

Abstract

A wide variety of bench-stable potassium heteroaryltrifluoroborates were prepared and general reaction conditions were developed for their cross-coupling to aryl and heteroaryl halides. The cross-coupled products were obtained in good to excellent yields. This method represents an efficient and facile installation of heterocyclic building blocks onto preexisting organic substructures.
Introduction
Heterobiaryls are ubiquitous in natural products, pharmacologically active compounds, polymers, and other useful materials. Transition metal-catalyzed cross-coupling reactions are often utilized for the construction of these compounds because they can directly assemble valuable motifs in a highly convergent fashion. Among the various cross-coupling reactions, the Suzuki-Miyaura reaction is the most extensively used method to prepare these compounds owing to the relatively low toxicity of the boron byproducts, the tolerance of a wide range of functional groups, and the ready availability of organoboron compounds.1,2
Although the cross-coupling of arylboronic acids with aryl and heteroaryl halides has been studied extensively, until recently the cross-coupling of heteroarylboronic acid counterparts had received less attention, and only isolated examples had been reported. The relative lack of concerted effort in this area might be attributed in part to the difficult preparation and isolation of heteroarylboronic acids.3 Fortunately, remarkable progress has recently been made by numerous groups in the development of highly active catalyst systems that promote the cross-coupling of a broad range of heteroarylboronic acids with aryl and heteroaryl halides using a single set of reaction conditions.4,5,6,7,8 For example, Buchwald and coworkers reported that palladium precatalysts {[Pd2(dba)3] and Pd(OAc)2}, sterically bulky, electron-rich phosphine ligands (SPhos and XPhos),9 and K3PO4 in n-butanol at 100 °C constituted an efficient system to cross-couple a wide range of heteroarylboronic acids with aryl and heteroaryl halides.4 Additionally, Fu and coworkers utilized [Pd2(dba)3], PCy3, and K3PO4 in dioxane/H2O at 100 °C to facilitate the cross-coupling of a broad range of nitrogen-containing heterocycles with aryl and heteroaryl halides.5 Both of these recent improvements in heteroaryl crosscouplings focused on the development of efficient catalyst systems and employed what is to date the most readily available nucleophilic organoboron reagents (i.e., boronic acids).
Because of their commercial availability, boronic acids are most often utilized in the Suzuki-Miyaura cross-coupling reactions. However, boronic acids are far from ideal, and they exhibit several limitations that make them unattractive nucleophilic coupling partners. Boronic acids are not monomeric materials, but rather exist in equilibrium with dimers and cyclic trimers (boroxines).10 Consequently, many boronic acids are waxy solids that are difficult to purify. Most importantly, many boronic acids, and especially electron-deficient heteroarylboronic acids, have a short shelf-life owing to their tendency to undergo facile protodeboronation. This instability often requires their storage at low temperatures. The tendency to protodeboronate quite often manifests itself during cross-coupling reactions carried out in polar protic solvents.7a The protodeboronation influences the stoichiometry of the reaction, requiring practitioners to use excess boronic acids to ensure that an adequate amount of this nucleophilic partner is available in cross-coupling reactions. In fact, with a few exceptions,11 virtually every study focusing on heteroarylboronic acid coupling employs excess organoboron reagent (as high as 250%) to achieve satisfactory yields.
Easily prepared, monomeric organoboron reagents that would be more resistant to protodeboronation would eliminate many of the problems encountered with the cross-coupling of heteroarylboronic acids. Heteroarylboronate esters could partially circumvent the problems associated with heteroarylboronic acids, but they themselves suffer from limitations that make them less appealing alternatives, including a lack of atom economy and problematic purification steps.
In recent years, there has been increased interest in the use of potassium organotrifluoroborates as coupling partners for the Suzuki-Miyaura reaction. 12 Unlike their tricoordinate organoboron counterparts, these tetracoordinate species are less prone to undergo protodeboronation. Consequently, near stoichiometric amounts of the nucleophilic partner are employed for cross-coupling reactions. The trifluoroborates are easily prepared from a wide variety of organoboron reagents by the addition of inexpensive potassium hydrogen fluoride (KHF2).12 The resulting organotrifluoroborates are air and moisture stable and can be stored indefinitely at room temperature without any precaution. Additionally, the vast majority of these salts are easily purified via recrystallization, precipitation, or Soxhlet extraction.
We envisioned that the union of highly active catalyst systems with potassium heteroaryltrifluoroborates as nucleophilic coupling partners would be an ideal platform for the cross- coupling of aryl and heteroaryl systems. Toward this end, we sought unified, general reaction conditions that could be broadly applied to virtually all heterocyclic nucleophiles and a wide range of electrophilic coupling partners. For this study, we specifically employed heteroaryltrifluoroborates for which the corresponding boronic acids were highly problematic. Herein, we describe the preparation of a wide variety of these bench-stable heteroaryltrifluoroborates and disclose their efficient cross-couplings to a broad range of aryl and heteroaryl halides using a single set of reaction conditions that include near stoichiometric amounts of the nucleophilic coupling partner.
Results and Discussions
In a previous communication, our group disclosed the preparation of a limited number of heteroaryltrifluoroborates and demonstrated their cross-coupling with aryl and heteroaryl halides.13 Slightly modifying the previous procedure, a large number of potassium heteroaryltrifluoroborates were prepared, including furanyl-, thiophenyl-, pyrrolyl-, pyrazinyl-, isoxazolyl-, pyridinyl-, pyrimidinyl-, indolyl-, benzothiophenyl-, benzofuranyl-, and benzopyridinyl derivatives from commercially available heteroarylboronic acids (Table 1). The reactions were monitored by 11B NMR spectroscopy, and in all cases the transformations were complete in less than 10 minutes.
