

Abstract
Both the R and S enantiomers of the amphibian alkaloid noranabasamine were prepared in > 30% overall yield with 80% ee and 86% ee, respectively. An enantioselective iridium catalyzed N-heterocyclization reaction with either (R)- or (S)-1-phenylethylamine and 1-(5-methoxypyridin-3-yl)-1,5-pentanediol was employed to generate the 2-(pyridin-3-yl)-piperidine ring system in 69–72% yield.
The pharmacology of amphibian alkaloids has generated significant interest in these molecules over the past decade. Many of these compounds have aided in the elucidation of biological mechanisms and the development of lead compounds for the treatment of a wide variety of pathologies mediated by nicotinic acetylcholine receptors (nAChRs) and corresponding ion channels.1 However, the paucity of useful quantities of isolated amphibian alkaloids has led to a flurry of synthetic activity to make these compounds available for biological study. While many of the amphibian alkaloids possess unique chemical structures,1 the similarity between noranabasamine (1) isolated from the columbian poison dart frog Phyllobates terribilis,2 and plant alkaloids isolated from the tobacco species Nicotian tabacum [e.g. nicotine (2), anabasine (3)]3, as well as the central asian shrub Anabasis aphylla [anabasamine (4)]4 is noteworthy. The plant derived piperidine alkaloids 2 and 3 are widely known to elicit their pharmacological effects via nAChRs.5 Anabasamine (4) has been much less studied but has been reported to inhibit acetylcholine esterase and exhibit anti-inflammatory activity.6
Our interests in the development of new pharmacotherapies for nAChR mediated disorders and disease states7 prompted an investigation into the synthesis of the enantiomers of noranabasamine (1). It was our aim to develop an efficient synthesis of 1, that would provide sufficient quantities for biological evaluation. In addition, it was envisaged that the preparation of both antipodes of 1 would aid in the confirmation of the absolute configuration of the natural product which has yet to be unequivocally established.4 Herein we describe the first enantioselective syntheses of both enantiomeric forms of noranabasamine.
Our retrosynthetic analysis illustrated in Scheme 1 focused on the disconnection of the terminal pyridyl group (ring C) to afford a 2-substituted piperidine fragment 5 as our initial target. There are a variety methods for the enantioselective construction of 2-subustituted piperidines8 but the iridium complex catalyzed N-heterocyclization of primary amines with diols recently reported by Yamaguchi and co-workers seemed to be exceptionally well suited for the construction of the AB-ring system of 5 and has not been explored for the preparation of natural products.9 A diastereoselective N-heterocyclization with the appropriate chiral primary amine was envisaged for introduction of the single stereogenic carbon atom of the noranabasamine skeleton. The approach was deemed not only straightforward but offered the flexibility for the preparation of various derivatives and analogues if structure-activity studies were warranted in the future.
Scheme 1.

Retrosynthetic analysis of noranabasamine (1)
As illustrated in Scheme 2, the required diol 9 was prepared from 5-bromo-2-methoxypyridine (7). Treatment of 7 with n-butyllithium followed by addition of δ-valerolactone to the lithiated pyridine solution afforded the ketone 8 in 98% yield. The ring-opening reaction proceded regioselectively to give 8 without further nucleophilic addition to the carbonyl. The carbonyl group of 8 was then reduced to the hydroxyl moiety with BH3•SMe2 to furnish the diol 9 in 88% yield.
Scheme 2.

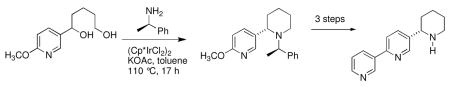
With the diol 9 in hand, our attention focused on the enantioselective construction of the piperidine ring using N-heterocyclization chemistry (Scheme 3). The diol 9 was heated at 110 °C in toluene with (R)-1-phenylethylamine10 in the presence of a catalytic amount (1.5 mol%) of (Cp*IrCl2)2 in a sealed reaction tube. The N-heterocyclization then proceeded diastereoselectively to provide the 2-substituted piperidine 10ab in 72% (10a:10b, dr, 95:5). The diastereoisomers were easily separated by column chromatography. The 2-substituted piperidine 11ab was prepared in similar fashion from diol 9 using (S)-1-phenylethylamine (Scheme 3). The piperidine 11ab was obtained in 69% yield with a diastereomeric ratio of 11a:11b equal to 95:5. Based upon the work of Fujita and coworkers we initially assigned the stereochemistry at C2 of 10a as possessing an S-configuration and 11a as having an R-configuration.
Scheme 3.

