

Abstract
Membrane proteins present major challenges for structural biology. In particular, the production of suitable crystals for high-resolution structural determination continues to be a significant roadblock for developing an atomic-level understanding of these vital cellular systems. The use of detergents for extracting membrane proteins from the native membrane for either crystallization or reconstitution into model lipid membranes for further study is assumed to leave the protein with the proper fold with a belt of detergent encompassing the membrane-spanning segments of the structure. Small-angle x-ray scattering was used to probe the detergent-associated solution conformations of three membrane proteins, namely bacteriorhodopsin (BR), the Ste2p G-protein coupled receptor from S. cerevisiae, and the E. coli porin OmpF. The results demonstrate that, contrary to the traditional model of a detergent-associated membrane protein, the helical proteins BR and Ste2p are not in the expected, compact conformation and associated with detergent micelles, while the beta-barrel OmpF is indeed embedded in a disk-like micelle in a properly-folded state. The comparison provided by the BR and Ste2p, both members of the 7TM family of helical membrane proteins, further suggests that the inter-helical interactions between the transmembrane helices of the two proteins differ, such that BR, like other rhodopsins, can properly refold to crystallize, while Ste2p continues to prove resistant to crystallization from an initially detergent-associated state.
Keywords: membrane proteins, detergent, G-protein coupled receptor, small-angle x-ray scattering
Membrane proteins comprise roughly 30% of most genomes sequenced to date, and serve a broad array of functions, including membrane transport and transmembrane signaling. Yet despite intense interest and study, membrane proteins continue to pose significant scientific and technical challenges, particularly for high-resolution structure determination. The number of atomic-resolution membrane protein structures makes up a very small fraction of the total number of structures deposited in the Protein Data Bank1. Membrane protein purification involves the use of detergents to extract the protein from the native membrane. Care must be taken to ensure that the detergent is efficient at recovering the membrane protein, which is often difficult to express in quantities comparable to soluble proteins. Further, the detergent must not irreversibly denature the membrane protein, thereby precluding refolding for further structural or functional studies.
It is generally assumed that a detergent-solubilized membrane protein remains properly folded with the detergent encompassing the trans-membrane portions of the structure2, as has been observed for the detergent visible in high-resolution structures of membrane proteins. For example, the crystal structure of the E. coli porin OmpF shows a one molecule thick belt of surfactant molecules around the membrane-spanning face of the protein3. Similarly, high-resolution crystal structures of bacteriorhodopsin (BR) show a limited number of detergent4 and lipid5 molecules surrounding the protein. Crystal structures of rhodopsin, a G-protein coupled receptor (GPCR), also have detergent molecules6-8. The detergent is not always well-ordered, leading to its exclusion from the reported structure. Prince and coworkers utilized neutron diffraction to visualize the low-resolution structure of the detergent around the light harvesting complex LH2 complex from R. acidophila, again showing a clear belt of detergent around the hydrophobic portion of the protein9. While these structures provide important insight into the interaction between membrane-proteins and detergent or lipids in the native membrane, the results provide little insight into the solution behavior of the detergent-solubilized proteins.
Detergent interactions with proteins have been studied by small-angle scattering techniques, most commonly using soluble proteins. The detergent micelles bind to hydrophobic portions of the protein sequence, disrupting the hydrophobic interactions that maintain the properly-folded tertiary structure of the protein. This structure, in which micelles decorate the unfolded protein, is often referred to as “beads-on-a-string” or a necklace model 10,11. The small-angle scattering signal from such a structure can be described analytically by treating it as fractal scattering. Little work has been done to study membrane proteins12,13, as they tend to be much harder to produce in sufficient quantities for study by small-angle scattering techniques, and detergent is required to dissolve the membrane protein in solution. Characterization of the structures of detergent-associated membrane proteins would validate assumptions using in functional studies and provide insight into the challenges encountered in crystallization of membrane proteins.
Here, a study of the solution structures of three different membrane protein-detergent complexes using circular dichroism (CD) spectroscopy and small-angle x-ray scattering (SAXS) is presented. The porin OmpF was studied in complex with the detergent octyl polyoxyethylene (OPOE), the detergent used in crystallization3. Bacteriorhodopsin, a 7TM protein, was studied in octyl glucoside (OG), also used for crystallization4. As a third model system, the Ste2p GPCR from S. cerevisiae, another member of the 7TM family of proteins, was studied in complex with dodecyl maltoside (DM). Unlike the other two proteins, the high-resolution structure of Ste2p has not been determined. The results demonstrate that detergent associates with OmpF in solution in the expected, belt-like state. In contrast, the two 7TM proteins are found to exist in partially unfolded states. The results have important implications for biophysical characterization of detergent-solubilized membrane proteins and for membrane protein crystallization from a detergent-solubilized state.