Table 1.
Preparation of Diverse Potassium Heteroaryltrifluoroboratesa
 | |||||
|---|---|---|---|---|---|
| entry | HetAr-BF3 | isolated yield % | entry | HetAr-BF3 | isolated yield % |
| 1 |
 2a 2a
|
90 | 13 |
 2m 2m
|
90 |
| 2 |
 2b 2b
|
59 | 14 |
 2n 2n
|
70 |
| 3 |
|
61 | 15 |
 2o 2o
|
74 |
| 4 |
 2d 2d
|
85 | 16 |
 2p 2p
|
67 |
| 5 |
 2e 2e
|
95 | 17 |
 2q 2q
|
71 |
| 6 |
 2f 2f
|
69(95)b | 18 |
 2r 2r
|
79 |
| 7 |
 2g 2g
|
74 | 19 |
 2s 2s
|
58 |
| 8 |
 2h 2h
|
31 | 20 |
 2t 2t
|
76 |
| 9 |
 2i 2i
|
49 | 21 |
 2u 2u
|
73 |
| 10 |
 2j 2j
|
72 | 22 |
 2v 2v
|
84 |
| 11 |
 2k 2k
|
94 | 23 |
 1 1
|
34(62)c |
| 12 |
 2l 2l
|
71 | |||
Commerically available heteroarylboronic acids were used.
4-Methylthiophen-2-ylboronic acid was recrystallized from CHCl3 immediately prior to use.
The isoquinolin-4-yltrifluoroborate was prepared directly from 4-bromoisoquinoline.
The conversion of heteroarylboronic acids to the corresponding heteroaryltrifluoroborates gave the products in yields ranging from 31–95% (Table 1). Immediate conversions of commercially received heteroarylboronic acids provided the heteroaryltrifluoroborates in good to excellent yields (Table 1, entries 1, 4–5, 7, 10–15, 17–18, 21–22). As a testament to their instability, the heteroarylboronic acids that were stored at −28 °C for several weeks and then converted to trifluoroborates gave the products in moderate to low yields (Table 1, entries 2–3, 6, 8–9, 16, 19, 23). Analysis of these commercial samples by 11B NMR spectroscopy revealed that the heteroarylboronic acids had partially protodeboronated (boric acid was observed at δ~18 ppm). To confirm that the loss of a considerable amount of material was from protodeboronation, we investigated two of the low yielding trifluoroborates [isoquinolin-4- yltrifluoroborate (1) and 4-methylthiophen-2-yltrifluoroborate (2f)] more deeply. In the first investigation, 1 was prepared from the corresponding 4-bromoisoquinoline in a sequential process of lithium-halogen exchange, boration using triisopropyl borate and quenching with KHF2 (eq 1).14 The one-pot, unoptimized reaction provided the isoquinolin-4-yltrifluoroborate (1) in 62% overall yield compared to a 34% yield from the one step procedure using commercially available heteroarylboronic acid (Table 1, entry 23).
 |
(1) |
In the second study, recrystallization of the commercially obtained 4-methylthiophen-2-ylboronic acid from CHCl3 and subsequent conversion to the corresponding trifluoroborate afforded the product 2f in 95% yield compared to a 69% yield achieved from the boronic acid used as received (Table 1, entry 6). Of special note, several heteroaryltrifluoroborates (2a, 2i, 2j, 2l) that were stored at ambient temperature for three months displayed tremendous stability, as they showed no sign of protodeboronation over this period of time as determined by 11B NMR spectroscopy.
With the requisite heteroaryltrifluoroborates in hand, general conditions were sought to accomplish their cross-coupling. The cross-coupling of a few heteroaryltrifluoroborates using conditions reported in the literature gave the cross-coupled products in modest yields,13,15 and therefore additional screening was undertaken to obtain more effective reaction conditions. We chose furan-2-yltrifluoroborate (2a) as a model nucleophilic partner because the corresponding furan-2-ylboronic acid is known to protodeboronate rapidly during cross-coupling events.4b,7a After an extensive screening process with 4- bromobenzonitrile as the electrophilic partner, 1 mol % of Pd(OAc)2, 2 mol % of RuPhos,16 and 2 equiv of Na2CO3 in ethanol at 85 °C were determined to be the most effective coupling conditions.17
Among the five-membered heterocycles, furan is one of the most extensively studied because numerous natural products, pharmaceutically active compounds, flavors, and fragrances incorporate this motif.18 Even though the furan moiety is an important scaffold, the low stability19 of the boronic acid derivatives toward protodeboronation limits their use in Suzuki-Miyaura reactions. Nonetheless, a few research groups have reported the cross-coupling of furanboronic acid derivatives.6,7a,20 Recently, Plenio and Fleckstein demonstrated the efficient cross-coupling of furan-3-ylboronic acid with a broad range of activated N-heteroaryl chlorides using Na2PdCl4 and disulfonated dicyclohexylfluorenylphosphine ligand at elevated temperature (100 °C), but the cross-coupling of furan-2-ylboronic acid was not reported.7a
Using the reaction conditions developed herein, furan-2-yltrifluoroborate (2a) was efficiently coupled with a variety of aryl halides. Initially, 2a was cross-coupled with various aryl bromides (Table 2, entries 1–8). As shown in Table 2, the reaction conditions worked equally well for electron-withdrawing (Table 2, entries 1, 3, 4, 6, 8), electron-donating (Table 2, entries 2, 5, 7, 9–10), and sterically hindered bromides (Table 2, entry 5). Next, attention was turned to other electrophiles, including aryl chlorides, - iodides, and -triflates. All classes of electrophiles studied provided the products in good yields (Table 2, entry 1). Because aryl chlorides are more readily available and inexpensive compared to other electrophiles, these substrates were examined in some detail with 2a. As was the case with bromides, electron-withdrawing (Table 2, entries 1, 3), electron-donating (Table 2, entries 2, 5, 10), and sterically hindered chlorides (Table 2, entries 5, 9) all reacted smoothly, affording the products in good to excellent yields. We also demonstrated that reducing the catalyst/ligand loading by half [0.5 mol % of Pd(OAc)2 and 1 mol % of RuPhos, respectively], the reaction could be scaled to 5 mmol, providing the cross-coupled product in 91% yield (Table 2, entry 1).
Table 2.
Cross-Coupling of Potassium Furan-2-yltrifluoroborate with Diverse Aryl Halidesa
 | |||
|---|---|---|---|
| entry | aryl halide | product | isolated yield % |
| 1 |

