

Abstract
The aim of the present study was to investigate the feasibility of the inclusion of a water-insoluble drug (diazepam, DZ) into solid lipid nanoparticles (SLNs), which offer combined advantages of rapid onset and prolonged release of the drug. This work also describes a new approach to prepare suppositories containing DZ-loaded SLN dispersions, as potential drug carrier for the rectal route. Modified high-shear homogenization and ultrasound techniques were employed to prepare SLNs. The effect of incorporation of different concentrations of Compritol® ATO 888 or Imwitor® 900K and Poloxamer 188 or Tween 80 was investigated. Results showed that varying the type or concentration of lipid matrix or surfactant had a noticeable influence on the entrapment efficiencies, particle size, and release profiles of prepared SLNs. Differential scanning calorimetry and X-ray diffraction measurements showed that the majority of SLNs possessed less ordered arrangements of crystals than the corresponding bulk lipids, which was favorable for increasing the drug loading capacity. Transmission electron microscopy and laser diffractometry studies revealed that the prepared nanoparticles were round and homogeneous and 60% of the formulations were less than 500 nm. Additionally, SLN formulations showed significant (P < 0.05) prolonged release than DZ solution. The subsequent step encompassed the preparation and evaluation of SLN-based suppositories utilizing SLN formulations that illustrated optimal release profiles. The in vitro release of DZ from the suppositories prepared using DZ-loaded SLN dispersions (equivalent to 2 mg DZ) was significantly (P < 0.05) extended compared to suppositories containing 2 mg DZ free drug.
Key words: diazepam, formulation, high-shear homogenization, SLN-loaded suppositories, solid lipid nanoparticles
INTRODUCTION
Although childhood seizures are a common disorder, their occurrence remains a frightening event; parents of children with even simple benign seizures think their child is dying, and this represents a frequent cause of emergency department visits. Early termination of seizures, by initiating therapy as soon as possible, preferably at home, has been increasingly emphasized as a key to minimize morbidity of these seizures (1–3). Therefore, a main goal for the treatment is rapid termination of seizures activity because the longer the episode of status epilepticus is untreated, the more difficult it is to control and the greater the risk of permanent brain damage (4,5).
Benzodiazepines (BZDs) are considered the treatment of choice for acute management of severe seizures. BZDs are active against a wide range of seizure types, have a rapid onset of action once delivered into the central nervous system, and are safe. Diazepam (DZ) is a benzodiazepine that is widely used as an anticonvulsant in the treatment of anxiety and insomnia and for induction and maintenance of anesthesia. DZ could be administered via different routes: orally, IV injections, rectal solutions, rectal gels, and suppositories (6,7). Although IV administration is probably the most rapid way of seizure suppression, an alternative and more convenient approach is highly needed when IV administration is not immediately available, for instance, because of the delay in transferring patient to hospital or arriving of emergency medical personnel. In addition, the administration of DZ parenterally may be accompanied by pain of injection and inflammation at the injection site (5,8,9).
Generally, oral administration of DZ is the route of choice in the daily practice of pharmacotherapy. Yet, when the patient is actively having seizure convulsions that might lead to severe spasms in the faciomandibular area causing aspiration and respiratory problems, the oral route is difficult, impossible, or hazardous. In addition, absorption following the oral administration of DZ tablets is usually erratic and slower and accompanied by acid hydrolysis as well as extensive liver metabolism; therefore, in some circumstances when rapid sedation is necessary or in the case of active convulsions, the oral route is not the route of choice (6,10–12). Thus, an effective route of administration would be rectally. When administered rectally, DZ, having high lipid solubility, results in prompt absorption and rapid entry to the central nervous system (6,13).
Solid lipid nanoparticles (SLNs) were developed at the beginning of 1990s as an alternative carrier system to traditional colloidal systems, such as emulsions, liposomes, and polymeric microparticles and nanoparticles (8,14–16). SLNs have attracted increasing attention as a potential drug delivery carrier owing to their numerous advantages such as the possibility of controlled drug release and drug targeting, the increased drug stability, the high drug payload, the possibility of incorporation of lipophilic and hydrophilic drugs, the biocompatibility of the lipid carrier, the avoidance of organic solvents, as well as the feasibility of large-scale production by a high-pressure homogenization technique (9,16,17). Due to the characteristic configuration of SLNs that consists of a biocompatible lipid core and an amphiphilic surfactant as an outer shell, the amount of drug in the outer shell and on the particle surface is released in the form of a burst and thus rapid onset of action is firstly observed. On the other hand, the drug incorporated into the particle core is released in a prolonged way. This makes the controlled release from these carriers possible which becomes an important tool when it is necessary to supply the drug over a prolonged period of time (8,16–18).
The aim of the present study was to investigate the feasibility of the inclusion of the water-insoluble drug (DZ) into SLNs that offer combined rapid onset and prolonged release of the drug, thus, providing early termination of seizures, protection from brain damage, as well as long-acting protection against further seizures or case complications. Modified high-shear homogenization and ultrasound techniques were employed to prepare the SLNs; the physicochemical properties of obtained DZ-loaded SLNs, such as particle size, drug loading capacity, drug entrapment efficiency, and in vitro drug release behavior, were evaluated. The following step encompassed the preparation of suppositories containing the selected DZ-loaded SLN dispersions using two different suppository bases. Finally, the in vitro release profile was used to evaluate the prepared prolonged release suppositories.
