided by a standard dissolution paddle rotating at 50 rpm. Samples were withdrawn at predetermined time intervals (2, 4, 6, 8, 10, 15, 20, 30, 45, and 60 min) for carvedilol analysis and replaced with equal volumes of fresh 0.1 N HCl.
The absorbances of the collected samples were measured spectrophotometrically at wavelength of 285 nm. The time required for emulsification was chosen as the time at which 90% carvedilol was measured. This test was also performed in phosphate buffer (pH 6.8) to detect the behavior of the formed emulsion upon entering the intestine.
Formulation dispersion–drug precipitation test
Formulation dispersion–drug precipitation test can be considered as one of the in vitro tests developed to help in the forecasting of dilution effect on the prepared carvedilol SEDDS. Formulation dispersion–drug precipitation test is carried out to predict whether precipitation is likely to occur upon dilution in the gastrointestinal tract through the determination of equilibrium solubility of the drug in the lipid formulation after dilution (34). Fifty milligrams of different type (A) systems (Table I) containing 12.5% W/W carvedilol were introduced into a vial containing 10 ml of 0.1 N HCl. Vials were capped and placed in a shaker with thermostatically controlled water bath at 37°C. One milliliter was filtered through 0.22 μm Millipore® membrane filter and analyzed for carvedilol content after 1, 2, and 3 days. As previously mentioned, cellulosic polymers may increase drug dispersion and prevent its precipitation. Accordingly, HPMC 5cp, HPMC 15cp, and MC 15cp were added in a concentration of 5%, 10%, or 15% W/W polymer/SEDDS to test their effect on drug precipitation.
Preparation and Evaluation of Simple Liquisolid Powders
Formulation of simple liquisolid powders from the prepared SNEDDS
For each simple liquisolid powder, the quantities of the chosen ingredients per unit dose multiplied by the number of prepared tablets were calculated. The following equation is used to calculate the amount of carrier materials (35):
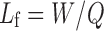 |
Lf is the liquid loading factor; W is the liquid medication weight; Q is the carrier material weight.
The excipient ratio (R) is the ratio between the carrier and coating materials as presented by the following equation (35):
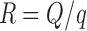 |
R is the excipient ratio or carrier to coating ratio; q is the coating material weight.
Excipient ratio (R) of 20 was used throughout this study to prepare liquisolid powders. Microcrystalline cellulose was used as a carrier (Q) and Aerosil® was used as a coating material (q). The previous equations were used to calculate the weight of tablet to be prepared.
SNEDDS-9 (system 9, Table I) containing 12.5% carvedilol with or without 5% HPMC 5cp, HPMC 15cp, or MC 15cp was prepared. Fifty milligrams of SNEDDS-9 or 52.5 mg of SNEDDS-9 containing 5% HPMC 5cp (SNEDDS-9/5% HPMC 5cp), 5% HPMC 15cp (SNEDDS-9/5% HPMC 15cp), or 5% MC 15cp (SNEDDS-9/5% MC 15cp) were loaded onto the carrier material (microcrystalline cellulose). After mixing, the resulting wet mixture was blended with the precalculated amount of the coating material (Aerosil®). The powder was then blended with the calculated quantities of the disintegrant (5% Ac-Di-Sol®), Table II.
Table II.
Composition of Different Simple Liquisolid Powders (Lf = 0.20-0.23)
| Liquisolid Powder | Liquid vehicle | Liquid load factor (Lf) | Unit dose weight (mg) |
|---|---|---|---|
| LS9-20 | SNEDDS-9 | 0.20 | 328.9 |
| LS9-21 | SNEDDS-9 | 0.21 | 316.5 |
| LS9-22 | SNEDDS-9 | 0.22 | 303.9 |
| LS9-23 | SNEDDS-9 | 0.23 | 292.9 |
| LSH5-20 | SNEDDS-9/5% HPMC 5cp | 0.20 | 331.4 |
| LSH15-20 | SNEDDS-9/5% HPMC 15cp | 0.20 | 331.4 |
| LSH15-21 | SNEDDS-9/5% HPMC 15cp | 0.21 | 319.0 |
| LSH15-22 | SNEDDS-9/5% HPMC 15cp | 0.22 | 306.4 |
| LSH15-23 | SNEDDS-9/5% HPMC 15cp | 0.23 | 295.4 |
| LSM15-20 | SNEDDS-9/5% MC 15cp | 0.20 | 331.4 |
| LSM15-21 | SNEDDS-9/5% MC 15cp | 0.21 | 319.0 |
Formulation of liquisolid powders containing Aeroperl®
Fixed-weight tablets (200 mg) containing 1% magnesium stearate, 5% Ac-Di-Sol®, and 94% adsorbent were prepared. The adsorbent was composed of Aeroperl®: microcrystalline cellulose in 1:20, 1:9, 3:17, 1:4, or 1:3 weight ratios, Table III.