|
 3a 3a
|
Br = 91 Cl = 84 (86b, 91c) I = 80 OTf = 82 |
| 2 |

|
 3b 3b
|
Br = 85 Cl = 75 |
| 3 |

|
 3c 3c
|
Br = 94 Cl = 75 |
| 4 |

|
 3d 3d
|
Br = 93 Cl = 87 |
| 5 |

|
 3e 3e
|
Br = 94 Cl = 62 |
| 6 |

|
 3f 3f
|
Br = 89 |
| 7 |

|
 3g 3g
|
Br = 91 |
| 8 |

|
 3h 3h
|
Br = 92 |
| 9 |

|
 3i 3i
|
Cl = 78 |
| 10 |

|
 3j 3j
|
Cl = 99 |
All reactions were carried out using 0.25 mmol of aryl halide and 0.26 mmol of furan-2-yltrifluoroborate.
1 mmol scale.
5 mmol scale; reduced catalyst/ligand loading to 0.5 mol % of Pd(OAc)2 and 1 mol % of RuPhos.
To expand the scope of the general reaction conditions further, attention was turned to the crosscoupling of various heteroaryltrifluoroborates with 4-halobenzonitriles.
Five-Membered Ring Heterocycles
As previously mentioned, furan-2-ylboronic acid is a challenging coupling partner,4b and using the protocols developed herein, 2a was efficiently cross-coupled to aryl bromides and -chlorides. The conditions established were utilized to examine the coupling of substituted furan-2-yltrifluoroborate derivatives. Toward this end, 5-methylfuran-2-yltrifluoroborate (2b) was coupled with 4- bromobenzonitrile, providing the product 4a in excellent yield (Table 3, entry 1). However, the coupling of 2b with 4-chlorobenzonitrile afforded the product 4a in moderate yield (Table 3, entry 1). Additionally, the coupling of 5-formylfuran-2-yltrifluoroborate (2c) with 4-bromobenzonitrile gave the desired product 4b in only 39% yield (Table 3, entry 2).
Table 3.
Cross-Coupling of Five-Membered Potassium Heteroaryltrifluoroborates with 4-Halobenzonitrilea
 | |||
|---|---|---|---|
| entry | HetAr-BF3K | product | isolated yield % |
| 1 |
 2b 2b
|
 4a 4a
|
Br = 95 Cl = 58 |
| 2 |
 2c 2c
|
 4b 4b
|
Br = 39b |
| 3 |
 2d 2d
|
 4c 4c
|
Br = 93 Cl = 96 |
| 4 |
 2e 2e
|
 4d 4d
|
Br = 98 Cl = 83 |
| 5 |
 2f 2f
|
 4e 4e
|
Cl = 74 |
| 6 |
 2g 2g
|
 4f 4f
|
Br = 90 Cl = 52c |
| 7 |
 2h 2h
|
 4g 4g
|
Cl = 20d(37)e |
| 8 |
 2i 2i
|
 4h 4h
|
Cl = 26d(84)e |
| 9 |
 2j 2j
|
 4i 4i
|
Cl = 71 |
All reactions were carried out using 0.25 mmol of aryl
halide and 0.26 mmol of heteroaryltrifluoroborate.
Used 3 mol % Pd(OAc)2 and 6 mol % RuPhos
Recovered 45% of deprotected product.
Heated reaction for 48 h.
Used 5 mol % Pd(OAc)2, 10 mol % RuPhos, and 3 equiv Na2CO3, 48 h
Thiophenes find extensive use in materials science and in medicinal chemistry. 21 Although thiophenylboronic acids are more stable than furanylboronic acids,7a they themselves also suffer from protodeboronation under protic conditions, with thiophen-2-ylboronic acid being more prone to protodeboronation than thiophen-3-ylboronic acid.22 Until recently, few examples had been reported for the cross-coupling of thiophenylboronic acid derivatives.4b,6,7a,20 Doucet and Santelli reported the crosscoupling of thiophen-3- and thiophen-2-ylboronic acids with aryl halides6a and heteroaryl bromides6b using the [Pd(C3H5)Cl]2/Tedicyp23 catalytic system. These cross-coupling reactions were performed at elevated temperatures (130 °C) using excess boronic acids.
Given the differential sensitivity of isomeric thiophenylboronic acids to protodeboronation, we were pleased that the position of the trifluoroborate moiety (at the 3- or 2-position) did not influence the coupling with 4-bromobenzonitrile, as the desired products 4c-d were obtained in comparable yields, 93% and 98%, respectively (Table 3, entries 3–4). However, for the coupling of 4-chlorobenzonitrile, differences were observed wherein thiophen-3-yltrifluoroborate (2d) was coupled in 96% yield, while thiophen-2-yltrifluoroborate (2e) coupled in a slightly lower 83% yield. These conditions also proved to be effective for the coupling of 4-methylthiophen-2-yltrifluoroborate (2f) with 4-chlorobenzonitrile, affording the product 4e in 74% yield (Table 3, entry 5).
Pyrroles receive significant attention because they are frequently found in natural products and have use in pharmaceuticals, molecular recognition, and materials science.24 Several conditions for the crosscoupling of pyrroleboronic acid derivatives have been reported in the literature.4a–b,5,25 The relatively limited number of citations might be attributed to difficulties associated with protodeboronation, as well as the propensity of the boronic acids to homocouple.3 The homocoupling can be avoided by protection of the nitrogen with either tert-butoxycarbonyl (Boc), triisopropylsilyl (TIPS), or phenysulfonyl groups. Buchwald and coworkers recently reported that N-protected pyrroleboronate esters serve as a better alternative than the corresponding boronic acids for the cross-coupling reactions.4a
To avoid homocoupling, we examined the couplings of N-Boc-pyrrol-2-yltrifluoroborate (2g). When 4-bromobenzonitrile was used as the electrophile, the desired product 4f was isolated in 90% yield (Table 3, entry 6). However, when 4-chlorobenzonitrile was used as the electrophile, the cross-coupled product 4f was obtained in 52% yield along with the corresponding deprotected product in 45% yield (Table 3, entry 6). The removal of the Boc group under cross-coupling conditions is not unusual and has been previously reported.5