MATERIALS AND METHODS
Materials
DZ and Poloxamer 188 (triblock copolymer of polyoxyethylene and polyoxypropylene) were obtained from Sigma (St. Louis, MO, USA); Compritol® ATO 888 (US/NF: glyceryl behenate, is a mixture of monoglycerides, diglycerides, and triglycerides of behenic acid) was purchased from Gattefossè, France. Imwitor® 900K (glyceryl monostearate) was kindly gifted from Sasol, Germany. Tween 80 (PEG Sorbitan mono-oleate) was purchased from Adwic, El-Nasr Pharmaceutical Chemicals Co., Egypt. For the preparation of suppositories, Witepsol W35 and S58 were obtained from Condea Chemie GmbH, Germany, to be used as suppository bases.
Preparation of DZ-Loaded SLNs
DZ-loaded SLN dispersions consisted of 5–10% matrix (lipid–drug mixture), stabilized by 2.5–5% surfactant (Poloxamer 188 or Tween 80). Compritol® ATO 888 or Imwitor® 900K were used as the lipid materials for SLNs; in addition, Imwitor® 900K acted as nonionic emulsifier and stabilizer (14). DZ was incorporated in a percentage of 5%. The amount of the drug to be added (in grams) was calculated as a percentage of the solid matrix as follows: 100 g of a 10% SLNs dispersion loaded with 5% drug contained 10-g solid consisting of 9.5-g lipid and 0.5-g drug (18). An overview of the w/w percent composition of SLN formulations is given in Table I.
Table I.
Composition of the Investigated Diazepam-Loaded SLN Dispersions (% w/w)
| Formula number | Drug | Lipid | Emulsifying agent | Water | ||
|---|---|---|---|---|---|---|
| (% w/w)a | Type | (% w/w)a | Type | (% w/w)a | (% w/w)a | |
| 1 | 0.25 | Compritol® ATO 888 | 4.75 | Tween 80 | 2.5 | 92.5 |
| 2 | 0.25 | 4.75 | 5 | 90 | ||
| 3 | 0.25 | 4.75 | Poloxamer 188 | 2.5 | 92.5 | |
| 4 | 0.25 | 4.75 | 5 | 90 | ||
| 5 | 0.5 | 9.5 | Tween 80 | 2.5 | 87.5 | |
| 6 | 0.5 | 9.5 | 5 | 85 | ||
| 7 | 0.5 | 9.5 | Poloxamer 188 | 2.5 | 87.5 | |
| 8 | 0.5 | 9.5 | 5 | 85 | ||
| 9 | 0.25 | Imwitor® 900K | 4.75 | Tween 80 | 2.5 | 92.5 |
| 10 | 0.25 | 4.75 | 5 | 90 | ||
| 11 | 0.25 | 4.75 | Poloxamer 188 | 2.5 | 92.5 | |
| 12 | 0.25 | 4.75 | 5 | 90 | ||
| 13 | 0.5 | 9.5 | Tween 80 | 2.5 | 87.5 | |
| 14 | 0.5 | 9.5 | 5 | 85 | ||
| 15 | 0.5 | 9.5 | Poloxamer 188 | 2.5 | 87.5 | |
| 16 | 0.5 | 9.5 | 5 | 85 | ||
| 17 | 0.25 | Compritol® ATO 888 | 4.75 | Tween 80: Poloxamer 188 (1:1) | 5 | 90 |
| 18 | 0.5 | 9.5 | 5 | 85 | ||
| 19 | 0.25 | Imwitor® 900K | 4.75 | 5 | 90 | |
| 20 | 0.5 | 9.5 | 5 | 85 | ||
aPercentage of the final SLN dispersion
DZ-loaded SLN dispersions were prepared by high-shear homogenization and ultrasound techniques as described previously (9,14,19,20); however, this method has been modified to be adopted to this work. The lipid–drug mixture was heated and maintained at 80°C, which exceeds the lipid melting temperature. An aqueous phase was prepared by dissolving Poloxamer 188 or Tween 80 (2.5% or 5% w/w) in distilled water and heated to the same temperature of oil phase. Hot aqueous phase was added to lipid phase and homogenization was carried out using Diax 900 homogenizer (Heidolph, Germany) at 21,000 rpm for 10 min. Coarse hot oil in water emulsion obtained was ultrasonicated using probe sonicator (ultrasonic processor, GE130, probe CV18, USA) for 10 min. DZ-loaded SLN dispersions were finally obtained by allowing hot nanoemulsion to cool to room temperature.
Characterization of DZ-Loaded SLNs
Determination of DZ Entrapment Efficiency %EE
The entrapment efficiencies of prepared systems were determined by measuring the concentration of free drug in the dispersion medium. The unentrapped DZ was determined by adding 100 μl of the nanosuspension to 9.9 ml ethanol (95%) in order to dissolve the unentrapped drug; the obtained suspension was centrifuged for 45 min at 6,000 rpm. The supernatant was separated and then filtered through hydrophilic acrylic copolymer membrane 0.2-μm filter (Versapor, German Sciences) (15). The filtrate was diluted using ethanol and measured spectrophotometrically (Shimadzu, model UV-1601 PC, Kyoto, Japan). The amount of free drug was detected in the filtrate and the amount of incorporated drug was determined as a result of the initial drug minus the free drug. The entrapment efficiency was calculated using the following equation (14,15):
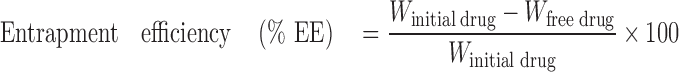 |
Where “Winitial drug” is the mass of initial drug used and the “Wfree drug” is the mass of free drug detected in the supernatant after centrifugation of the aqueous dispersion.