Table III.
Composition of Different Liquisolid Powders Containing Aeroperl®
| Formula | Aeroperl®: MCC |
|---|---|
| ALS-1Aa | 1:20 |
| ALS-2Aa | 1:9 |
| ALS-3Aa | 3:17 |
| ALS-4Aa | 1:4 |
| ALS-5Aa | 1:3 |
| ALS-1Mb | 1:20 |
| ALS-2Mb | 1:9 |
| ALS-3Mb | 3:17 |
| ALS-4Mb | 1:4 |
| ALS-5Mb | 1:3 |
aTablets prepared by adding Aeroperl® followed by MCC as adsorbent
bTablets prepared by adding MCC followed by AeroperlT® as adsorbent
For each formula, the quantities of the chosen ingredients per unit dose multiplied by the number of prepared tablets were calculated. Accordingly, SNEDDS containing 5% HPMC 15cp (SNEDDS-9/5% HPMC 15cp) were loaded first onto the microcrystalline cellulose or Aeroperl®. After mixing, the resulting mixture was blended with the precalculated amount of either Aeroperl® or microcrystalline cellulose. Liquisolid powders were then blended with the calculated quantities of the disintegrant (5% Ac-Di-Sol®) and glidant (1% magnesium stearate).
Characterization and determination of liquisolid powders physical properties
Angle of repose (θ) was measured using a protractor for the heap of granules formed by passing 10 g of sample through a funnel onto a horizontal surface. As general guide powders having θ > 50° have unsatisfactory difficult flow properties, 25–40° have reasonable flow potential, whereas minimum angles close to 25° correspond to very good flow properties (36).
The bulk density (ρbulk) and the tapped density (ρtap) were determined (14,37). For each sample, the compatibility index or Carr index (CI) was calculated according to the following equation: 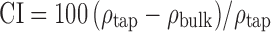 and Hausner ratio (HR) was calculated according to the following equation:
and Hausner ratio (HR) was calculated according to the following equation: 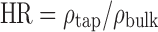 . Powders with a CI between 5% and 18% are suitable for producing tablets, and those with an HR below 1.25 are of good flowability (37–39).
. Powders with a CI between 5% and 18% are suitable for producing tablets, and those with an HR below 1.25 are of good flowability (37–39).
Preparation and Evaluation of Liquisolid Tablets
Preparation of carvedilol liquisolid tablets
Liquisolid powders showing acceptable physical properties were compressed into tablets of desired weight using a single punch tablet press machine (single punch machine, Royal Artist, India).
Evaluation of liquisolid tablets
To verify the uniformity and conformity of the tablets within each batch (40). The mean weight is expressed in milligram ± SD. The friability was determined using a friabilator (digital test apparatus, model DFI-1, India) at 25 rpm for 4 min. The friability is expressed in terms of weight loss and is calculated in percentage (40). The uniformity of content was determined by crushing tablets from each formula and determining the content of each tablet individually. Stage one criterion according to the European Pharmacopoeia for 6.25-mg carvedilol tablets is that carvedilol content must fall in the range of ≥5.31 to ≤7.19 mg with mean carvedilol content of 5.94–6.56 mg (41,42). A hardness tester (Monsanto, USA) was used to measure the crushing strength of the tablets. The mean hardness was calculated and expressed in kilograms (kg ± SD). A hardness of 3–7 kg was acceptable (42). Finally, the mean disintegration time of tablets from each lot was determined in minutes (min ± SD) using the disintegration test apparatus (Pharma Test, Type PTZ3). The medium used was 0.1 N HCl at 37°C. The tubes were raised and lowered at a constant frequency of 30 cycles per minute (43).
The dissolution of carvedilol from liquisolid tablets was performed in 500 ml of 0.1 N HCl at 37 ± 0.5°C using the USP Dissolution Tester (Testpharm, Germany), Apparatus II (rotating paddle), at a rotation of 50 rpm. Aliquots from the dissolution medium were withdrawn at 5, 10, 15, 20, 30, 45, and 60 min. The withdrawn samples were then filtered and adequately analyzed for carvedilol content by measuring the absorbencies at λmax 285 nm.
Particle size analysis
Each liquisolid tablet was gently mixed with 10 ml of 0.1 N HCl, filtered, and subjected to particle size analysis test using a laser diffraction particle size analyzer (Zetasizer 1000HS, Malvern, UK). Light scattering was monitored at 90°C and 25°C. The date was analyzed in terms of volume. Particle size was compared to that of SNEDDS-9 after dilution with 0.1 N HCl.