Pyrazoles have extensive use in the pharmaceutical26 and agrochemical industries as heterocyclic building blocks. As with the case of pyrroleboronic acid derivatives, the cross-coupling of pyrazoleboronic acid derivatives is also difficult without protecting groups.5,27 Fu and coworkers recently attempted the cross-coupling of unprotected 1H-pyrazol-4- and 1H-pyrazol-5-ylboronic acid, but the products were obtained in < 21% yields. Not surprisingly, the protection of pyrazol-4- and pyrazol-5-ylboronic acid improved the yields dramatically.5
The cross-coupling of unprotected 1H-pyrazol-4- and 1H-pyrazol-5-yltrifluoroborate under conditions developed herein provided the desired heterobiaryls, 4h-g in low yields (20% and 26%), after 48 h (Table 3, entries 7–8). Upon increasing the catalyst/ligand loading to 5 mol % of Pd(OAc)2 and 10 mol % of RuPhos using 3 equiv of Na2CO3 and heating the reaction for 48 h, the cross-coupling of 1HPage pyrazol-5-yltrifluoroborate (2i) gave the product 4h in 84% yield (Table 3, entry 8). Disappointingly, the cross-coupling of 1H-pyrazol-4-yltrifluoroborate (2h) only improved slightly to give the crosscoupled product 4g in 37% yield (Table 3, entry 7).
Among the five-membered heterocycles, isoxazoles have been studied the least, which is perhaps due to the lack of target compounds containing this structural scaffold.28 Scattered examples have been reported for the cross-coupling of isoxazoleboronic acid derivatives with aryl bromides, and the couplings typically required 5–10 mol % of catalyst loading.29
The unified conditions developed herein proved to be effective for the coupling of the 3,5- dimethylisoxazol-4-yltrifluoroborate (2j) with 4-chlorobenzonitrile, affording the product 4i in 71% yield (Table 3, entry 9).
Six-Membered Ring Heterocycles and Benzannulated Heterocycles
Pyridines are prevalent heterocycles found in natural products and bioactive compounds, and therefore their efficient incorporation onto organic molecules as building blocks is highly desirable.30 However, the installation of pyridyl derivatives via the Suzuki-Miyaura reaction has been challenging owing to their low stability, electron-deficiency, and the resulting reduced nucleophilicity of the organoboron species.31 Recently, important progress has been made in the use of pyridinyl derivatives for cross-coupling reactions. 4, 5, 7b, 13,15a,32
For the cross-coupling of pyridinyltrifluoroborates, the catalyst/ligand loading was increased to 3 mol % of Pd(OAc)2 and 6 mol % of RuPhos because the original reaction conditions developed required a longer reaction time (>24 h) for the cross-coupling. The increase in catalyst/ligand loading is not surprising; as mentioned above, pyridinyl derivatives are electron-deficient, less nucleophilic and thereby transmetalate more slowly.31
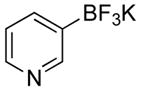
The cross-coupling of pyridin-4-yltrifluoroborate (2k) with 4-bromo- and 4-chlorobenzonitrile was examined. The desired product, 5a, was obtained in excellent yields (Table 4, entry 1). We observed a slight decrease in yields with the coupling of pyridin-3-yltrifluoroborate (2l) with 4-bromo- and 4- chlorobenzonitrile (Table 4, entry 2). Next, we examined the coupling of substituted pyridyl derivatives, 2-fluoropyridin-3- and 6-fluoropyridin-3-yltrifluoroborate (Table 4, entries 3–4). The coupling of 2- fluoropyridin-3-yltrifluoroborate (2m) with 4-bromobenzonitrile afforded the product 5c in 73% yield, while the coupling with 4-chlorobenzonitrile afforded the product 5c in only 49% yield (Table 4, entry 3). Surprisingly, for the cross-coupling of 6-fluoropyridin-3-yltrifluoroborate (2n), the opposite trend was observed, wherein the coupling with 4-chlorobenzonitrile provided the product 5d in a higher yield than 4-bromobenzonitrile (Table 4, entry 4). Of particular note, the coupling of pyridin-2- yltrifluoroborate with 4-chlorobenzonitrile was attempted, but unfortunately none of the cross-coupled product was obtained.
Table 4.
Cross-Coupling of Six-Membered Potassium Heteroaryltrifluoroborates with 4-Halobenzonitrilea
 | |||
|---|---|---|---|
| entry | HetAr-BF3K | product | isolated yield % |
| 1 |
 2k 2k
|
 5a 5a
|
Br = 90 Cl = 96 |
| 2 |
 2l 2l
|
 5b 5b
|
Br = 75 Cl = 78 |
| 3 |
 2m 2m
|
 5c 5c
|
Br = 73 Cl = 49 |
| 4 |
 2n 2n
|
 5d 5d
|
Br = 61 Cl = 77 |
| 5 |
 2o 2o
|
 5e 5e
|
Br = 88 Cl = 86 |
| 6 |
 2p 2p
|
 5f 5f
|
Cl = 96 |
All reactions were carried out using 0.25 mmol of aryl halide and 0.26 mmol of heteroaryltrifluoroborate.
Pyrimidine derivatives, including the nucleobase uracil, have extensive use in the pharmaceutical industry.33 In particular, 5-substituted uracil derivatives are potential antiviral agents, and one possible method for their preparation is by Suzuki-Miyaura cross-coupling, followed by a reduction.34 In 2005, Seley and coworkers reported the preparation of a 5-substituted uracil derivative via cross-coupling of 2,4-dibenzyloxypyrimidin-5-ylboronic acid with an appropriate electrophile in the presence of 5.5 mol % of Pd(PPh3)4, followed by a reduction with ammonia in butanol. The desired product was isolated in 78% yield over two steps.35