Diastereoselective N-Heterocyclization
Presumably N-heterocyclization proceeds through the formation of various imine and enamine intermediates.9 To assure ourselves that epimerization/racemization of the two stereogenic centers had not occurred during the ring generating process it would be necessary to establish the enantiomeric integrity of the piperidine ring. Crooks and coworkers recently reported a procedure for the determination of the enantiopurity of anabasine and related alkaloids using NMR spectroscopy and the chiral shift reagent 1,1′-binaphthyl-2,2′-diylphosphoric acid (BNPPA).11 With this technique in mind, it was envisaged that the issue of the enantiopurity of the piperidine ring system would be more easily resolved with 1, due to the similar secondary amine structure of noranabasamine to the anabasine.
With the piperidine ring system 10a in hand, our attention was directed toward completing the synthesis of noranabasamine (1). Based upon the initial stereochemical assignment of 12, it was assumed that the N-heterocyclization product 10a, possessed the correct stereochemistry at C2. However, the conversion of 10a into 1 would require the manipulation of the methoxy group into a more suitable moiety to facilitate an aryl cross-coupling reaction. To this end, treatment of 10a with POCl3 furnished the 5-chloropyridin-3-yl derivative 12 in 92% yield (Scheme 4). The Suzuki-Miyaura coupling of 12 with 3-pyridineboronic acid could be achieved using several different types of palladium/ligand systems.12 Initially we employed the catalytic system reported by Nolan and coworkers for the coupling sequence.13 This furnished the tricyclic compound 13 in 65% isolated yield. Unfortunately, despite numerous attempts and various conditions to remove the N-phenylethyl auxiliary group, none were successful. The increased basicity of the molecule and the additional steric hinderance around the nitrogen atom completely shut down the hydrogenolysis of the N-1-phenylethyl group. High pressure, high temperature and extended reaction times either resulted in recovery of unreacted starting material or decomposition and formation of intractable mixtures.
Scheme 4.

To avoid the problematic hydrogenolysis of 13, an alternative sequence of reactions was devised to prepare 1 (Scheme 5). The methoxy derivative 10a was subjected to hydrogenolysis conditions to furnish 14 and concomitant treatment with POCl3 provided the chloro analogue 15 in 56% yield over the two-step procedure. This sequence was also applied to 11a and furnished the corresponding 17 in 61% yield.
Scheme 5.

Synthesis of Noranabasamine
For the final step, the Suzuki-Miyaura coupling conditions developed by Fu and coworkers were utilized.14 These conditions for the coupling of 3-pyridineboronic acid with 15 were found to be superior to other methods because of the ease of the work-up and purification steps. This afforded the (S)-noranabasamine (−)-1 in 84% yield, [α]D25 = −32.9 (c 0.33, CH3OH). The nmr data of (−)-1 was identical to the reported data of the isolated material and the optical rotation was also levorotatory.15 The synthesis of the (R)-noranabasamine (+)-1 {[α]D25 +34.6 (c 0.5, CH3OH) from 17 provided additional support of a 2S-configuration of the natural antipode of noranabasamine.
At this point we sought to establish the enantiopurity of the piperidine ring systems. As expected use of the chiral shift reagent BNPPA afforded baseline resolution of the proton signals for H2, H6 and H2″ of both the enantiomers of 1.16 From the NMR study it was quite clear that the enantiopurity of (−)-1 was greater than 86% ee, while that of (+)-1 was greater than 80% ee. From these results it can be inferred that the diastereoselective N-heterocyclization reactions that furnished 10a and 11a (Scheme 3) are highly enantioselective (>80% ee) and consistent with previous studies.9
In summary, we have shown that the iridium catalyzed N-heterocyclization reaction is a facile method for the efficient and enantioselective construction of 2-(pyridin-3-yl)-piperidine alkaloids. This reaction was a key step in the first total synthesis of both enantiomers of the amphibian alkaloid noranabasamine (1) in greater than 30% overall yield and has allowed us to establish the absolute configuration of the natural product as levorotatory. Additional studies with regard to the scope and limitations of this reaction system are ongoing and will be reported in due course. The biological evaluation of both enantiomeric forms of noranabasamine is currently under investigation and will be reported elsewhere.
Supplementary Material
Figure 1.