Materials and Methods
Octyl polyoxyethylene (OPOE) was purchased from Axxora LLC (San Diego, CA). Dodecyl maltoside (DM) and octyl glucoside (OG) were from Anatrace Inc. (Maumee, OH). Bacteriorhodopsin (BR) from H. salinarum was bought from Sigma (St. Louis, MO). Other chemicals were obtained from VWR (West Chester, PA) or Sigma (St. Louis, MO). All chemicals were used without further purification.
Purification and analysis of the S. cerevisiae Ste2p GPCR were recently described14. Briefly, Ste2p with a rhodopsin affinity tag, consisting of a nine amino acid residue sequence of rhodopsin, was constructed and used for purification. Yeast cells were homogenized and membranes were collected from soluble fraction using ultracentrifugation. Resulting membranes were solubilized in the presence of DM and the insoluble fraction was removed by ultracentrifugation. Solubilized membranes were mixed with monoclonal antibody (rhodopsin) 1D4-coupled Sepharose 4B and the resin slurry was batch-loaded onto a column. After extensive washing, the antibody bound receptor was eluted by excessive competition with the 9-amino acid rhopsin peptide.
The AD102/pNLF10 strain for wild type OmpF was a generous gift from Dr. Anne H. Delcour of the University of Houston. OmpF was expressed and purified as described previously15,16 with slight modifications. Briefly, the cells were grown in LB broth and induced with 1mM isopropyl-β-thiogalactose (IPTG). After being washed with cold 10 mM Hepes (pH 7.6), the cells were resuspended in 10 mM Hepes (pH 7.6). The cells were broken by French Press and the DNA was sheared by sonication. Unbroken cells were removed by low-speed centrifugation and the membrane pelleted at 200,000 × g for 2 hours at 20 °C. The membrane pellet was resuspended in 20 mM sodium phosphate buffer (pH 7.6) containing 10 mM NaCl and 1% OPOE with bath sonication. The suspension was then centrifuged at 200,000 × g for 60 min at 20 °C. A subsequent extraction used 1% OPOE and was followed by three additional extractions at 3% OPOE. OmpF was specifically extracted at 3% OPOE. After OPOE concentration was reduced to 0.5% by dialysis, the preparation was applied to an anion exchange column (Mono Q HR5/50; Pharmacia Corp.) equilibrated with 10 mM sodium phosphate buffer (pH 7.6), 10 mM NaCl, 0.5% OPOE. Proteins was eluted with a salt gradient between 250 mM to 500 mM NaCl.
Circular Dichroism Spectroscopy
CD spectra were recorded on a JASCO J-810 Spectropolarimeter (JASCO Inc., Easton, MD) at ambient temperature. A quartz cuvette with a path length of 0.2 cm was employed. Purified proteins reconstituted into detergent micelles were measured. OmpF was measured at a concentration of 0.2 mg/mL in phosphate buffer with 0.5% OPOE, and Ste2p was ~0.1 mg/mL with 0.05% DM, while the BR concentration was 0.1 mg/mL with 1% OG. Protein secondary structure content was evaluated using basis spectra from poly-lysine17 and basis spectra determined from proteins of known structure,18 which were obtained from the K2D19 webpages (http://www.embl-heidelberg.de/~andrade/k2d.html).
Small-angle x-ray scattering
SAXS experiments were performed in the Center for Structural Molecular Biology 4m SAXS facility20. In addition to collecting data for samples with protein, data were collected for the associated detergent-containing buffer solutions. The detergent-containing buffer solutions were reduced with respect to a detergent-free buffer and found to have very low signal relative to the protein-containing samples, indicating that using the detergent-containing buffer solutions as the background for the protein-detergent complex samples was reasonable. Protein concentrations were 9.0 mg/mL for OmpF-OPOE and 20.0 mg/mL for BR-OG, while the Ste2p-DM was collected at 1.0 mg/mL. Additional data sets were collected at 4.5 mg/mL for OmpF-OPOE and 4.6 mg/mL for BR-OG. No concentration-dependent effects were observed. Data were reduced and azimuthally averaged according to previously published procedures20 to the 1D intensity profile I(q) vs. q. Multiple data sets were collected for the protein-detergent complexes to check for artifacts due to radiation damage and no artifacts were observed in the data.
Small-angle x-ray scattering analysis and modeling
When dealing with an isotropic solution of particles, the one-dimensional scattering intensity can be written as follows.
| (1) |
where q = (4π sinθ)/λ, the momentum transfer, in which 2θ is the scattering angle from the incident beam and λ is the wavelength of the incident radiation (1.542 Å). Np is the number density of particles and the product ΔρV is the total excess scattering length of the particle relative to the solvent. The function F(q), called the form factor, results from the particle shape, while the function S(q), called the structure factor, results from interparticle interactions. When the sample is an isotropic, dilute solution of non-interacting particles, S(q) = 1 for all values of q. The time and ensemble average structure that exists within the sample volume is sampled during the measurement.