Study of P-glycoprotein Inhibition Activity
Effect of SNEDDS-9 and paclitaxel on the growth of HCT-116 cells was estimated by 3,(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide assay according to Hansen et al. (44). Three different experiments were carried out.
In the first experiment, HCT-116 colon cells were treated only with different concentrations of SNEDDS-9 (0.75–100 μg/ml) to calculate its IC10 value. In the second experiment, HCT-116 cells were treated only with different concentrations of paclitaxel (200–1,000 ng/ml) to calculate its IC50 value. In the third experiment, HCT-116 cells were treated with different concentrations of paclitaxel (25–1,000 ng/ml) in presence of the IC10 concentration of the formula (SNEDDS-9) to calculate the possible change in the IC50 value of paclitaxel in order to detect any P-gp inhibition effect (45). Linearized form of the median effect equation was used to calculate IC50 and IC10 (46–48):
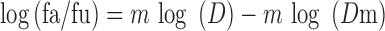 |
Where D is the dose; fa and fu are the fractions of the system affected and unaffected, respectively, by the dose D; Dm is the dose required to produce the median effect (IC50), and m is a Hill-type coefficient signifying the sigmoidicity of the dose–effect curve.
RESULTS AND DISCUSSION
Preparation and Evaluation of SEDDS
Construction of ternary-phase diagram
Upon examining the different combinations of HCO-40, Transcutol® HP, and MCT, no phase separation was observed upon storage for 72 h at ambient temperature. Visual observations of the dispersion experiment, using 0.1 N HCl, were demonstrated in the phase diagram, Fig. 1. From the results, it was deduced that an increase in HCO-40 content increased the clarity of the produced emulsion (14,31). Also, it showed that the particle size increased upon increasing MCT content (29). This could be explained by the fact that the surfactant stabilizes the O/W interface and its concentration increased at the interface upon decreasing the oily content. That attributed to a decrease in the emulsion particle size thus a more clear-appearing emulsion was produced (49).
Fig. 1.

Ternary-phase diagrams between HCO-40, Transcutol, and MCT
Figure (2) shows that increasing HCO-40 to Transcutol® ratio at a constant MCT percentage increased the emulsification time. This may be due to the increase in system viscosity and formation of gel-like structure that decreased water penetration into the system (31).
Fig. 2.

Time required for emulsification of systems (A and B), presented in minutes
Determination of carvedilol saturated solubility in different systems
It was noticed that increasing Transcutol® percentage resulted in an increase in carvedilol solubility (Fig. 3). There was a nonsignificant decrease in the solubility upon increasing MCT percent from 10% to 20% (p > 0.05). Type (A) systems were able to dissolve 3.125, 6.25, and up to 12.5 mg carvedilol per 100 mg system.
Fig. 3.

Saturated solubility of carvedilol in different systems composed of HCO-40, Transcutol®, and MCT in different ratios (white bars systems containing 10% MCT, gray bars systems containing 20% MCT)
Preparation and evaluation of carvedilol SEDDS
None of the prepared systems showed any precipitation within the storage period. Figure 1 shows the effect of 12.5% W/W carvedilol addition on systems classified as type (A). Systems with a high HCO-40 percentage produced emulsions having finer particle size, which indicates better stability. Also, it can be noted that systems containing 20% Miglyol® produced emulsion with larger particle size than systems containing 10% Miglyol®. Generally, it was observed that incorporation of 12.5% W/W carvedilol into systems classified as type (A) mostly increased particle size of the formed emulsions (33).
Time needed to measure 90% carvedilol was chosen as the emulsification time. Tested systems were emulsified within the first 2 min except systems 0, 1, and 2 (Fig. 4). These systems were considered as the most viscous systems, as they contained the highest percentage of HCO-40. After 4 min, detected carvedilol was higher from system 2 in comparison to system 1 (p = 0.023). This could be explained by the fact that presence of Transcutol® increased systems’ hydrophilicity and thus water penetration into the gel-like structure. No precipitation of carvedilol was observed throughout the experimental period.
Fig. 4.

Percentage of detected carvedilol in 0.1 N HCl as a function of time
Carvedilol, as a basic drug, dissolves in the acidic pH of the stomach (ionized form). However, precipitation in the higher pH environment of the small intestine (site of absorption) may occur, eradicating any advantage gained in dissolving the drug in the stomach (50–52). To verify the possibility of drug precipitation under intestinal conditions, emulsification test was done in pH 6.8 (53). There was efficient self-emulsification and dispersion of carvedilol within tested pHs (1.2 and 6.8). Within 2 min, medicated system 9 dissolved 89.4 ± 1.4% and 86.4 ± 0.7% carvedilol in pH 1.2 and 6.8, respectively (p = 0.115). The results of detected carvedilol upon testing system 9 in phosphate buffer (pH 6.8) showed a great improvement in comparison to carvedilol powder (0.2 ± 0.0%). This indicates that the improvement of carvedilol dispersion after incorporation within the SMEDDS is observed within acidic and basic media regardless its intrinsic solubility.