Transmission Electron Microscopy
The morphology of DZ SLNs (selected samples F1 and F10) as well as DZ pure drug was examined using an electronic transmission microscope (model JEM-1230, Jeol, Tokyo, Japan) at 70 kV. After 50-fold dilution with the original dispersion medium of the preparation, the samples were negatively stained with 1% (w/v) phosphotungstic acid for observation.
Particle Size Analysis
Particle size analysis of DZ-loaded SLNs was performed by laser diffraction (LD) using Malvern Mastersizer X laser scattering instrument (detection limit 0.1–2,000 μm; Malvern Instruments Ltd., Malvern, UK); all measurements and analysis setting were controlled using standard operation procedures. After diluting the SLN suspensions with deionized water, the aqueous nanoparticulate dispersion was added to the sample dispersion unit and the measurements were performed using a 45-mm focus objective and a beam length of 2.4 mm. The analysis was executed three times for each sample and the average values were taken.
X-ray Diffraction
The X-ray diffraction patterns were determined for the drug (DZ), the bulk lipids (Compritol® ATO 888 and Imwitor® 900K), and the prepared SLNs (F1 and F10). Samples were exposed to a monochromatic nickel-filtered copper radiation (45 kV, 40 mA) in a wide-angle X-ray diffractometer (advanced diffraction system, Scintag Inc., USA) with 2θ angle between 4° and 50°.
Differential Scanning Calorimetry
The thermotropic properties and phase transition behavior of DZ, Compritol® ATO 888, Imwitor® 900K, and SLN formulations F1 and F10 were evaluated by Shimadzu Differential Scanning Calorimeter (DSC)-50 (Shimadzu, Kyoto, Japan). Samples of about 5 mg were sealed in a 50-μl aluminum pans at a heating rate of 10°C/min throughout the analysis. Empty aluminum pans were used as references and the whole thermal behaviors were studied under a nitrogen purge.
In Vitro Release Studies of DZ from SLNs.
The in vitro release of DZ from different SLN formulations was determined using a modified dialysis membrane diffusion technique (21–23). An accurately weighed amount of DZ-loaded SLN dispersions equivalent to 2 mg DZ was transferred to a glass cylinder having the length of 6 cm and diameter of 2.5 cm fitted at its lower end with presoaked cellulose membrane (Spectra/Por dialysis membrane 12,000–14,000 Mwt cutoff). The glass cylinder was then suspended in the dissolution flask of a USP dissolution apparatus I (Pharma Test, Germany) containing 400-ml phosphate buffer (pH 7.4) and it was allowed to rotate at a constant speed (25 rpm) at 37°C ± 0.5 (23). Aliquots were withdrawn and replaced with fresh buffer and then the drug content was determined spectrophotometrically (Shimadzu, model UV-1601 PC, Kyoto, Japan) at predetermined time intervals for a total period of 8 h. The results were the mean values of three runs. The resulting data were analyzed by using the software SPSS 14.0 (SPSS Inc., Chicago, IL, USA) applying one-way analysis of variance (ANOVA) test followed by post hoc multiple comparisons using least significant difference (LSD). Differences between formulations were considered to be significant at P < 0.05.
Preparation and Evaluation of DZ SLN-Based Suppositories
The SLN formulations that showed optimal results, regarding loading capacity and in vitro release, were selected to be incorporated in the two different suppository bases (Witepsol W35 and Witepsol S58). DZ SLN-based suppositories were prepared according to the fusion method (24). Briefly, an accurate weight of suppository bases (Witepsol W35 or Witepsol S58) calculated to prepare 2-mg suppositories was melted and allowed to cool. Prior to the congealing point of the bases, accurately weighed DZ-loaded SLN dispersions (F1 and F10), equivalent to 2 mg DZ/suppository, were added to the melted bases. Homogeneous dispersions were formed in melted base and then molded. After the suppositories were cooled at room temperature, they were stored at 4°C to avoid the development of cracking. Percent compositions (w/w) of DZ-loaded SLN suppositories as well as suppositories prepared using DZ pure drug are shown in Table II. Formulations B1 and B2 (prepared by the same procedure but contained free DZ equivalent to 2 mg per suppository) were used as control.
Table II.
Percent Composition of the Investigated Diazepam-Loaded SLN Suppositories (%, w/w)
| Formula | Diazepam | Diazepam-loaded SLNs | Witepsol W35 | Witepsol S58 | |
|---|---|---|---|---|---|
| F 1 | F 10 | ||||
| B1 | 0.1 | – | – | 99.9 | – |
| B2 | 0.1 | – | – | – | 99.9 |
| B3 | – | 40 | – | 60 | – |
| B4 | – | 40 | – | – | 60 |
| B5 | – | – | 40 | 60 | – |
| B6 | – | – | 40 | – | 60 |
In order to evaluate the prepared DZ-loaded SLN suppositories compared to the control suppositories prepared by loading the drug directly into the suppository base, the in vitro release profiles of the suppositories were performed using USP basket method (Pharma Test, Germany) (24). Four hundred milliliters of phosphate buffer (pH 7.4) were used as the dissolution media stirred at a rate of 25 rpm. The suppositories were placed in the phosphate buffer and maintained at 37°C for a period of 8 h. At appropriate predetermined time intervals, samples were taken, filtered, and assayed by UV spectrophotometer (Shimadzu, model UV-1601 PC, Kyoto, Japan). The mean of three determinations was used to calculate the drug release from each of the formulation.