Using the conditions developed herein, the efficient cross-coupling of 2,4-dimethoxypyrimidin- 5-yltrifluoroborate (2o) was demonstrated with 4-bromo- and 4-chlorobenzonitrile in 88% and 86% yields, respectively (Table 4, entry 5). Additionally, 2-methoxypyrimidin-4-yltrifluoroborate (2p) was successfully coupled to 4-chlorobenzonitrile in excellent yield (Table 4, entry 6).
Indoles are among the most important heterocycles because they are found in myriad natural products, pharmaceutical agents, and polymers.36 Because they are highly desirable targets, numerous methods to incorporate this heterocyclic core into organic molecules have been developed. As with pyrrolyl- and pyrazinylboronic acid derivatives, indolylboronic acid derivatives usually necessitate protection for an efficient cross-coupling. The Suzuki-Miyaura cross-coupling of substituted indolylboronic acid derivatives with aryl and heteroaryl halides has been reported in the literature.4a–b,5,7b,37 Recently, indolyltrifluoroborates have been shown to be better nucleophilic partners for the cross-coupling reactions, as they provided the products in higher yields. 15c,38
To expand the utility of the general reaction conditions to indoles, we cross-coupled 1H-indol-6- and 1H-indol-5-yltrifluoroborate, obtaining the products 6a-b in excellent yields (Table 5, entries 1–2). During the cross-coupling of N-Boc-indol-2-yltrifluoroborate (2s), the Boc group was cleaved under the reaction conditions, and the deprotected product 6c was obtained in 81% yield (Table 5, entry 3).
Table 5.
Cross-Coupling of Benzannulated Potassium Heteroaryltrifluoroborates with 4-Halobenzonitrilea
 | ||||
|---|---|---|---|---|
| entry | HetAr-BF3K | product | isolated yield % | |
| 1 |
 2q 2q
|
 6a 6a
|
Br = 96 Cl = 83 |
|
| 2 |
 2r 2r
|
 6b 6b
|
Cl = 98 | |
| 3 |
 2s 2s
|
 6c 6c
|
Cl = 81 | |
| 4 |
 2t 2t
|
|
Cl = 82 | |
| 5 |
 2u 2u
|
|
Cl = 92 | |
| 6 |
 2v 2v
|
 6f 6f
|
Br = 88b Cl = 86b |
|
| 7 |
 1 1
|
 6g 6g
|
Cl = 85b | |
All reactions were carried out using 0.25 mmol of aryl halide and 0. 26 mmol of heteroaryltrifluoroborate.
Used 3 mol % of Pd(OAc)2 and 6 mol % of RuPhos.
Although benzothiophenes and benzofurans are less common heterocycles than indoles, they still find use in the pharmaceutical industry.39 The cross-coupling of benzothiophenyl- and benzofuranylboronic acid derivatives has been reported, generally with aryl bromides. 40 We demonstrated that 4- chlorobenzonitrile smoothly cross-coupled to benzothiophen-2-yltrifluoroborate (2t) and benzofuran-2- yltrifluoroborate (2u) in 82% and 92% yields, respectively (Table 5, entries 4, 5).
Benzopyridine derivatives, including quinoline and isoquinoline, are most often found in naturally occurring alkaloids and other pharmacologically active compounds. Even though the first synthesis of quinolin-3-ylboronic acid dates back to 1959,3 only a few examples have been reported in the literature of their incorporation into an organic molecule by means of the Suzuki-Miyaura cross-coupling reaction.11a,41
The cross-coupling of quinolin-3-yltrifluoroborate (2v) with 4-bromo- and 4-chlorobenzonitrile afforded the heterobiaryl product 6f in comparable yields (Table 5, entry 6). The cross-coupling of isoquinolin-4-yltrifluroborate (1) with 4-chlorobenzonitrile provided the product 6g in 82% yield (Table 5, entry 7).
Heterobiaryls
One of the biggest challenges in cross-coupling reactions occurs when both coupling partners are heterocycles. Until recently, no general conditions had been developed to enable this useful C-C bond formation.4a,5,6b,7 To expand the scope of the general reaction conditions further, attention was turned to the coupling of heterobiaryls. Because these are difficult couplings, 3 mol % of Pd(OAc)2 and 6 mol % of RuPhos were employed. Initially, we examined the cross-coupling of 2-chloropyridine with 2a, because both are known to be demanding coupling partners.7a We were able to achieve coupling, albeit in modest yield (Table 6, entry 1). Subsequently, the coupling of 2-chloropyridine with 2j and 2l was successful, affording the desired coupled products 7b–c in 95% and 82% yield, respectively (Table 6, entries 2–3). The coupling of 3-chloropyridine with 2q afforded the product 7d in excellent yield (Table 6, entry 4), but the coupling of 2s provided the deprotected product 7e in only moderate yield (Table 6, entry 5). Additionally, 2a coupled to 2-chloropyrimidine, 2-acetyl-5-chlorothiophene, 2-formyl-5- chlorothiophene, and 2-formyl-5-chlorofuran in moderate to excellent yields (Table 6, entries 6–10).
Table 6.
Cross-Coupling of Various Potassium Heteroaryltrifluoroborates with Various Heteroaryl Chloridesa
 | |||||
|---|---|---|---|---|---|
| entry | HetAr-Cl | HetAr-BF3K | isolated yield % | ||
| 1 |