Formulation dispersion–drug precipitation test
Presence of high percentage of co-solvent (Transcutol®) in systems 20, 27, and 35 increased carvedilol precipitations in the performed test regardless their classification as type A, B, or C, respectively (Fig. 5). Drug precipitation upon dilution could be attributed to the relationship between drug solubility and co-solvent concentration, which is commonly approximated to a logarithmic relationship (7,8). It was noticed that systems containing 20% oil were more susceptible to carvedilol precipitation than systems containing 10% oil (1 vs. 2, 4 vs. 5, 8 vs. 9, and 13 vs. 14 having p = 0.001, p < 0.001, p < 0.001 and p < 0.001, respectively). Consequently, it was concluded that maintaining carvedilol within the emulsion structure is privileged to co-solvent presence under constant surfactant content. It should be mentioned that carvedilol precipitation may not only be due to the decrease in its solubility upon SEDDS dilution but also it may be due to phase separation of the formed emulsion (54).
Fig. 5.

Formulation dispersion–drug precipitation test for different SEDDS
Systems 4, 8, 13, 14, 20, 27, and 35 were excluded from this study as they retained less than 70% of the initial drug content at the end of the formulation dispersion–drug precipitation test, while systems 0, 1, and 2 needed more than 2 min for the emulsification process. On the other hand, system 5 contains more surfactant than system 9 which is expected to be more toxic (5). Accordingly, in order to improve carvedilol equilibrium solubility, system 9 was chosen for further studies.
Each of the selected polymers (HPMC 5cp, HPMC 15cp, and MC 15cp) was added to system 9 in 5%, 10%, or 15% (W/W). Addition of such polymers enabled the retainment of carvedilol in the aqueous media by increasing its stability (77% increased to 98–102% according to the polymer type and concentration). Similar results were obtained with Itoh, Gao, and co-workers who proved that the addition of polymeric materials to SEDDS inhibited drug crystallization (30,54,55).
Preparation and Evaluation of Simple Liquisolid Powders and Tablets
Liquisolid powders having an angle of repose (θ) close to 25°, an HR less than 1.25, and a CI between 5% and 18% were considered as powders with acceptable physical properties. Liquisolid powders LS9-22, LSH15-22, and LSM15-20 showed acceptable physical properties. They were chosen to be compressed into liquisolid tablets, using 11-mm concave punch, as they passed the lowest weights of unit dose within powders with acceptable physical properties in the following types of loaded liquid: SNEDDS-9, SNEDDS-9/5% HPMC 15cp, and SNEDDS-9/5% MC 15 cp, respectively.
The effect of polymer type on the physical properties of liquisolid powders depends on different mechanisms such as:
Changing the molecular weight that would affect their adsorption (MWt of MC < HPMC 5cp < HPMC 15cp ((56–58); http://www.dow.com/methocel/build/resource/chem.htm, 2008 January 13).
Hydrogen bonding between adsorbed polymer molecules would be affected by the amount of hydroxyl group per certain weight (MC > HPMC 5cp > HPMC 15cp (56–58); http://www.dow.com/methocel/build/resource/chem.htm, 2008 January 13).
Accordingly, different polymers changed the physical properties of liquisolid powders differently. Liquisolid powders adsorbing HPMC 15cp showed better physical properties than MC 15 cp followed by HPMC 5 cp.
Tested simple liquisolid tablets confirmed the accepted criterion for weight variation, drug content, friability, hardness, and disintegration. Tested liquisolid tablets dissolved about 90% of carvedilol within 45 min (Fig. 6). It was noticed that the dissolution profile of carvedilol from the tested tablets was improved if compared to that of the powder form. Liquisolid tablets improved carvedilol dissolution profile as they introduced carvedilol into the dissolution media in a nanoemulsion. Presence of carvedilol within the nanoemulsion increased the release surface area thus increasing its diffusion into the dissolution media (5,11).
Fig. 6.