RESULTS AND DISCUSSION
Characterization of DZ-Loaded SLNs
DZ Entrapment Efficiency %EE
The prerequisite to obtain a sufficient loading capacity was a sufficiently high solubility of the drug in the melted lipid. The amount of drug to be incorporated into the delivery system is dependent on the physicochemical properties of drug and the preparation process (25,26).
In order to attain optimal DZ encapsulation efficiency, several factors were varied including the type and concentration of surfactant as well as the type and concentration of lipid material used. The entrapment efficiencies of all SLN formulations are presented in Fig. 1. It is observed that varying the type and concentration of either the surfactant or the lipid material used had a noticeable influence on the entrapment efficiencies of prepared SLNs. For SLN formulations prepared using either Compritol® ATO 888 (F1- F8) or Imwitor® 900K (F9- F16) as lipid material, when the amounts of the emulsifiers increased, the entrapment efficiency decreased. This could be attributed to the increase in the solubility of DZ in the aqueous phase as the percentage of surfactant increased, due to the solubilization effect of the emulsifier (23,27). This result was obtained for all formulations with the exception of F3 and F4 as well as F9 and F10, where an increase in surfactant concentration led to an increase in the amount of DZ entrapped. For F3 and F4, prepared using Compritol® ATO 888, it could be suggested that, in the presence of this lipid material, at its lower concentration (5%), the increase in Poloxamer concentration increased the entrapment as it acted as a steric stabilizer (16). While for F9 and F10, containing Imwitor® 900K as lipid material, the increase in entrapment upon increasing Tween 80 concentration could be attributed to the added emulsification and stabilization effect of this lipid material in the presence of Tween 80 (14).
Fig. 1.

Entrapment efficiency (%EE) of diazepam in different diazepam-loaded SLN formulations
It could also be noted that for formulations prepared using either Compritol® ATO 888 or Imwitor® 900K as lipid matrix, increasing the lipid concentration from 5% to 10% consequently resulted in a decrease in the amount of DZ entrapped. The decrease in %EE suggests that the crystallization of the lipid phase produces a partial expulsion of the drug on the particle surface (28). Furthermore, the high viscosity at the interface produced by higher lipid concentration will cause a decrease of solvent diffusion and hence fewer lipid molecules will be carried into the aqueous phase. Therefore, the formation and stabilization of lipid aggregates at these higher concentrations are reduced (29).
For SLN formulations prepared using a mixture of surfactants F17–F20, it could be observed that Compritol® ATO 888 as a lipid matrix resulted in higher drug entrapment than Imwitor® 900K (8). Compritol® ATO 888 as a mixture of monoglycerides, diglycerides, and triglycerides produces less ordered lipid crystals than Imwitor® 900K which is composed solely of glycerol monostearate. In the nanoparticle structure, the lipid that results in a highly crystalline structure with a perfect lattice would lead to drug expulsion. On the other hand, the imperfection (lattice defects) of the lipid structure could offer more loading space to accommodate drugs. As a result, the structure of less ordered arrangement in the nanoparticles would be beneficial to the drug loading capacity (14,30).
Transmission Electron Microscopy and Particle Size Analysis
Figure 2 shows the shape of the nanoparticles entrapping DZ; the particles investigated reveal round and homogeneous shading; the micrographs also confirmed that the prepared SLNs were less than 500 nm in size. However, in order to obtain more precise information on the size distribution, more careful analyses including LD were performed and the results for which are given hereunder.
Fig. 2.

Transmission electron micrographs of a pure diazepam, b diazepam-loaded SLN dispersion formula F1, c diazepam-loaded SLN dispersion formula F10, d SLN micrograph representing the core and shell theory
Figure 2d illustrates the presence of a layer surrounding the particles which is characteristic in the case of DZ SLNs as reported by Sznitowska et al. (12). In this study, the authors confirmed by TEM that some of the drug was on the surface-forming DZ layer around the particles; such layer was not present in the unloaded SLNs. This is also in agreement with other investigations, which postulate a drug-enriched shell around a core, explained by drug expulsion due to increase in lipid crystallinity with time (8,18,31). The authors also estimated that this fraction contained about 15% of the total DZ while the rest of the drug is incorporated in a lamellar lattice structure of the lipid core and this explains the fast release of the drug-enriched shell with the prolonged release of DZ incorporated in the core.
Laser diffractometric results for particle size analysis revealed that 60% of the prepared SLN formulations possessed particle sizes below 500 nm at room temperature (Fig. 3). A sufficient high-energy input is necessary to break down the droplets into the nanometer range. A finer dispersion can be obtained by using higher energy derived from elevated production temperature, pressure, higher stirring rate, longer emulsification time, stronger ultrasound power…etc. Besides production parameters, the lipid matrix, the surfactant blend as well as the viscosity of lipid and aqueous phase influence the outcome of the procedure. Similarly, surfactant concentration showed a profound influence on the particle size and distribution (26,30,32).