Data were fit either according to Guiner21, or using a power-law, as appropriate. Structural models were produced from the SAXS data using different approaches. When data were consistent with a dilute solution of identical particles, models were constructed using an existing high-resolution structure embedded in a detergent micelle with two regions of different densities representing the hydrophobic core and a shell to represent the polar groups of the surfactants, as shown in Figure 1, using software developed at Oak Ridge National Laboratory. The parameters for the modeling were the surfactant disk radius R, the half thickness T and the core half-thickness C. Disk radii and the thicknesses of the core and shell were tested using a Monte Carlo search procedure to find a model having a SAXS profile consistent with the data. The intensity profiles were calculated from the models using the software ORNL_SAS22. The 25 best models found by the search, as judged by the χ2 parameter23 of the model intensity profile against the data, were retained in a sorted list for use in assessing the reproducibility and variability of the models24. If such a model was inappropriate, triaxial ellipsoid and triaxial core-shell ellipsoid models were generated using the software ELLSTAT24.
Figure 1.
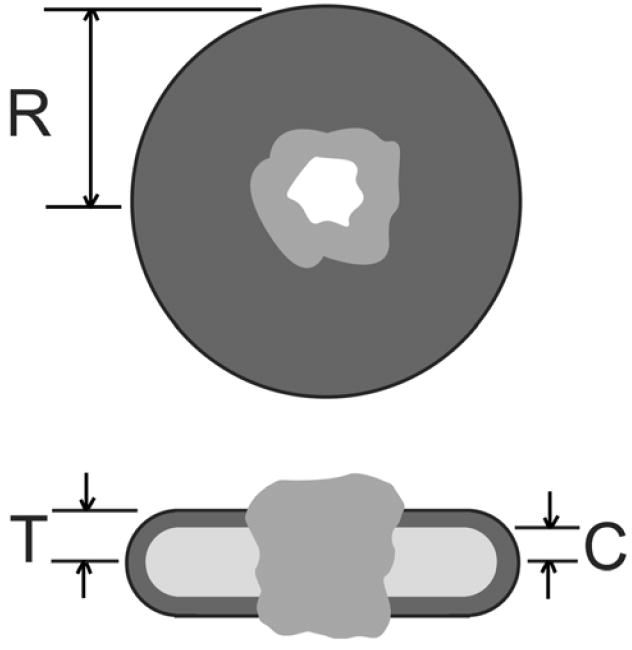
Schematic of the structure used for modeling a properly folded membrane protein in a membrane-like detergent micelle. The upper view shows the detergent disk (dark grey) of radius R penetrated by the structure of the membrane protein (grey) with a water channel through the structure (white). The lower figure shows a cut through the disk with the polar head group region of the detergent (dark grey) and nonpolar core of the bilayer (light grey), with thickness 2C. The micelle thickness is 2T.
The final modeling approach considered the possibility that the detergent had denatured the protein, resulting in a “beads-on-a-string” structure displaying fractal scattering10,11. In the “beads-on-a-string” model the polypeptide chain is in an unfolded, random state in solution with detergent micelles distributed along the polypeptide chain. The intensity was modeled as described in Equation 1. The F(q) of the micelles was that of a core-shell ellipsoid of revolution, which has an analytical form25. In the “beads-on-a-string” model, S(q) accounts for interparticle correlations that exist between the micelles. Previous workers derived an analytical function for S(q), which is given in Equation 210,11.
| (2) |
D is the fractal dimension of the packing of the micelles. If D = 3, then the particles are in a compact arrangement. For values of D less than 3, the micelles are in a more open structure. ξ is the correlation length of the fractal system. The term r0 is the equivalent micelle radius. In the model, the interaction between the micelles is assumed to have a finite range and therefore provides a measure of the extent of unfolding of the polypeptide chain. Γ(x) is the gamma function. The free parameters for the model fitting were the ellipsoidal semi-axes and the D and ξ parameters of S(q). Again, the quality of the fit was evaluated using the reduced χ2 parameter23. The micelle sizes and polar shell thicknesses for BR and OG were allowed to range around the values previously determined for free micelles of the surfactants26.
The electron density of the proteins for the modeling was assigned to an average value for proteins (~0.44 electrons/Å3), while the solvent was 0.34 electrons/Å3. The detergent electron densities were divided into two regions corresponding to the hydrocarbon chains and the polar region corresponding to the rest of the molecules. For DM, the electron density of the chains is 0.276 electrons/Å3 while that of the dry polar region is 0.527 electrons/Å3. In the case of OG, the electron density of the chains is 0.267 electrons/Å3 while that of the dry polar region is 0.538 electrons/Å3. Wet densities for the polar regions of DM and OG were taken from a previously published study26 of protein-free detergent micelles, being 0.520 electrons/Å3 for DM. A value of 0.500 electrons/Å3, being within the range of reported values26, was used for OG. OPOE is actually a mixture of different polar groups attached to the hydrocarbon chain with the same electron density as the chain of OG. The electron density of the OPOE polar group was estimated at 0.37 electrons/Å3.