Dissolution profiles of carvedilol from different liquisolid tablets compared to carvedilol powder
Particle size analysis is presented in Fig. 7. SNEDDS-9 demonstrated a monodisperse system with a mean diameter of 17.9 nm. Simple liquisolid tablet particle size analysis revealed two peaks; one corresponds to the nanoemulsion system while the other represents some insoluble nanoparticles in the tablet components. Liquisolid tablets LS9-22, LSH15-22, and LSM15-20 produced a nanoemulsion with mean diameter of 26.6, 17.0, and 27.4 nm, respectively. Processing SNEDDS into liquisolid tablet did not affect the nanoparticle size after emulsification in 0.1 N HCl.
Fig. 7.

Particle size distribution of emulsified system 9 and liquisolid tablets dispersed in 0.1 N HCl
Preparation and Evaluation of Liquisolid Powders and Tablets Containing Aeroperl®
Powders containing adsorbent of Aeroperl®: MCC in 3:17, 1:4, and 1:3 weight ratios showed acceptable physical properties regardless the order of adsorbent addition. Powders designed to contain 52.5 mg of SNEDDS-9/5% HPMC 15cp within 200-mg unit weight were successfully prepared. They were compressed using single punch machine having 9-mm concave or flat punch. Flat punch produced significant increase in tablets hardness at 95% level of significance. Accordingly, flat punch was used during further study.
Tested tablets had shown acceptable hardness and disintegration time. After mixing Aeroperl® with SNEDDS-9/5% HPMC 15cp, mixing with MCC did not produce homogenous mixture easily. Tablets prepared by adding MCC as an adsorbent followed by Aeroperl® were chosen for further study as they were easier to formulate. They showed acceptable weight variation, drug content, friability, and dissolution profile (Fig. 6). Nevertheless, ALS-5M was superior to ALS-4M in its disintegration time (4.7 ± 0.3 min) and hardness (4.0 ± 0.4 kg).
Liquisolid tablet, ALS-5M, particle size analysis revealed three peaks; one corresponded to the nanoemulsion system while the others represented some insoluble nanoparticles in the tablet components (Fig. 7). It produced a nanoemulsion with a mean diameter of 28.4 nm. Processing SNEDDS into liquisolid tablets containing granulated SiO2 was also able to retain the nanoparticle size of the nanoemulsion.
Study of P-gp inhibition Activity
The calculated IC10 value of SNEDDS was 0.60 μg/ml, while IC50 value of paclitaxel was 435.32 ng/ml. The concomitant treatment with both formula and paclitaxel resulted in an inhibition in the cell growth and a potential downshift in the dose–response curve of paclitaxel, in a way that depressed the IC50 value of paclitaxel to 306.54 ng/ml. That decrease in IC50 indicated presence of P-gp inhibition activity which may lead to a decrease in carvedilol efflux from the intestinal cells with consequent improvement in its absorption.
CONCLUSIONS
In this study, combination of HCO-40, Transcutol® HP, and MCT in variable ratios showed rapid emulsification in aqueous media. Meanwhile, systems which form nanoemulsion and are able to retain the drug in a solubilized form after dispersion in aqueous media will be preferred as a carrier for poorly water-soluble drugs. The amount of self-nanoemulsifying system used to dissolve carvedilol was less than that used in previous studies (59). Furthermore, addition of cellulosic polymers to SNEDDS is expected to improve its efficiency upon dilution in the gastrointestinal tract. Liquisolid tablets were able to carry SNEDDS and possess acceptable pharmacopeia requirements for tablets. In addition, they were able to introduce the SNEDDS into the dissolution media where it was efficiently transformed into nanoemulsion by the gentle agitation provided in the dissolution experiment. Modifying silicon dioxide physical form from amorphous into granulated improved the physical properties of both liquisolid powders and tablets. It enhanced the adsorption power and compressibility of liquisolid tablets. Successfully, SNET were able to introduce carvedilol in a unique immediate-release dosage form.
Acknowledgement
I am very grateful for the Sasol, Gattefosse, and Colorcon companies and Mohamed H. AbouGhaly for providing the required chemicals for my research. I am grateful to Assoc. Prof. Amira M. Gamal-Eldeen, Associate Professor of Genetic Engineering and Biotechnology Division, National Research Center, for her kind help in the toxicological experimental work in this paper.