Fig. 3.

The mean particle sizes of different diazepam-loaded SLN formulations (0 day)
As shown in Fig. 3, as surfactant concentration is increased, a consequent decrease in particle size of all SLN formulations is observed. The decrease in nanoparticles size at high surfactant concentrations is due to effective reduction in interfacial tension between the aqueous and lipid phases, leading to the formation of emulsion droplets of smaller size, which on cooling results in smaller nanoparticles. Furthermore, high surfactant concentrations effectively stabilize the particles created by forming a steric barrier on the particle surface, thereby protecting the particles from coagulation (9,16,33).
It could also be noted that, for formulations F17–F20, composed of a mixture of surfactants, Compritol® ATO 888 as a lipid material produced SLNs which were larger in size than those produced using Imwitor® 900K. It was suggested that glycerol monostearate is not only used as a lipid matrix but also as a surfactant that facilitates emulsification and formation of SLNs of smaller particle sizes (14,34).
Differential Scanning Calorimetry and X-ray Diffraction
DSC experiments are useful to understand solid dispersions like solid solutions, simple eutectic mixtures, or, as in this case, drug and lipid interactions. It is a tool that gives an insight into the melting and recrystallization behavior of crystalline materials like SLNs (14,30). The DSC thermograms of pure DZ, Compritol® ATO 888, Imwitor® 900K as well as SLN formulations F1 and F10 are shown in Fig. 4. The thermal curves of Compritol® ATO 888 and Imwitor® 900K bulk lipids showed endothermic peaks at 70.4°C and 60.6°C, respectively. The melting endothermic peaks of these lipids in the prepared SLNs F1 and F10 were slightly shifted to a lower temperature (58.6°C for F1 and 58.3°C for F10). The decrease in melting temperature of glyceride lipid nanoparticles compared with bulk lipids has been attributed to their small size (nanometer range), the dispersed state of the lipid, and the presence of surfactants (16,18,35). Furthermore, the lower melting enthalpy values should suggest less ordered lattice arrangement of the lipid within nanoparticles compared to the bulk materials (14). DSC thermograms of DZ-loaded SLNs F1 and F10 also show depressed peaks of DZ compared to the pure drug, which suggests that DZ could be present in an amorphous form in the SLNs; this behavior is expected to improve the solubility of the drug in water resulting in a better bioavailability (36).
Fig. 4.

DSC thermograms of A pure diazepam, B Compritol® ATO 888, C diazepam-loaded SLN formula F1 prepared using 5% Compritol® ATO 888 and 2.5% Tween 80, D Imwitor® 900K, and E diazepam-loaded SLN formula F10 prepared using 5% Imwitor® 900K and 5% Tween 80. The curves have been displaced vertically for better visualization
X-ray diffraction data listed in Fig. 5 were in good agreement with the results obtained by DSC measurements. The diffraction pattern of the bulk lipid matrix showed remarkable difference from those of the nanoparticles, where the bulk lipids had sharp peaks that are almost absent in the diffractograms of the SLNs. This indicates the less ordered crystal arrangements in the SLN formulations compared to the pure lipid materials, such an amorphous state would contribute to the higher drug loading capacity. Additionally, the observed disappearance of DZ peaks in both formulations indicates that it does not exist in the crystalline state in the SLNs (14,37).
Fig. 5.

X-ray diffraction pattern of A pure diazepam, B bulk Compritol® ATO 888, C diazepam-loaded SLN formula F1 prepared using 5% Compritol® ATO 888 and 2.5% Tween 80, D bulk Imwitor® 900K, and E diazepam-loaded SLN formula F10 prepared using 5% Imwitor® 900K and 5% Tween 80. The curves have been displaced vertically for better visualization
In Vitro Release from SLNs
The in vitro release results of certain SLN formulations are shown in Fig. 6. It could be noted that certain formulations (F1, F2, F4, F7, F10, and F11) were chosen in order to evaluate their in vitro drug release; this choice was based upon the entrapment efficiencies of the formulations, where a %EE of ∼70% or higher was an inclusion criterion for the release study. The percentage of drug released (% ± SD) after 2 h (Q2 h) and 8 h (Q8 h) from the prepared SLNs are shown in Table III.
Fig. 6.

Release profiles of diazepam-loaded SLNs in 7.4 phosphate buffer at 37 ± 0.5°C; each is the mean of three experiments
Table III.