Results
The SDS-PAGE analysis and Western blot indicated that Ste2p was purified to near homogeneity (Figure 2a). The majority of the Ste2p exists in a monomeric state, although the gel shows a weak band near 100 kDa, suggestive of a small population of Ste2p oligomers. The higher molecular weight band, which is consistent with dimers of Ste2p, is estimated to be ~11% of the population. SDS-PAGE gel (Figure 2b) shows that OmpF proteins in a detergent OPOE solution were dominated by a single band at 64 kDa. This band represents the trimeric OmpF proteins15 with certain secondary structures. No monomeric or dimeric OmpF can be observed from the SDS-PAGE gel.
Figure 2.
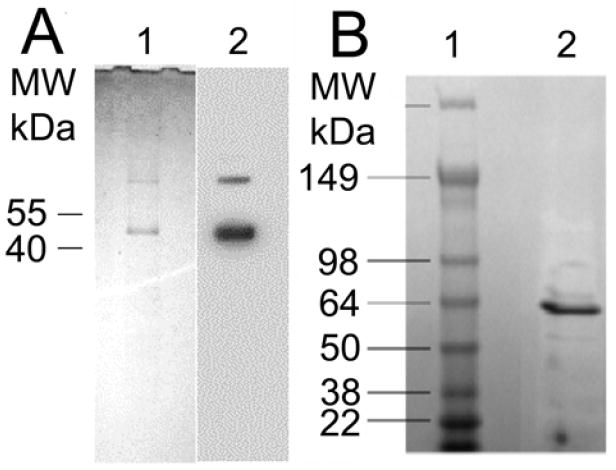
A. Purification of a yeast GPCR, Ste2p. (1) Ste2p was purified and analyzed in SDS-PAGE. Purified sample was stained with Coomassie Blue after SDS-PAGE. A tear in the gel that occurred during drying is visible near 30 kDa. (2) Western blot analysis of the purified receptor using 1D4 antibody is also shown. B. Purification SDS PAGE of OmpF in 4-20% gel. Lane 1 is the mass marker and lane 2 is purified OmpF after FPLC.
The CD scans of OmpF, BR and Ste2p are shown in Figure 3. The results indicate that the trimeric OmpF proteins in 0.5% OPOE solution are rich in β-structure, similar to previously published spectra27,28, although the proteins appear to be less folded than the native trimeric OmpF based on the zero-crossing of the spectrum27,29. The spectrum was fit far better by the set of basis spectra determined from proteins of known structure, yielding 19% α-helix, 74% β-structure and 7% random coil. The CD spectrum of the BR-OG complex is suggestive of a predominantly α-helical structure, having two minima near 208 nm and 222 nm. Fitting using the poly-lysine basis spectra produced better results and show the protein secondary structure to consist of 84% α-helical structures and 16% β-structures. The CD spectrum of Ste2p in 0.5% DM solution indicates that the structure is not strongly α-helical. Instead, the data, which was best fit by the basis spectra determined from known proteins, indicates that the structure is 41% α-helix, 39% β-structures and 20% random coil, roughly consistent in helical content according to a secondary structure prediction using PSIPRED30,31 (49% α-helix, 9% β-structures and 42% random coils).
Figure 3.
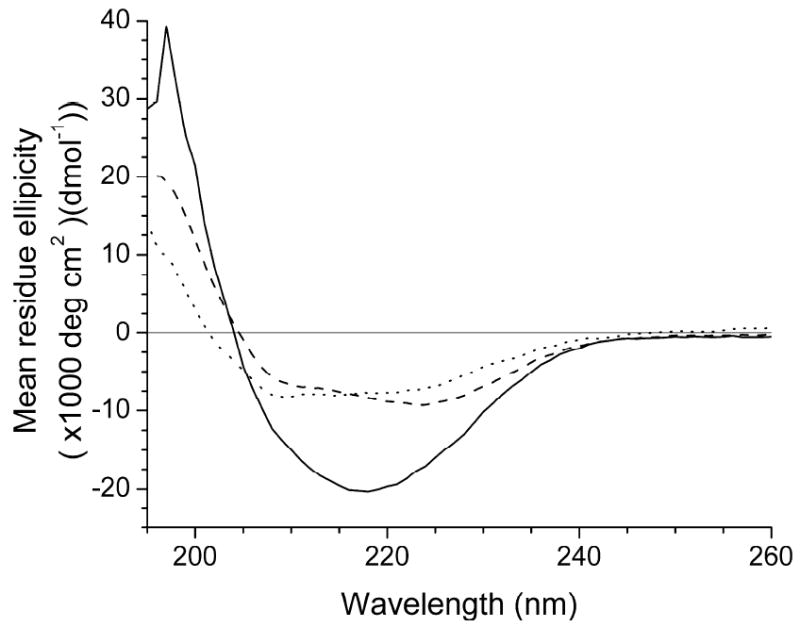
CD spectra of OmpF-OPOE ( ), BR-OG (
), BR-OG ( ) and Ste2p-DM (
) and Ste2p-DM ( ). The OmpF-OPOE spectra is consistent with a primarily beta-sheet structure, while the BR-OG suggests that the structure is predominantly α-helical. The Ste2p-DM spectra suggests that there is a mixture of secondary structures.