References
- 1.Weuts I., Kempen D., Decorte A., Verreck G., Peeters J., Brewster M., Van den Mooter G. Phase behaviour analysis of solid dispersions of loperamide and two structurally related compounds with the polymers PVP-K30 and PVP-VA64. Eur. J. Pharm. Sci. 2004;22:375–385. doi: 10.1016/j.ejps.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Ammar H. O., Salama H. A., Ghorab M., Mahmoud A. A. Implication of inclusion complexation of glimepiride in cyclodextrin-polymer systems on its dissolution, stability and therapeutic efficacy. Int. J. Pharm. 2006;320:53–57. doi: 10.1016/j.ijpharm.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Muhrer G., Meier U., Fusaro F., Albano S., Mazzotti M. Use of compressed gas precipitation to enhance the dissolution behavior of a poorly water-soluble drug: generation of drug microparticles and drug-polymer solid dispersions. Int. J. Pharm. 2006;308:69–83. doi: 10.1016/j.ijpharm.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Odeberg J. M., Kaufmann P., Kroon K. G., Hoglund P. Lipid drug delivery and rational formulation design for lipophilic drugs with low oral bioavailability, applied to cyclosporine. Eur. J. Pharm. Sci. 2003;20:375–382. doi: 10.1016/j.ejps.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Gershanik T., Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000;50:179–188. doi: 10.1016/S0939-6411(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 6.Gursoy R. N., Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004;58:173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Pouton C. W. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006;29:278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Pouton C. W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997;25:47–58. doi: 10.1016/S0169-409X(96)00490-5. [DOI] [Google Scholar]
- 9.Newton M., Petersson J., Podczeck F., Clarke A., Booth S. The influence of formulation variables on the properties of pellets containing a self-emulsifying mixture. J. Pharm. Sci. 2001;90:987–995. doi: 10.1002/jps.1051. [DOI] [PubMed] [Google Scholar]
- 10.Tuleu C., Newton M., Rose J., Euler D., Saklatvala R., Clarke A., Booth S. Comparative bioavailability study in dogs of a self-emulsifying formulation of progesterone presented in a pellet and liquid form compared with an aqueous suspension of progesterone. J. Pharm. Sci. 2004;93:1495–1502. doi: 10.1002/jps.20068. [DOI] [PubMed] [Google Scholar]
- 11.Franceschinis E., Voinovich D., Grassi M., Perissutti B., Filipovic-Grcic J., Martinac A., Meriani-Merlo F. Self-emulsifying pellets prepared by wet granulation in high-shear mixer: influence of formulation variables and preliminary study on the in vitro absorption. Int. J. Pharm. 2005;291:87–97. doi: 10.1016/j.ijpharm.2004.07.046. [DOI] [PubMed] [Google Scholar]
- 12.Newton J. M., Bazzigialuppi M., Podczeck F., Booth S., Clarke A. The rheological properties of self-emulsifying systems, water and microcrystalline cellulose. Eur. J. Pharm. Sci. 2005;26:176–183. doi: 10.1016/j.ejps.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Newton J. M., Godinho A., Clarke A. P., Booth S. W. Formulation variables on pellets containing self-emulsifying systems. Pharm. Technol. Eur. 2005;17:29–32. [Google Scholar]
- 14.Nazzal S., Nutan M., Palamakula A., Shah R., Zaghloul A. A., Khan M. A. Optimization of a self-nanoemulsified tablet dosage form of ubiquinone using response surface methodology: effect of formulation ingredients. Int. J. Pharm. 2002;240:103–114. doi: 10.1016/S0378-5173(02)00130-8. [DOI] [PubMed] [Google Scholar]
- 15.Kasim N. A., Whitehouse M., Ramachandran C., Bermejo M., Lennernas H., Hussain A. S., Junginger H. E., Stavchansky S. A., Midha K. K., Shah V. P., Amidon G. L. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 16.Talbert R. L. Pharmacokinetics and pharmacodynamics of beta blockers in heart failure. Heart Fail. Rev. 2004;9:131–137. doi: 10.1023/B:HREV.0000046368.08825.20. [DOI] [PubMed] [Google Scholar]
- 17.Frishman W. H. Carvedilol. Drug Therapy. 1998;339:1759–1765. doi: 10.1056/NEJM199812103392407. [DOI] [PubMed] [Google Scholar]