The Percentage of Diazepam Released after 2 h (Q 2 h) and 8 h (Q 8 h) from the Prepared SLNs, DZ Solution (S), DZ-SLN-based Suppositories (B3–B6), and DZ Suppositories (B1 and B2; % w/w ± SD)
| Formula | Q 2 h a (% ± SD) | Q 8 h a (% ± SD) |
|---|---|---|
| F1 | 55.70 ± 2.57* | 89.9 ± 4.18 |
| F2 | 24.95 ± 2.47* | 35.5 ± 3.37* |
| F4 | 11.32 ± 1.74* | 28.12 ± 2.49* |
| F7 | 34.45 ± 2.43* | 75.6 ± 4.27* |
| F10 | 50.15 ± 4.39* | 95.17 ± 6.29 |
| F11 | 37.28 ± 3.97* | 73.32 ± 4.83* |
| S | 90.69 ± 3.2 | 99.62 ± 5.33 |
| B1 | 97.71 ± 3.12 | 99.32 ± 3.27 |
| B2 | 98.98 ± 2.80 | 99.69 ± 3.81 |
| B3 | 38.51 ± 3.55** | 74.81 ± 3.97** |
| B4 | 40.44 ± 2.04*** | 81.30 ± 3.25*** |
| B5 | 47.60 ± 3.68** | 89.92 ± 4.13 |
| B6 | 53.75 ± 3.89*** | 92.14 ± 4.44 |
*P < 0.05, compared with the corresponding percent released (Q 2 h or Q 8 h) from DZ solution S; **P < 0.05, compared with the corresponding percent released (Q 2 h or Q 8 h) from B1; ***P < 0.05, compared with the corresponding percent released (Q 2 h or Q 8 h) from B2
aEach value represents mean ± SD of three determinations
In order to compare the drug release profile from the prepared SLN formulations, Q2 h and Q8 h were statistically analyzed using one-way ANOVA followed by post hoc multiple comparisons using LSD test.
Generally, all SLN formulations showed significant slower release than DZ solution (S; P < 0.05). This confirms that a sink condition for DZ release was accomplished and that the dialysis membrane used in the dissolution procedure did not limit DZ release.
Additionally, in all tested SLN formulations, a rapid release was observed in the first 2 h that reached about 50% of the overall DZ released from each formulation, which could be due to the drug-enriched shell around the particles. This was followed by prolonged DZ release due to slow diffusion of the lipophilic drug from the lipid matrix.
Although the release rate of SLNs could be influenced by complex factors, it was reported that among the factors that contribute to a fast release are the large surface area, a high diffusion coefficient due to small molecular size, low viscosity in the matrix, and a short diffusion distance for the drug (i.e., release from outer surface region of the nanoparticle) (18).
By inspection of the data in Table III, it could be concluded that the release extents from formula F1 is significantly higher (P < 0.05) than F2 (Q2 h = 55.7% and 24.9%, Q8 h = 89.9% and 35.5% for F1 and F2, respectively). This difference could be attributed to the increase in surfactant concentration (Tween 80) from 2.5% to 5%. In fact, the mechanism of action of surfactants is complex and is not fully understood due to the large variety of effects they can produce (38,39). Nevertheless, it has been reported that the increase in Tween 80 concentration results in a consequent increase in the viscosity of the lipid-solvent phase (5,29), which in turn leads to a slower release rate in the SLNs prepared using higher Tween 80 content.
By comparing the release results of SLN formulations F2 and F4 in Fig. 6 and Table III, F2 exhibited higher release extents than F4 (Q2 h = 24.9% and 11.3% (P < 0.05), Q8 h = 35.5% and 28.1% for F2 and F4, respectively), which could be due to the smaller particle size of F2 formulation (328 nm) than that of F4 (1,080 nm). In fact, as the particle size decreases, the available surface area increases and the release is increased, consequently (18).
The effect of the type of lipid matrix used on the release of SLNs is illustrated by formulations F2 and F10, where the formulation (F2) containing Compritol® ATO 888 exhibited significantly more sustained release than F10 (P < 0.05 for corresponding values of Q2 h and Q8 h), containing Imwitor 900K. As mentioned above, Compritol® ATO 888 produces less ordered lipid crystals than Imwitor® 900K, leading to lower drug expulsion from the imperfect lattice, contributing to the prolonged release of the lipophilic drug (14,30).
Evaluation of the DZ SLN-Based Suppositories
The in vitro release results of the prepared DZ SLN-based suppositories as well as suppositories prepared using DZ pure drug are shown in Fig. 7. The percentage of drug released (% ± SD) after 2 h (Q2 h) and 8 h (Q8 h) from the prepared suppositories are shown in Table III. The release from B1 and B2 containing the pure drug is significantly faster (P < 0.05) than the SLN-containing suppository formulations (B3–B6). B1 and B2 exhibited ∼100% release of the drug after 1.5 h while B3–B6 continued to release their DZ contents over the entire 8-h period. Figure 7 also demonstrates that the release of DZ from the Witepsol W35 suppositories (B1) was slightly slower (P > 0.05) than from those prepared using Witepsol S58 (B2); this could be explained by the fact that Witepsol W35 possesses a slightly higher melting point than Witepsol S58. The overall release results prove that the release of DZ from the SLN-based suppositories (B3–B6) was consistent with the in vitro release of DZ from the SLN formulations F1 and F10. It has been shown that the release of DZ from F1 SLN formulation was slower than that of F10; this was reflected on the suppositories’ release where formulations B3 and B4 illustrated slower release than B5 and B6.
Fig. 7.