|
 2a 2a
|
67 (7a) | ||
| 2 |

|
 2j 2j
|
95 (7b) | ||
| 3 |

|
 2l 2l
|
82 (7c) | ||
| 4 |

|
 2q 2q
|
93 (7d) | ||
| 5 |

|
 2s 2s
|
57b (7e) | ||
| 6 |

|
 2a 2a
|
92 (7f) | ||
| 7 |

|
 2a 2a
|
52 (7g) | ||
| 8 |

|
 2a 2a
|
77 (7h) | ||
| 9 |

|
 2a 2a
|
64 (7i) | ||
| 10 |

|
 2a 2a
|
81 (7j) | ||
All reactions were carried out using 0.25 mmol of heteroaryl halide and 0.26 mmol of heteroaryltrifluoroborate.
Yield of Boc deprotected cross-coupled product.
In conclusion, over twenty structurally diverse five-membered, six-membered, and benzannulated heteroaryltrifluoroborate derivatives were prepared from commercially available boronic acids. We determined that these organotrifluoroborates are much less prone to protodeboronation when compared to their heteroarylboronic acid counterparts, and thus they can be stored indefinitely at ambient temperatures. By combining the electron-rich, monodentate ligand, RuPhos, with heteroaryltrifluoroborates as the nucleophilic coupling partners, general, mild, and efficient reaction conditions for cross-coupling were developed. Of note, essentially stoichiometric ratios of the heteroaryltrifluoroborates can be employed in these reactions, which take place at lower temperatures than previously developed protocols when using comparable catalyst loading (as low as 0.5 mol %). Using a unified set of reaction conditions, a broad range of aryl and heteroaryl halides were cross-coupled in good to excellent yields. Furthermore, the reaction conditions developed are scalable and economically viable because inexpensive aryl chlorides, Na2CO3, and ethanol are utilized in the cross-coupling reactions.
Experimental Section
General Experimental Procedure for the Preparation of Potassium Heteroaryltrifluoroborates. Preparation of Potassium Furan-2-yltrifluoroborate (2a).42
To a solution of furan-2-ylboronic acid (8.93 mmol, 1.0 g) in MeOH (2.6 mL, 3.5 M or enough MeOH to give a free flowing suspension) under N2 was added KHF2 (2.09 g, 26.8 mmol) in one portion at 0 °C. To the suspension, H2O was added dropwise (5.95 mL, 4.5M) at 0 °C. The ice-water bath was removed and the reaction was stirred at rt until 11B NMR indicated completion of the reaction (~2 min). The crude mixture was concentrated and dried overnight in vacuo. The crude solid was purified using continuous Soxhlet extraction (4 h) with acetone (60 mL) (unless otherwise specified). The collected solvent was concentrated, and then redissolved in a minimal amount of acetone (5 mL). The addition of ether (30 mL) led to the precipitation of the product. The product was filtered, concentrated, and dried in vacuo to afford the pure compound in 90% yield (1.08 g, 8.03 mmol) as a light orange solid mp: > 200 °C 1H NMR (500 MHz, DMSO-d6) δ 7.40 (m, 1H), 6.18 (m, 1H), 6.06 (d, 1H, J = 2.6 Hz). 13C NMR (125.8 MHz, DMSO-d6) δ 141.0, 110.2, 108.5. 19F NMR (470.8 MHz, DMSO-d6) δ -139.1. 11B NMR (128.4 MHz, DMSO-d6) δ 0.3. FT-IR (KBr) 3428, 3022, 2921, 2822, 1496, 1126, 1088, 1066, 1002 cm− 1. HRMS (ESI) m/z calcd. for C4H3BF3O (M-K) 135.0235, found 135.0234.
General Experimental Procedure for Suzuki-Miyaura Cross-Coupling Reaction of Aryl and Heteroaryl Electrophiles with Heteroaryltrifluoroborates. Preparation of 4-(Furan-2-yl)benzonitrile (3a). 43
A Biotage microwave vial was charged with Pd(OAc)2 (3.4 mg, 0.015 mmol), RuPhos (14 mg, 0.03 mmol) 4-bromobenzonitrile (46.0 mg, 0.25 mmol), potassium furan-2-yltrifluoroborate (46.0 mg, 0.26 mmol) and Na2CO3 (53.0 mg, 0.5 mmol). The test tube was sealed with a cap lined with a disposable Teflon septum, evacuated and purged (x 3). Ethanol (0.18 M, 1.4 mL) was added via syringe and the reaction was heated at 85 °C for 12 h. The reaction mixture was allowed to cool to rt and filtered through a thin pad of silica gel (elution with 25% methanol in EtOAc). The solvent was removed in vacuo and the crude product was purified by silica gel column chromatography (elution with hexane/EtOAc 7:1) to yield the pure product in 91% yield (38.49 mg, 0.23 mmol) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.72 (d, 1H, J = 8.5 Hz), 7.63 (d, 1H, J = 8.5 Hz), 7.52 (m, 1H), 6.80 (d, 1H, J = 3.5 Hz), 6.52 (dd, 1H, J = 1.8, 3.4 Hz). 13C NMR (125.8 MHz, CDCl3) δ 152.1, 143.8, 134.8, 132.7, 124.1, 119.0, 112.3, 110.4, 108.3.
Supplementary Material
Experimental procedures, spectral characterization, and copies of 1H, 13C, 11B, and 19F spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was generously supported by the National Institutes of Health (General Medical Sciences), and Merck Research Laboratories. We also acknowledge Aldrich, BoroChem, and Frontier Scientific for their donation of the heteroarylboronic acids.
References