- 18.Giessmann T., Modess C., Hecker U., Zschiesche M., Dazert P., Kunert-Keil C., Warzok R., Engel G., Weitschies W., Cascorbi I., Kroemer H. K., Siegmund W. CYP2D6 genotype and induction of intestinal drug transporters by rifampin predict presystemic clearance of carvedilol in healthy subjects. Clin. Pharmacol. Ther. 2004;75:213–222. doi: 10.1016/j.clpt.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Wen X., Tan F., Jing Z., Liu Z. Preparation and study the 1:2 inclusion complex of carvedilol with β-cyclodextrin. J. Pharm. Biomed. Anal. 2004;34:517–523. doi: 10.1016/S0731-7085(03)00576-4. [DOI] [PubMed] [Google Scholar]
- 20.Bogman K., Erne-Brand F., Alsenz J., Drewe J. The role of surfactants in the reversal of active transport mediated by multidrug resistance proteins. J. Pharm. Sci. 2003;92:1250–1261. doi: 10.1002/jps.10395. [DOI] [PubMed] [Google Scholar]
- 21.Yang S., Gursoy R. N., Lambert G., Benita S. Enhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitors. Pharm. Res. 2004;21:261–270. doi: 10.1023/B:PHAM.0000016238.44452.f1. [DOI] [PubMed] [Google Scholar]
- 22.Shen H., Zhong M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J. Pharm. Pharmacol. 2006;58:1183–1191. doi: 10.1211/jpp.58.9.0004. [DOI] [PubMed] [Google Scholar]
- 23.Gershanik T., Haltner E., Lehr C. M., Benita S. Charge-dependent interaction of self-emulsifying oil formulations with Caco-2 cells monolayers: binding, effects on barrier function and cytotoxicity. Int. J. Pharm. 2000;211:29–36. doi: 10.1016/S0378-5173(00)00591-3. [DOI] [PubMed] [Google Scholar]
- 24.Tayrouz Y., Ding R., Burhenne J., Riedel K. D., Weiss J., Hoppe-Tichy T., Haefeli W. E., Mikus G. Pharmacokinetic and pharmaceutic interaction between digoxin and Cremophor RH40. Clin. Pharmacol. Ther. 2003;73:397–405. doi: 10.1016/S0009-9236(03)00059-6. [DOI] [PubMed] [Google Scholar]
- 25.Porter C. J., Charman W. N. In vitro assessment of oral lipid based formulations. Adv. Drug. Deliv. Rev. 2001;1(Suppl 50):S127–S147. doi: 10.1016/S0169-409X(01)00182-X. [DOI] [PubMed] [Google Scholar]
- 26.Cornaire G., Woodley J., Hermann P., Cloarec A., Arellano C., Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int. J. Pharm. 2004;278:119–131. doi: 10.1016/j.ijpharm.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Grovea M., Müllertzb A., Nielsenc J. L., Pedersen P. Bioavailability of seocalcitol II: development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur. J. Pharm. Sci. 2006;28:233–242. doi: 10.1016/j.ejps.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Cui S., Zhao C., Tang X., Chen D., He Z. Study on the bioavailability of puerarin from Pueraria lobata isoflavone self-microemulsifying drug-delivery systems and tablets in rabbits by liquid chromatography–mass spectrometry. Biomed. Chromatogr. 2005;19:375–378. doi: 10.1002/bmc.460. [DOI] [PubMed] [Google Scholar]
- 29.Hong J. Y., Kim J. K., Song Y. K., Park J. S., Kim C. K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control Release. 2006;110:332–338. doi: 10.1016/j.jconrel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Gao P., Rush B. D., Pfund W. P., Huang T., Bauer J. M., Morozowich W., Kuo M. S., Hageman M. J. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 2003;92:2386–2398. doi: 10.1002/jps.10511. [DOI] [PubMed] [Google Scholar]
- 31.Khoo S.-M., Humberstone A. J., Porter C. J. H., Edwards G. A., Charman W. N. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int. J. Pharm. 1998;167:155–164. doi: 10.1016/S0378-5173(98)00054-4. [DOI] [Google Scholar]
- 32.Kang B. K., Lee J. S., Chon S. K., Jeong S. Y., Yuk S. H., Khang G., Lee H. B., Cho S. H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004;274:65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 33.Li P., Ghosh A., Wagner R. F., Krill S., Joshi Y. M., Serajuddin A. T. Effect of combined use of nonionic surfactant on formation of oil-in-water microemulsions. Int. J. Pharm. 2005;288:27–34. doi: 10.1016/j.ijpharm.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 34.Hasan N., Moss S. H., Pouton C. W. Through, C. W. Pouton Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006;29:278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 35.Spireas S., Sadu S., Grover R. In vitro release evaluation of hydrocortisone liquisolid tablets. J. Pharm. Sci. 1998;87:867–872. doi: 10.1021/js970346g. [DOI] [PubMed] [Google Scholar]
- 36.Staniforth J. N. Pharmaceutics. In: Aulton M. E., editor. The science of dosage form design. Livingstone: Churchill; 1988. p. 610. [Google Scholar]
- 37.Sinko P. J., editor. Martin’s physical pharmacy and biopharmaceutical sciences. 5. Philadelphia: Lippincott Williams &Wilkins; 2006. p. 558. [Google Scholar]