). The OmpF-OPOE spectra is consistent with a primarily beta-sheet structure, while the BR-OG suggests that the structure is predominantly α-helical. The Ste2p-DM spectra suggests that there is a mixture of secondary structures.
Small-angle x-ray scattering profiles for the three systems studied are shown in Figure 4. The OmpF-OPOE profile has a clearly different character than the other two systems studied. The particle is compact with a radius of gyration of 50.2 ± 2.2 Å. In contrast, the scattering profiles for the BR-OG and Ste2p-DM samples display power-law behavior, with slopes of -2.4 and -2.5, respectively, over a q-range of 0.02 to 0.08 Å-1. A slope of this magnitude is consistent with mass fractals, and the q-range over which the behavior is displayed includes length scales consistent with the size of the native trimeric form of BR4,5. Neither of the helical protein data sets contain a region suitable for Guinier fitting (q·Rg < 1.3). If the data were the result of aggregation of the properly folded monomeric or native oligomer of either protein encompassed by a detergent micelle belt, one would expect to see evidence of the scattering characteristic of the detergent-associated trimeric form in the case of BR, or the minimal oligomer in the case of the Ste2p, with higher order aggregates. The data would have a character similar to that of the OmpF-OPOE shown in Figure 4. A small population of lower-order oligomers (i.e. dimers or trimers), such as is indicated by the SDS-PAGE gel (Figure 2), would have a limited impact on the data, being most noticeable as slightly increased intensity over the minimal structure at the lowest q-values. The data at larger q-values of slightly aggregated samples would tend toward the intensity profile expected for the minimal oligomer. The fractal character of the data precludes reliable estimation of the amount of oligomers in the sample from the SAXS data.
Figure 4.
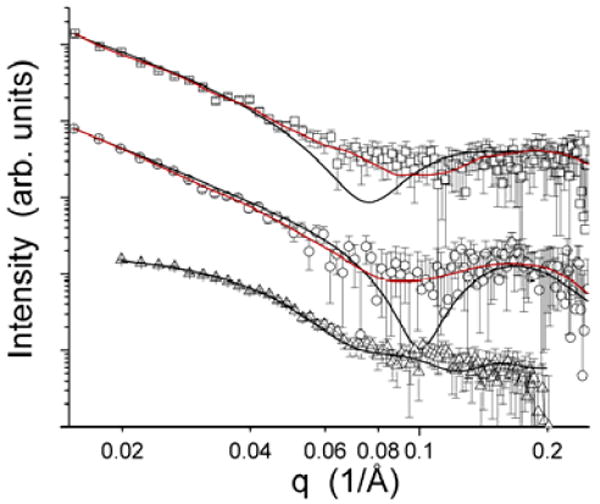
Small-angle x-ray scattering profiles for the three membrane protein-detergent complexes studied: BR-OG (□), Ste2p-DM (○) and OmpF-OPOE (△). The black curves shown with the data are the best model fits for each of the complexes, being the beads-on-a-string model10,11 for BR-OG and Ste2p-DM and the OmpF crystal structure3 embedded in a membrane-like detergent micelle for the OmpF-OPOE complex. The red curves are triaxial core-shell ellipsoidal model best fits generated using ELLSTAT24.
The model fits to the measured scattering profiles are also shown in Figure 4. In the case of the OmpF-OPOE complex, the best-fit model intensity profile, which has a χ2 of 0.798, results from a relatively small detergent micelle surrounding the intact trimer structure, having a radius R of 57.0 Å. The best fit model had T = 26.6 Å and C = 15.5 Å. If one looks at the set of best-fit models with χ2 values within 5% of the best value, the structural parameters R, T and C average, 58.0 Å, 26.6 Å and 15.4 Å, with standard deviations of 0.6 Å, 0.6 Å and 0.2 Å, respectively.