- 1.For reviews on palladium-catalyzed Suzuki-Miyaura reactions see: Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483.Suzuki A. J Organomet Chem. 1999;576:147–168.Little AF, Fu GC. Angew Chem, Int Ed. 2002;41:4177– 4211.
- 2.Li JJ, Gribble GW. Palladium in Heterocyclic Chemistry. Pergamon Press; Amsterdam: 2000. [Google Scholar]
- 3.Tyrell E, Brookes P. Synthesis. 2004:469–483. [Google Scholar]
- 4.(a) Billingsley K, Anderson KW, Buchwald SL. Angew Chem, Int Ed. 2006;45:3484–3488. doi: 10.1002/anie.200600493. [DOI] [PubMed] [Google Scholar]; (b) Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]; (c) Billingsley K, Buchwald SL. Angew Chem, Int Ed. 2008;47:4695–4698. doi: 10.1002/anie.200801465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kudo N, Perseghini M, Fu GC. Angew Chem, Int Ed. 2006;45:1282–1284. doi: 10.1002/anie.200503479. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kondolff I, Doucet H, Santelli M. Synlett. 2005:2057–2061. [Google Scholar]; (b) Kondolff I, Doucet H, Santelli M. J Mol Catal A: Chem. 2007;269:110–118. [Google Scholar]
- 7.(a) Fleckenstein CA, Plenio H. J Org Chem. 2008;73:3236–3244. doi: 10.1021/jo8001886. [DOI] [PubMed] [Google Scholar]; (b) Fleckenstein CA, Plenio H. Eur J Org Chem. 2008:4267–4279. doi: 10.1002/chem.200701877. [DOI] [PubMed] [Google Scholar]
- 8.(a) Smith AE, Clapman KM, Batsanov AS, Bryce MR, Tarbit B. Eur J Org Chem. 2008:1458–1463. [Google Scholar]; (b) Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ. Org Lett. 2006;8:1787–1789. doi: 10.1021/ol060268g. [DOI] [PubMed] [Google Scholar]; (c) Guram AS, Wang X, Bunel EE, Faul MM, Larsen RD, Martinelli MJ. Org Lett. 2007;72:5104–5112. doi: 10.1021/jo070341w. [DOI] [PubMed] [Google Scholar]
- 9.SPhos: 2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl; XPhos: 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
- 10.(a) Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]; (b) Onak T. Organoborane Chemistry. Academic Press; New York: 1975. [Google Scholar]
- 11.(a) Baxendale IR, Griffiths-Jones CM, Ley SV, Tranmer GK. Chem Eur J. 2006;12:407–4416. doi: 10.1002/chem.200501400. [DOI] [PubMed] [Google Scholar]; (b) Parry PP, Bryce MR, Tarbit B. Synthesis. 2003;7:1035–1038. [Google Scholar]; (c) Parry PP, Bryce MR, Tarbit B. Org Biomol Chem. 2003;1:1447–1449. doi: 10.1039/b302767h. [DOI] [PubMed] [Google Scholar]
- 12.For reviews on potassium organotrifluoroborates, see: Darses S, Genêt JP. Eur J Org Chem. 2003:4313–4327.Molander GA, Figueroa R. Aldrichim Acta. 2005;38:49–56.Molander GA, Ellis N. Acc Chem Res. 2007;40:275–286. doi: 10.1021/ar050199q.Stefani HA, Cella R, Adriano S. Tetrahedron. 2007;63:3623–3658.Darses S, Genêt JP. Chem Rev. 2008;108:288–305. doi: 10.1021/cr0509758.
- 13.Molander GA, Biolatto B. J Org Chem. 2003;68:4302–4314. doi: 10.1021/jo0342368. [DOI] [PubMed] [Google Scholar]
- 14.See Supporting Information for the preparation of isoquinolin-4-yltrifluoroborate from the corresponding 4-bromoisoquinoline.
- 15.(a) Barder TE, Buchwald SL. Org Lett. 2004;6:2649–2652. doi: 10.1021/ol0491686. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Fumagalli T. J Org Chem. 2006;71:5743–5747. doi: 10.1021/jo0608366. [DOI] [PubMed] [Google Scholar]; (c) Pagano N, Maksimoska J, Bregman H, Williams DS, Webster RD, Xue F, Meggers E. Org Biomol Chem. 2007;5:1218–1227. doi: 10.1039/b700433h. [DOI] [PubMed] [Google Scholar]
- 16.RuPhos: 2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl.
- 17.Variables changed in coupling optimization included: catalyst/ligand ratios (5/10 mol %, 3/6 mol %]; solvents: (DME, n-butanol, sec-butanol and tert-amyl alcohol) and bases (Cs2CO3, K2CO3, K3PO4, Et3N), base equiv (3, 1, and 0).
- 18.(a) Liu Y, Zhang S, Abreu PJM. Nat Prod Rep. 2006;23:630–651. doi: 10.1039/b604586c. [DOI] [PubMed] [Google Scholar]; (b) Zviely M. Perfum Flavor. 2006;31:20–35. [Google Scholar]; (c) Sanz C, Czerny M, Cid C, Schieberle P. Eur Food Res Technol. 2002;214:299–302. [Google Scholar]
- 19.Florentin D, Fournie-Zaluski MC, Callanquin M, Roques BP. J Heterocycl Chem. 1976;13:1265–1272. [Google Scholar]
- 20.(a) Li JH, Zhu QM, Xie YX. Tetrahedron. 2006;62:10888–10895. [Google Scholar]; (b) Li JH, Deng CL, Xie YX. Synth Commun. 2007;37:2433–2448. [Google Scholar]
- 21.(a) Joule JA, Mills K, editors. Heterocyclic Chemistry. Blackwell Science; Malden, MA: 2000. [Google Scholar]; (b) Press JB. In: Thiophene and its Derivatives. Gronowitz S, editor. Wiley; New York: 1985. pp. 345–677. [Google Scholar]
- 22.Brown RD, Buchanan AS, Humffray A. Aust J Chem. 1965;18:1521–1525. [Google Scholar]
- 23.Tedicyp: cis,cis,cis-1,2,3,4-tetrakis(diphenylphosphinomethyl)cyclopentane.
- 24.Banwell MG, Goodwin TE, Ng S, Smith JA, Wong DJ. Eur J Org Chem. 2006:3043–3060. and references cited therein. [Google Scholar]
- 25.Alvarez A, Guzman A, Ruiz A, Velarde E. J Org Chem. 1992;57:1653–1656. [Google Scholar]