- 38.Abdelbary G., Prinderre P., Eouani C., Joachim J., Reynier J. P., Piccerelle P. The preparation of orally disintegrating tablets using a hydrophilic waxy binder. Int. J. Pharm. 2004;278:423–433. doi: 10.1016/j.ijpharm.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 39.Chateau M.-E., Galet L., Soudais Y., Fages J. Processing a detergent powder formulation: direct compression, and high shear wet granulation followed by compression. Powder Technol. 2005;157:191–198. doi: 10.1016/j.powtec.2005.05.026. [DOI] [Google Scholar]
- 40.British Pharmacopoeia Commission . British pharmacopoeia 2002. London: Her Majesty's Stationary Office; 2002. [Google Scholar]
- 41.Council of Europe . European pharmacopoeia. 5. Strasbourg: European Directorate for the Quality of Medicines; 2005. [Google Scholar]
- 42.Smith J. C., Tarocco G., Merazz F., Salzmann U. Are generic formulations of carvedilol of inferior pharmaceutical quality compared with the branded formulation? Curr. Med. Res. Opin. 2006;22:709–720. doi: 10.1185/030079906X96461. [DOI] [PubMed] [Google Scholar]
- 43.US Pharmacopoeia Convention . United States Pharmacopoeia. 3. Rockville: US Pharmacopoeia Convention; 2002. [Google Scholar]
- 44.Hansen M. B., Nielsen S. E., Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods. 1989;12:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 45.Wang S. -W., Monagle J., McNulty C., Putnam D., Chen H. Determination of P-glycoprotein inhibition by excipients and their combinations using an integrated high-throughput process. J. Pharm. Sci. 2004;93:2755–2767. doi: 10.1002/jps.20183. [DOI] [PubMed] [Google Scholar]
- 46.Chou T. C., Talalay P. Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme. Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 48.Chou T. C., Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis–Menten kinetic systems. J. Biol. Chem. 1977;252:6438–6442. [PubMed] [Google Scholar]
- 49.Levy M. Y., Benita S. Drug release from submicronized o/w emulsion: a new in vitro kinetic evaluation model. Int. J. Pharm. 1990;66:29–37. doi: 10.1016/0378-5173(90)90381-D. [DOI] [Google Scholar]
- 50.Kostewicz E. S., Wunderlich M., Brauns U., Becker R., Bock T., Dressman J. B. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J. Pharm. Pharmacol. 2004;56:43–51. doi: 10.1211/0022357022511. [DOI] [PubMed] [Google Scholar]
- 51.Nolte K., Backfisch G., Neidlein R. In vitro absorption studies with carvedilol using a new model with porcine intestine called BM-RIMO (Boehringer–Mannheim ring model) Arzneimittelforschung. 1999;49:745–749. doi: 10.1055/s-0031-1300495. [DOI] [PubMed] [Google Scholar]
- 52.Stegemanna S., Leveillerb F., Franchic D., de Jongd H., Lindén H. When poor solubility becomes an issue: from early stage to proof of concept. Eur. J. Pharm. Sci. 2007;31:249–261. doi: 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- 53.Galia E., Nicolaides E., Horter D., Lobenberg R., Reppas C., Dressman J. B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998;15:698–705. doi: 10.1023/A:1011910801212. [DOI] [PubMed] [Google Scholar]
- 54.Itoh K., Tozuka Y., Oguchi T., Yamamoto K. Improvement of physicochemical properties of N-4472 part I formulation design by using self-microemulsifying system. Int. J. Pharm. 2002;238:153–160. doi: 10.1016/s0378-5173(02)00085-6. [DOI] [PubMed] [Google Scholar]
- 55.Gao P., Guyton M. E., Huang T., Bauer J. M., Stefanski K. J., Lu Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturable formulations. Drug Dev. Ind. Pharm. 2004;30:221–229. doi: 10.1081/DDC-120028718. [DOI] [PubMed] [Google Scholar]
- 56.Cisneros-Zevallos L., Krochta J. M. Dependence of coating thickness on viscosity of coating solution applied to fruits and vegetables by dipping method. J. Food Sci. 2003;68:503–510. doi: 10.1111/j.1365-2621.2003.tb05702.x. [DOI] [Google Scholar]
- 57.S. M. Grillo, R. M. Steffenino, D. C. Kunkle, and K. Saraceni. Aqueous maltodextrin and cellulosic polymer film coatings. WO 91/15548. (1991).
- 58.Naruenartwongsakul S., Chinnan M. S., Bhumiratana S., Yoovidhya T. Pasting characteristics of wheat flour-based batters containing cellulose ethers. Lebensm. Wiss. Technol. 2004;37:489–495. doi: 10.1016/j.lwt.2003.12.001. [DOI] [Google Scholar]
- 59.Wei L., Sun P., Nie S., Pan W. Preparation and evaluation of SEDDS and SMEDDS containing carvedilol. Drug Dev. Ind. Pharm. 2005;31:785–794. doi: 10.1080/03639040500216428. [DOI] [PubMed] [Google Scholar]