Release profiles of diazepam from suppositories in 7.4 phosphate buffer at 37 ± 0.5°C; each is the mean of three experiments
CONCLUSIONS
In this study, a poorly water-soluble drug DZ was successfully incorporated into SLNs by modified high-shear homogenization and ultrasound techniques. The effects of different formulation variables on %EE and physicochemical properties were investigated. The in vitro release tests confirmed the prolongation of the drug release time, where SLN formulations showed slower release compared to the diffusion rate of the DZ solution. The type and concentration of surfactant as well as the lipid matrix used had a great influence on the physicochemical characterization of SLNs and the in vitro drug release. SLN formulations F1 and F10 composed of Tween 80 as a surfactant and lower concentration of the lipid matrix (5% Compritol ATO 888 for F1 and 5% Imwitor 900 K for F10) showed the best results in view of the entrapment efficiency as well as in vitro drug release. The SLN-based suppositories as final dosage form would provide means by which the DZ- loaded SLNs would be administered rectally, which is the most suitable route in most epileptic cases as previously discussed. The in vitro release results from the DZ SLN-based suppositories were consistent with the release of DZ from the SLN formulation F1 and F10. It appears that SLNs as well as the prepared suppositories offer a promising delivery system for the prolongation of DZ release through the rectal route. However, further investigation is necessary to establish diazepam in vitro–in vivo relationship for the sedation effect following rectal delivery and the clinical relevance of these findings.
References
- 1.Alldredge B., Gelb A., Isaacs S., Corry M., Allen F., Ulrich S., Gottwald M., O’Neil N., Neuhaus J., Segal M., Lowenstein D. A comparison of lorazepam, DZ, and placebo for the treatment of out-of-hospital status epilepticus. N. Engl. J. Med. 2001;345:631–637. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- 2.O’Dell C. What do we tell parents of a child with simple or complex febrile seizures? In: Baram T. Z., Shinnar S., editors. Febrile seizures. San Diego: Academic; 2002. pp. 305–316. [Google Scholar]
- 3.O’Dell C., Shinnar S., Ballaban-Gil K., Hornick M., Sigalova M., Kang H., Moshé S. Rectal DZ gel in the home management of seizures in children. Pediatr. Neurol. 2005;33:166–172. doi: 10.1016/j.pediatrneurol.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 4.McNamara J. O. Drugs effective in the therapy of the epilepsies. In: Gilman A. G., editor. The pharmacological basis of therapeutics. New York: McGraw Hill; 1996. pp. 478–485. [Google Scholar]
- 5.Li L., Nandi I., Kim K. Development of an ethyl laurate-based microemulsion for rapid-onset intranasal delivery of DZ. Int. J. Pharm. 2002;237:77–85. doi: 10.1016/S0378-5173(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 6.Kriel R., Cloyd J., Pellock J., Mitchell W., Cereghino J., Rosman N. Rectal DZ gel for treatment of acute repetitive seizures. Pediatr. Neurol. 1999;20:282–288. doi: 10.1016/S0887-8994(98)00156-8. [DOI] [PubMed] [Google Scholar]
- 7.Carceles M., Ribó A., Dávalos R., Martinez T., Hernández J. Effect of DZ on adenosine 3, 5-cyclic monophosphate (cAMP) plasma levels in anesthetized patients. Clin. Ther. 2004;26:737–743. doi: 10.1016/S0149-2918(04)90073-2. [DOI] [PubMed] [Google Scholar]
- 8.Müller R. H., Mader K., Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery—a review of the state of the art. Eur. J. Pharm. Biopharm. 2000;50:161–177. doi: 10.1016/S0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 9.Mehnert W., Mader K. Solid lipid nanoparticles: production, characterization and applications. Adv. Drug. Deliv. Rev. 2001;47(2–3):165–196. doi: 10.1016/S0169-409X(01)00105-3. [DOI] [PubMed] [Google Scholar]
- 10.Parfitt K. Martindale, the complete drug reference. 32. London: Pharmaceutical Press; 1999. pp. 661–668. [Google Scholar]
- 11.MacDonald A., Michaelis A., Senkowski B. Analytical profiles of drug substances and excipients. San Diego: Academic; 1972. pp. 79–99. [Google Scholar]
- 12.Sznitowska M., Gajewska M., Janicki S., Radwanska A., Lukowski G. Bioavailability of DZ from aqueous-organic solution, submicron emulsion and solid lipid nanoparticles after rectal administration in rabbits. Eur. J. Pharm. Biopharm. 2001;52:159–163. doi: 10.1016/S0939-6411(01)00157-6. [DOI] [PubMed] [Google Scholar]
- 13.Cloyd J., Lalonde R., Beniak T., Novack G. A single blind, crossover comparison of the pharmacokinetics and cognitive effects of a new rectal DZ gel with intravenous DZ. Epilepsia. 1998;39:520–526. doi: 10.1111/j.1528-1157.1998.tb01415.x. [DOI] [PubMed] [Google Scholar]
- 14.Hou D., Xie C., Huang K., Zhu C. The production and characteristics of solid lipid nanoparticles (SLNs) Biomaterials. 2003;24:1781–1785. doi: 10.1016/S0142-9612(02)00578-1. [DOI] [PubMed] [Google Scholar]