- 26.Elguero J. In: Comprehensive Heterocyclic Chemistry. Katrizky AR, Rees CW, Scriven EFV, editors. Pergamon Press; Oxford: 1996. [Google Scholar]
- 27.(a) Gérard AL, Bouillon A, Mahasekake C, Collot V, Rault S. Tetrahedron Lett. 2006;47:4665–4669. [Google Scholar]; (b) Gérard AL, Mahasekake C, Collot V, Rault S. Tetrahedron Lett. 2007;48:4123–4126. [Google Scholar]; (c) Browne DL, Helm MD, Plant A, Harrity JPA. Angew Chem, Int Ed. 2007;46:8656–8658. doi: 10.1002/anie.200703767. [DOI] [PubMed] [Google Scholar]
- 28.Schnürch M, Flasik R, Khan AF, Spina M, Mihovilovic MD, Stanetty P. Eur J Org Chem. 2006:3283–3307. [Google Scholar]
- 29.Goh WK, Black DS, Kumar N. Tetrahedron Lett. 2007;48:9008–9011. [Google Scholar]; (b) Kumar JSD, Ho MM, Leung JM, Toyokumi T. Adv Synth Catal. 2002;344:1146–1151. [Google Scholar]; (c) Davies MW, Wybrow RAJ, Johnson CN, Harrity JPA. Chem Commun. 2001:1558–1559. doi: 10.1039/b103319k. [DOI] [PubMed] [Google Scholar]; (d) Moore JE, Davies MW, Goodenough KN, Wybrow RAJ, York M, Johnson CN, Harrity JPA. Tetrahedron. 2005;61:6707–6714. [Google Scholar]
- 30.(a) Pozharskii AF, Soldartenko AT, Katritzky A. Heterocycles in Life and Society. Wiley; New York: 1997. [Google Scholar]; (b) Bonnet V, Mongin F, Trecourt F, Breton F, Marsais F, Knochel P, Queguiner G. Synlett. 2002:1008–1010. [Google Scholar]
- 31.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 32.(a) Batey RA, Quach TD. Tetrahedron Lett. 2001;42:9099–9103. [Google Scholar]; (b) Bouillon A, Lancelot JC, Collot V, Bovy PR, Rault S. Tetrahedron. 2002;58:3323–3328. [Google Scholar]; (c) Bouillon A, Lancelot JC, Bovy PR, Rault S. Tetrahedron. 2002;58:4369–4373. [Google Scholar]; (d) Saygili N, Batsanov AS, Bryce MR. Org Biomol Chem. 2004;2:852–857. doi: 10.1039/b314624n. [DOI] [PubMed] [Google Scholar]; (c) Hodgson P, Salingue F. Tetrahedron Lett. 2004;45:685–687. [Google Scholar]; (d) Thompson AE, Hughes G, Batsanov AS, Bryce MR, Parry PR, Tarbit B. J Org Chem. 2005;70:388–390. doi: 10.1021/jo0402226. [DOI] [PubMed] [Google Scholar]; (e) Thompson AE, Batsanov AS, Bryce MR, Saygili N, Parry PR, Tarbit B. Tetrahedron. 2005;61:5131–5135. [Google Scholar]; (f) Yamamoto Y, Takizawa M, Yu XQ, Miyaura N. Angew Chem. 2008;47:928–931. doi: 10.1002/anie.200704162. [DOI] [PubMed] [Google Scholar]
- 33.Clapman KM, Smith AE, Batsanov AS, McIntyre L, Pountney A, Bryce MR, Tarbit B. Eur J Org Chem. 2007:5712–5716. and references cited therein. [Google Scholar]
- 34.(a) Peters D, Hörnfeldt AB, Gronowitz S. J Heterocycl Chem. 1990;27:2165–2173. [Google Scholar]; (b) Wellmar U, Hörnfeldt AB, Gronowitz S. J Heterocycl Chem. 1995;32:1159–1163. [Google Scholar]
- 35.Seley K, Zhang L, O’Daniel PI. J Org Chem. 2004;70:1612–1619. doi: 10.1021/jo048218h. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey GR, Kuethe JT. Chem Rev. 2006;106:2875–2911. doi: 10.1021/cr0505270. [DOI] [PubMed] [Google Scholar]
- 37.(a) Yang Y, Martin AR. Heterocycles. 1992;34:1395–1398. [Google Scholar]; (b) Prieto M, Zurita E, Rosa E, Munoz L, Lloyd-Williams P, Giralt E. J Org Chem. 2004;69:6812–6820. doi: 10.1021/jo0491612. [DOI] [PubMed] [Google Scholar]; (c) Payack JF, Vazquez E, Matty L, Kress MH, McNamara J. J Org Chem. 2005;70:175–178. doi: 10.1021/jo048334k. [DOI] [PubMed] [Google Scholar]; (d) Kher S, Lake K, Sircar I, Pannala M, Bakir F, Zapf J, Xu K, Zhang SH, Liu J, Morera L, Sakurai N, Jack R, Cheng JF. Bioorg Med Chem Lett. 2007;17:4442–4446. doi: 10.1016/j.bmcl.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 38.Mizuta M, Seio K, Miyata K, Sekine M. J Org Chem. 2007;72:5046–5055. doi: 10.1021/jo070206j. [DOI] [PubMed] [Google Scholar]
- 39.Arzel E, Swahn B, Wensbo D. Chem Abstr. Vol. 149. p. 332192. WO 2008108730, September 12, 2008. [Google Scholar]
- 40.(a) Kitamura Y, Sako S, Udzu T, Tsutsui A, Maegawa T, Monguchi Y, Sajiki H. Chem Commun. 2007:5069–5071. doi: 10.1039/b712207a. [DOI] [PubMed] [Google Scholar]; (b) DiMauro EF, Vitullo JR. J Org Chem. 2006;71:3959–3962. doi: 10.1021/jo060218p. [DOI] [PubMed] [Google Scholar]; (c) Wang Z, Elokdah H, McFarlene G, Pan S, Antane M. Tetrahedron Lett. 2006;47:3365–3369. [Google Scholar]; (d) Kumar S, Kim TY. J Org Chem. 2000;65:3883–3884. doi: 10.1021/jo991933k. [DOI] [PubMed] [Google Scholar]
- 41.Rewinkel J, Enthoven M, Golstein I, van der Rijst M, Scholten A, van Tilborg M, de Weys D, Wisse J, Hamersma H. Bioorg Med Chem. 2008;16:2753–2763. doi: 10.1016/j.bmc.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020–3027. [Google Scholar]
- 43.Majita T, Pac C, Nakasone A, Sakurai H. J Am Chem Soc. 1981;103:4499–4508. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, spectral characterization, and copies of 1H, 13C, 11B, and 19F spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org.