Noranabasamine (1) and related plant alkaloids.
Acknowledgments
We thank Prof. Edwin Vedejs at the University of Michigan for the assistance provided to Mr. Lei Miao and we thank Prof. Gregory Fu at MIT for the assistance provided to Ms. Hong Shu during the aftermath of Hurricane Katrina. This research was funded by the National Institute on Drug Abuse (DA11528) and the University of New Orleans.
Footnotes
Supporting Information Available: Full experimental details and characterization data of all synthetic products. The material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For a review see (a) Daly JW, Spande TF, Garraffo HM. J Nat Prod. 2005;68:1556. doi: 10.1021/np0580560.Daly JW. J Med Chem. 2003;46:445. doi: 10.1021/jm0204845.Gomes A, Giri B, Saha A, Mirsha R, Dasgupta SC, Debnath A, Gomes A. Ind J Exp Biol. 2007;45:579.Arneric SP, Brioni JD, editors. Neuronal Nicotinic Receptors: Pharmacology and Therapeutic Opportunities. Wiley-Liss Inc.; New York: 1999.
- 2.Tokuyama T, Daly JW. Tetrahedron. 1983;39:41. [Google Scholar]
- 3.Leete E, Mueller ME. J Am Chem Soc. 1982;104:6440. [Google Scholar]
- 4.Lovkova MY, Nurimov E. Isv Akad Nauk SSSR Ser Biol. 1978:545. [PubMed] [Google Scholar]
- 5.Crooks PA, Dwoskin LP. Biochem Pharmacol. 1997;54:743. doi: 10.1016/s0006-2952(97)00117-2. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tilyabaev Z, Abduvakhabov AA. Chemistry of Natural Compounds (translation of Khimiya Prirdnykh Sodenenii) 1998;34:295. [Google Scholar]; (b) Tilyabaev Z, Aabd Mukhamedzhanova KhS. Dokl Akad Nauk UzSSR. 1984;8:45. [Google Scholar]; (c) Mukhamedzhanova KhS. Dokl Akad Nauk UzSSR. 1983;7:47. [Google Scholar]
- 7.(a) Cheng J, Izenwasser S, Zhang C, Zhang S, Wade D, Trudell ML. Bioorg Med Chem Lett. 2004;14:1775. doi: 10.1016/j.bmcl.2004.01.025. [DOI] [PubMed] [Google Scholar]; (b) Nishiyama T, Gyermek L, Trudell ML, Hanaoka K. Eur J Pharm. 2003;470:27. doi: 10.1016/s0014-2999(03)01784-9. [DOI] [PubMed] [Google Scholar]; (c) Cheng J, Zhang C, Stevens ED, Izenwasser S, Wade D, Chen S, Paul D, Trudell ML. J Med Chem. 2002;45:3041. doi: 10.1021/jm0103561. [DOI] [PubMed] [Google Scholar]; (d) Cheng J, Izenwasser S, Wade D, Trudell ML. Med Chem Res. 2001;10:356. [Google Scholar]
- 8.(a) Hande SM, Kawai N, Uenishi J. J Org Chem. 2009;74:244. doi: 10.1021/jo801926g. [DOI] [PubMed] [Google Scholar]; (b) Spangenberg T, Breit B, Mann A. Org Lett. 2009;11:261. doi: 10.1021/ol802314g. [DOI] [PubMed] [Google Scholar]; (c) Castro A, Ramírez J, Juárez J, Terán JL, Orea L, Galindo A, Gnecco D. Heterocycles. 2007;71:2699. [Google Scholar]; (d) Amat M, Bassas O, Llor N, Cantó M, Peréz M, Molins E, Bosch J. Chem Eur J. 2006;12:7872. doi: 10.1002/chem.200600420. [DOI] [PubMed] [Google Scholar]; (e) Ayers JT, Xu Rui, Dwoskin LP, Crooks PA. AAPS J. 2005;7:E752. doi: 10.1208/aapsj070375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Amat M, Cantó M, Llor N, Bosch J. Chem Commun. 2002;5:526. doi: 10.1039/b200020m. [DOI] [PubMed] [Google Scholar]; (g) Felpin F, Girard S, Vo-Thanh G, Robins RJ, Villieras J, Lebreton J. J Org Chem. 2001;66:6305. doi: 10.1021/jo010386b. [DOI] [PubMed] [Google Scholar]; (h) Felpin F, Vo-Thanh G, Robins RJ, Villieras J, Lebreton J. Synlett. 2000;11:1646. [Google Scholar]; (i) Hattori K, Yamamoto H. Tetrahedron. 1993;49:1749–1760. [Google Scholar]; (j) Kunz H, Pfrengle W. Angew Chem Int Ed Eng. 1989;101:1041. [Google Scholar]; (k) Pfrengle W, Kunz H. J Org Chem. 1989;54:4261. [Google Scholar]; (l) Giovannini A, Savoia D, Umani-Ronchi A. J Org Chem. 1989;54:228. [Google Scholar]
- 9.Fujita K, Fujii T, Yamaguchi R. Org Lett. 2004;6:3525. doi: 10.1021/ol048619j. [DOI] [PubMed] [Google Scholar]
- 10.Commercially available from Alfa Aesar Chemical Co. with enantiopurity of 99% ee. The (S)-enantiomer was available in 99.5% ee.
- 11.Ravard A, Crooks PA. Chirality. 1996;8:295. [Google Scholar]
- 12.For a review see: Li JJ, Gribble GW. Palladium in Heterocyclic Chemistry. Pregamon; Amstedam: 2000. pp. 191–7.
- 13.Viciu MS, Germaneau RF, Navarro-Fernandez O, Stevens ED, Nolan SP. Organometallics. 2002;21:5470. [Google Scholar]
- 14.Kudo N, Perseghini M, Fu GC. Angew Chem Int Ed. 2006;45:1282. doi: 10.1002/anie.200503479. [DOI] [PubMed] [Google Scholar]
- 15.The specific rotation for the natural material was reported as [α]D −14.4 ° (CH3OH). See reference 2.
- 16.See Supporting Information for details and spectra.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.