- 15.Souto E. B., Wissing S. A., Barbosa C. M., Müller R. H. Development of a controlled release formulation based on SLN and NLC for topical clotrimazole delivery. Int. J. Pharm. 2004;278:71–77. doi: 10.1016/j.ijpharm.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 16.Reddy L. H., Murthy R. S. Etoposide-loaded nanoparticles made from glyceride lipids: formulation, characterization, in vitro drug release, and stability evaluation. AAPS PharmSciTech. 2005;6((2)):Article 24. doi: 10.1208/pt060224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim B., Na K., Choi H. Preparation and characterization of solid lipid nanoparticles (SLN) made of cacao butter and curdlan. Eur. J. Pharm. Sci. 2005;24:199–205. doi: 10.1016/j.ejps.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 18.zur Muhlen A., Schwarz C., Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery–drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998;45(2):149–155. doi: 10.1016/S0939-6411(97)00150-1. [DOI] [PubMed] [Google Scholar]
- 19.Wissing S. A., Kayser O., Müller R. H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug. Deliv. Rev. 2004;56:1257–1272. doi: 10.1016/j.addr.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Manjunath K., Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J. Control. Rel. 2005;107:215–228. doi: 10.1016/j.jconrel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 21.El-Gazayerly O. N., Hikal A. H. Preparation and evaluation of acetazolamide liposomes as an ocular delivery system. Int. J. Pharm. 1997;158:121–127. doi: 10.1016/S0378-5173(97)00186-5. [DOI] [Google Scholar]
- 22.Devaraj G. N., Parakh S. R., Devraj R., Apte S. S., Rao B. R., Rambhau D. Release studies on niosomes containing fatty alcohols as bilayer stabilizers instead of cholesterol. J. Colloid. Interface Sci. 2002;251:360–365. doi: 10.1006/jcis.2002.8399. [DOI] [PubMed] [Google Scholar]
- 23.Luo Y., Chen D., Ren L., Zhao X., Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J. Control. Release. 2006;114:53–59. doi: 10.1016/j.jconrel.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Hanaee J., Javadzadeh Y., Taftachi S., Farid D., Nokhodchi A. The role of various surfactants on the release of salbutamol from suppositories. IL FARMACO. 2004;59:903–906. doi: 10.1016/j.farmac.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 25.K. Westesen. Particles with modified physico-chemical properties, their preparation and uses, US Patent 6, 197, 349 (2001).
- 26.Li Y., Dong L., Jia A., Chang X., Xue H. Preparation and characterization of solid lipid nanoparticles loaded traditional Chinese medicine. Int. J. Biol. Macromol. 2006;38:296–299. doi: 10.1016/j.ijbiomac.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Venkateswarlu V., Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J. Control. Release. 2004;95:627–638. doi: 10.1016/j.jconrel.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Casadei M. A., Cerreto F., Cesa S., Giannuzzo M., Feeney M., Marianecci C., Paolicelli P. Solid lipid nanoparticles incorporated in dextran hydrogels: a new drug delivery system for oral formulations. Int. J. Pharm. 2006;325:140–146. doi: 10.1016/j.ijpharm.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 29.Quintanar-Guerrero D., Tamayo-Esquivel D., Ganem-Quintanar A., Allémanna E., Doelker E. Adaptation and optimization of the emulsification–diffusion technique to prepare lipidic nanospheres. Eur. J. Pharm. Sci. 2005;26:211–218. doi: 10.1016/j.ejps.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Jenning V., Thunemann A. F., Gohla S. H. Characterization of a novel solid lipid nanoparticle carrier system based on binary mixtures of liquid and solid lipids. Int. J. Pharm. 2000;199:167–177. doi: 10.1016/S0378-5173(00)00378-1. [DOI] [PubMed] [Google Scholar]
- 31.zur Muhlen A., Mehnert W. Drug release mechanism of prednisolone loaded solid lipid nanoparticles. Pharmazie. 1998;53:552–555. [Google Scholar]
- 32.Schwarz C., Mehnert W., Müller R. H. Influence of production parameters of solid lipid nanoparticles (SLN) on the suitability for intravenous injection. Eur. J. Pharm. Biopharm. 1994;40:24S. [Google Scholar]
- 33.Lim S., Kim C. Formulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all-trans retinoic acid. Int. J. Pharm. 2002;243:135–146. doi: 10.1016/S0378-5173(02)00269-7. [DOI] [PubMed] [Google Scholar]
- 34.Rowe R. C., Sheskey P. J., Weller P. J. Handbook of pharmaceutical excipients. 4. London: American Pharmaceutical Association, Pharmaceutical Press; 2003. [Google Scholar]
- 35.Bunjes H., Westesen K., Koch M. H. J. Crystallization tendency and polymorphic transitions in triglyceride nanoparticles. Int. J. Pharm. 1996;129:159–173. doi: 10.1016/0378-5173(95)04286-5. [DOI] [Google Scholar]
- 36.Cavalli R., Caputo O., Carlotti M., Trotta M., Scarnecchia C., Gasco M. R. Sterilization and freeze-drying of drug-free and drug-loaded solid lipid nanoparticles. Int. J. Pharm. 1997;148:47–54. doi: 10.1016/S0378-5173(96)04822-3. [DOI] [Google Scholar]
- 37.Chattopadhyay P., Shekunov B. Y., Yimb D., Cipolla D., Boyd B., Farr S. Production of solid lipid nanoparticle suspensions using supercritical fluid extraction of emulsions (SFEE) for pulmonary delivery using the AERx system. Adv. Drug. Deliv. Rev. 2007;59:444–453. doi: 10.1016/j.addr.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Gibaldi M., Feldman S. Mechanisms of surfactant effects on drug absorption. J. Pharm. Sci. 1970;59:579–589. doi: 10.1002/jps.2600590502. [DOI] [PubMed] [Google Scholar]
- 39.Rieger M. Surfactants. In: Lieberman H., Rieger M., Banker G., editors. Pharmaceutical dosage forms: disperse systems. New York: Marcel Dekker; 1988. pp. 285–366. [Google Scholar]