In the case of the BR-OG complex, both the beads-on-a-string models and the core-shell ellipsoidal models produced reasonable fits to the data, as can be seen in Figure 4. The beads-on-a-string model fit the BR-OG data reasonably well, having a χ2 of 2.08, although there are clear deviations present near 0.07 Å-1, where the local minima in model profile is located. For this best-fit model, D was found to be 1.04, ξ was 177.5 Å and the ellipsoidal semi-axes were (15.0 Å, 15.0 Å, 41.0 Å) with a shell thickness of 2.5 Å. Models having χ2 within 5% of the value of the best-fit model have structural parameters that are consistent with the best fit model. The core-shell ellipsoid model, in which the core and shell densities were assigned that of the protein-free detergent micelles, fits the data even better, having a χ2 of 0.89. The ellipsoid is large and flat, having semi-axes of (227.4 Å, 215.0 Å, 17.3 Å) and a shell thickness of 5.9 Å. An attempt to fit the data using a solid, triaxial ellipsoid (not shown) did not produce acceptable fits to the data.
Both modeling approaches also provided acceptable fits to the Ste2p-DM SAXS data (Figure 4). The beads-on-a-string model fit the BR-OG data reasonably well, having a χ2 of 1.02, with the strongest deviations near the minima in the model profile near 0.1 Å-1. For this best-fit model, D was found to be 1.41, ξ was 92.5 Å and the ellipsoidal semi-axes were (19.9 Å, 32.9 Å, 32.9 Å) with a shell thickness of 4.5 Å. Models having χ2 within 5% of the value of the best-fit model have structural parameters that are consistent with the best fit model. Again, the core-shell ellipsoid model having core and shell densities from the protein-free detergent micelles, fits the data very well, having a χ2 of 0.48. The ellipsoid is large and flat, having semi-axes of (249.0 Å, 127.5 Å, 20.8 Å) and a shell thickness of 5.9 Å. An attempt to fit the data using a solid, triaxial ellipsoid (not shown) did not fit the Ste2p-DM SAXS data as well as the core-shell model or the beads-on-a-string model.
Discussion
The results presented here demonstrate that a detergent-solubilized membrane protein is not necessarily properly folded with a belt of detergent encompassing the membrane-spanning portions of the structure. The beta-barrel membrane protein OmpF, which is unrelated to the other proteins studied, in complex with the detergent OPOE forms a compact particle that is consistent with a native trimer3 embedded in a membrane-like structure. The situation was considerably different for the two helical membrane proteins studied. While the CD spectroscopy suggests that the secondary structures are consistent with expectations of helical content, the SAXS data sets are not consistent with a natively folded protein surrounded by a belt of detergent. Previous studies found that BR in OG detergent solution can adopt a different conformation than both native and vesicle-associated states of the protein based on UV-Vis absorption and CD spectroscopic analysis32. Both an unfolded beads-on-a-string model10,11 and a large core-shell ellipsoid model were capable of fitting the data well, but the core-shell models consistently provided better fits to the data. Large, solid ellipsoidal models were not capable of fitting the data as well as either of these two approaches. Yet, the large core-shell model is not particularly physically realistic due to the need for a strong density difference between the core and shell. The protein-detergent ratio for both helical membrane proteins was roughly 1:50 (per monomer), meaning that such a large assembly should have a more uniform density than was required to obtain such good fits to the data due to the penetration of the helices across the ellipsoid along the narrow axis. An alternative possibility is that the helices are adsorbed into the polar region of the bilayer, lying parallel to the surface. Such a configuration would maintain the large scattering length density difference between the core and shell. The nature of the transmembrane structure of natively-folded BR makes such a configuration unlikely, as would be the case for Ste2p. The low detergent content of the complexes may also explain why the data lack a strong minima. A muted minima has been observed for bovine serum albumin complexes with sodium dodecyl sulfate at low concentrations33 and may be due to incomplete denaturing of the protein. The population of oligomers indicated in the Ste2p samples by the SDS-PAGE analyses (Figure 2a) would only flatten the minima significantly if the oligomers were properly folded. The data cannot exclude this possibility. Unfolded Ste2p oligomers would not have a significant impact on the data in this q-range, which is dominated by the micelle scattering. A state in which the individual helices of each polypeptide chain in BR and Ste2p are solubilized by detergent is the most likely model for the system, but no such model for scattering data has been developed. The results demonstrate that the solution state of a detergent-solubilized membrane protein cannot always be assumed to be properly folded and that relatively simple characterizations can be employed to verify the conformation of the protein-detergent complex before continuing on to functional studies or crystallization trials.
Other studies of membrane protein-detergent complexes suggest that both properly folded and denatured states of membrane proteins result from the interaction. An early SAXS with contrast variation study of rhodopsin in complex with dodecyldimethylamine oxide found compact, monomeric rhodopsin associated with detergent shielding the hydrophobic segments of the protein that suggested the detergent formed a belt around the membrane spanning portions of the structure13. Neutron scattering studies found there to be a significant distance between the centers of mass of detergent and rhodopsin within the complexes studied, but this separation was attributed to differential detergent association resulting from the amphiphilic nature of the protein34,35. A similarly compact and well-defined structure was observed for the light-harvesting complex LH2 from R. spheroides 2.4.136. In contrast, another study of the light-harvesting complex LH2 from R. acidophila suggests the possibility that the protein-detergent complex does not have the expected conformation37. While the small-angle scattering data from the LH2 of R. acidophila were analyzed assuming that the complex was correctly folded, the data displays a clear, broad peak near 0.14 Å-1, which can result from small ellipsoidal structures. Similar results were observed for the Wzz protein38. While the data in both studies were interpreted assuming that the particle was properly folded, the presence of the wide angle peak, suggestive of a micelle structure, raises the possibility that the two different protein-detergent complexes studied were also at least partially unfolded.
A recent study of detergent-solubilized photosystem-1 membrane protein complex from spinach suggested that the entire membrane protein complex may not be completely unfolded by the detergent used39. Instead, the results of the study suggested that a properly folded core of protein was surrounded by a cloud of unfolded polypeptide associated with detergent micelles. The photosystem-1 complex differs significantly from any of the proteins studied in the current work, being a great deal larger and composed of multiple, non-identical subunits. A considerable portion of the photosystem-1 complex is not exposed to the hydrophobic core of the membrane, making it possible that association of those portions of the complex with detergent is unfavorable relative to the association of any available detergent with the membrane-exposed portions of the structure. It is unlikely that such an arrangement exists for the much smaller helical proteins studied here because much more of the structure is normally exposed to the nonpolar core of the membrane.
Conclusions
This work demonstrates that detergent-associated membrane proteins need not fold properly with their hydrophobic segments encapsulated in a belt of detergent when in solution. Instead, a detergent, even one expected to be suitably gentle to retain protein functionality, can produce a non-native conformation. Small-angle scattering can play an important role in the preliminary characterization of protein-detergent complexes, particularly those that will be used as foundation material for crystallization work. The results from Ste2p further suggest that one of the causes of difficulties in obtaining crystals for high-resolution structure determination from detergent-solubilized membrane proteins may in fact be that the protein is too denatured to refold when in complex with detergent. The recent high-resolution structures of the engineered40,41 and antibody-bound42 β2-adrenergic GPCR demonstrate the importance of lipidic systems, rather than traditional detergent preparations, for crystallization of GPCRs that should also be applicable to crystallization of other membrane proteins because they better mimic the native membrane environment.
Acknowledgments
The authors would like to thanks Dr. Anne H. Delcour of the University of Houston for the generous gift of the OmpF clone. This work was supported by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed and operated by UT-Battelle, LLC for the U. S. Department of Energy under contract No. DE-AC05-00OR22725, the Joint Directed Laboratory Development Program from the University of Tennessee, and NIH GM-022087 from the National Institute of General Medical Sciences.
References
- 1.White SH. Protein Science. 2004;13:1948. doi: 10.1110/ps.04712004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gohon Y, Popot JL. Current Opinion in Colloid & Interface Science. 2003;8:15. [Google Scholar]
- 3.Cowan SW, Garavito RM, Jansonius JN, Jenkins JA, Karlsson R, Konig N, Pai EF, Pauptit RA, Rizkallah PJ, Rosenbusch JP, Rummel G, Schirmer T. Structure. 1995;3:1041. doi: 10.1016/s0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- 4.Belrhali H, Nollert P, Royant A, Menzel C, Rosenbusch JP, Landau EM, Pebay-Peyroula E. Structure. 1999;7:909. doi: 10.1016/s0969-2126(99)80118-x. [DOI] [PubMed] [Google Scholar]
- 5.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Journal of Molecular Biology. 1999;291:899. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Edwards PC, Burghammer M, Villa C, Schertler GFX. Journal of Molecular Biology. 2004;343:1409. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 7.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. Journal of Molecular Biology. 2004;342:571. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 8.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5982. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prince SM, Howard TD, Myles DAA, Wilkinson C, Papiz MZ, Freer AA, Cogdell RJ, Isaacs NW. Journal of Molecular Biology. 2003;326:307. doi: 10.1016/s0022-2836(02)01361-x. [DOI] [PubMed] [Google Scholar]
- 10.Chen SH, Teixeira J. Physical Review Letters. 1986;57:2583. doi: 10.1103/PhysRevLett.57.2583. [DOI] [PubMed] [Google Scholar]
- 11.Teixeira J. Journal of Applied Crystallography. 1988;21:781. [Google Scholar]
- 12.Lipfert J, Columbus L, Chu VB, Doniach S. Journal of Applied Crystallography. 2007;40:S235. [Google Scholar]
- 13.Sardet C, Tardieu A, Luzzati V. Journal of Molecular Biology. 1976;105:383. doi: 10.1016/0022-2836(76)90100-5. [DOI] [PubMed] [Google Scholar]
- 14.Lee BK, Jung KS, Son C, Kim H, Verberkmoes NC, Arshava B, Naider F, Becker JM. Protein Expression and Purification. 2007;56:62. doi: 10.1016/j.pep.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basle A, Iyer R, Delcour AH. Biochimica Et Biophysica Acta-Biomembranes. 2004;1664:100. doi: 10.1016/j.bbamem.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Simonet VC, Basle AM, Klose KE, Delcour AH. Biophysical Journal. 2003;84:533A. doi: 10.1074/jbc.M301202200. [DOI] [PubMed] [Google Scholar]
- 17.Yang JT, Wu CSC, Martinez HM. Methods in Enzymology. 1986;130:208. doi: 10.1016/0076-6879(86)30013-2. [DOI] [PubMed] [Google Scholar]
- 18.Chang CT, Wu CSC, Yang JT. Analytical Biochemistry. 1978;91:13. doi: 10.1016/0003-2697(78)90812-6. [DOI] [PubMed] [Google Scholar]
- 19.Andrade MA, Chacon P, Merelo JJ, Moran F. Protein Engineering. 1993;6:383. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 20.Woodward JD, Pickel JM, Anovitz LM, Heller WT, Rondinone AJ. Journal of Physical Chemistry B. 2006;110:19456. doi: 10.1021/jp062471z. [DOI] [PubMed] [Google Scholar]
- 21.Guinier A, Fournet G. Small-angle Scattering of X-rays. Wiley; New York: 1955. [Google Scholar]
- 22.Tjioe E, Heller WT. Journal of Applied Crystallography. 2007;40:782. [Google Scholar]
- 23.Taylor JR. An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements. 2. University Science Book; Sausalito, CA: 1997. [Google Scholar]
- 24.Heller WT. Journal of Applied Crystallography. 2006;39:671. [Google Scholar]
- 25.Pedersen JS. Advances in Colloid and Interface Science. 1997;70:171. [Google Scholar]
- 26.Lipfert J, Coltnnbus L, Chu VB, Lesley SA, Doniach S. Journal of Physical Chemistry B. 2007;111:12427. doi: 10.1021/jp073016l. [DOI] [PubMed] [Google Scholar]
- 27.Visudtiphole V, Thomas MB, Chalton DA, Lakey JH. Biochemical Journal. 2005;392:375. doi: 10.1042/BJ20051257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe Y. Journal of Chromatography A. 2002;961:137. doi: 10.1016/s0021-9673(02)00540-x. [DOI] [PubMed] [Google Scholar]
- 29.Perczel A, Park K, Fasman GD. Proteins-Structure Function and Genetics. 1992;13:57. doi: 10.1002/prot.340130106. [DOI] [PubMed] [Google Scholar]
- 30.McGuffin LJ, Bryson K, Jones DT. Bioinformatics. 2000;16:404. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 31.Jones DT. Journal of Molecular Biology. 1999;292:195. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 32.Swords NA, Wallace BA. Biochemical Journal. 1993;289:215. doi: 10.1042/bj2890215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santos SF, Zanette D, Fischer H, Itri R. Journal of Colloid and Interface Science. 2003;262:400. doi: 10.1016/S0021-9797(03)00109-7. [DOI] [PubMed] [Google Scholar]
- 34.Yeager M, Schoenborn B, Engelman D, Moore P, Stryer L. Biophysical Journal. 1976;16:A36. doi: 10.1016/0022-2836(80)90319-8. [DOI] [PubMed] [Google Scholar]
- 35.Osborne HB, Sardet C, Michelvillaz M, Chabre M. Journal of Molecular Biology. 1978;123:177. doi: 10.1016/0022-2836(78)90320-0. [DOI] [PubMed] [Google Scholar]
- 36.Hong XG, Weng YX, Li M. Biophysical Journal. 2004;86:1082. doi: 10.1016/S0006-3495(04)74183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du LC, Weng YX, Hong XG, Xian DC, Katsumi K. Chinese Physics Letters. 2006;23:1861. [Google Scholar]
- 38.Tang KH, Guo H, Yi W, Tsai MD, Wang PG. Biochemistry. 2007;46:11744. doi: 10.1021/bi701181r. [DOI] [PubMed] [Google Scholar]
- 39.O’Neill H, Heller WT, Helton KE, Urban VS, Greenbaum E. Journal of Physical Chemistry B. 2007;111:4211. doi: 10.1021/jp067463x. [DOI] [PubMed] [Google Scholar]
- 40.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 42.Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]