

Abstract
4-Hydroxynonenal (4-HNE) has been suggested to be involved in stress-induced signaling for apoptosis. In present studies, we have examined the effects of 4-HNE on the intrinsic apoptotic pathway associated with p53 in human retinal pigment epithelial (RPE and ARPE-19) cells. Our results show that 4-HNE causes induction, phosphorylation, and nuclear accumulation of p53 which is accompanied with down regulation of MDM2, activation of the pro-apoptotic p53 target genes viz. p21 and Bax, JNK, caspase3, and onset of apoptosis in treated RPE cells. Reduced expression of p53 by an efficient silencing of the p53 gene resulted in a significant resistance of these cells to 4-HNE-induced cell death. The effects of 4-HNE on the expression and functions of p53 are blocked in GSTA4-4 over expressing cells indicating that 4-HNE-induced, p53-mediated signaling for apoptosis is regulated by GSTs. Our results also show that the induction of p53 in tissues of mGsta4 (-/-) mice correlate with elevated levels of 4-HNE due to its impaired metabolism. Together, these studies suggest that 4-HNE is involved in p53-mediated signaling in in vitro cell cultures as well as in vivo that can be regulated by GSTs.
Keywords: 4-Hydroxynonenal, Oxidative stress, p53, Glutathione S-transferase, Lipid peroxidation, Apoptosis, Retinal pigment epithelial (RPE) cells
Introduction
Exposure of cells and tissues to xenobiotics or physico-chemical sources of oxidative stress (e.g. UV) causes the generation of reactive oxygen species (ROS), which in turn can cause damage by interacting with cellular macromolecules such as DNA, proteins, and lipids, as well as by forming secondary toxic products. ROS-initiated membrane lipid peroxidation (LPO) is highly damaging to cells because a single oxidative event can start a chain reaction and lead to the formation of large amounts of LPO products including toxic electrophiles such as 4-hydroxynonenal (4-HNE) [1]. In recent years, 4-HNE has been shown to be an important second messenger involved in signaling for cell cycle arrest, differentiation, apoptosis and regulation of the expression of a multitude of genes including p53, in cells of diverse origin [1-3]. P53 plays an important role in apoptosis, growth arrest, genomic stability, cell senescence, and differentiation. It is normally kept at a low concentration in cells by its relatively short half-life. Some cells may also have a latent form of p53 which is inactive for transcription [4-6]. Diverse cellular and stressful events such as DNA damage, hypoxia, and oxidative stress activate the p53 gene by causing it to accumulate rapidly through a posttranscriptional mechanism, generally in a proportion to the extent of DNA damage [4,7]. In addition to induction through stress signals, increased quantities of p53 protein in cells are found in cells modified by T antigen and E1a, the gene products encoded by the DNA viruses SV40 and adenovirus, respectively [8]. While the SV40 Tag is known to bind to the p53 protein [9], and extend the lifespan of human cells [10], more recent studies have demonstrated a functional p53 in the SV40-transformed human [9] and even the murine [11] cells.
Once activated by phosphorylation [12-14], the modified and stable p53 can induce up-regulation of the downstream target genes, p21 and Bax [15-18]. Expression of the p21 gene product inhibits the cyclin-dependent kinase activity and arrests cell cycle progression while p53-dependent Bax synthesis induces apoptosis by binding to Bcl-2 and antagonizing its non-apoptotic ability. Since generation of 4-HNE has been suggested to be a common denominator in mechanisms of apoptosis caused by diverse forms of oxidative stress [19,20], it is likely that it would also affect the expression and activation of p53. This idea is supported by our recent studies showing that alterations in the intracellular levels of 4-HNE in HLE B-3 cells affect the expression of a multitude of genes including p53 [21,22]. Thus, it is possible that 4-HNE-mediated activation of p53 may be one of the mechanisms responsible for 4-HNE-induced apoptosis reported in many cell types [19,23-27].
Retina is particularly susceptible to oxidative stress because not only it is bombarded constantly by ROS-producing UV and high-energy visible light [28], but also because retinal pigment epithelial (RPE) cells maintain and support the photoreceptors by phagocytosis and degradation of the photoreceptor outer segment membranes which are rich in polyunsaturated fatty acids [29-31]. It has been suggested that LPO products contribute to retinal pigment epithelial dysfunction, initiating retinal degenerative disorders including age-related macular degeneration (ARMD) which is the leading cause of blindness in the developed world [32]. Present studies were designed to study the effects of 4-HNE on the expression and activation of p53 in RPE cells focusing on the p53-mediated intrinsic pathway for apoptosis. Glutathione S-transferase A4-4 (GSTA4-4)-mediated metabolism of 4-HNE is one of the major determinants of the intracellular concentration of 4-HNE [21,22,33]. Therefore, we have also examined the possible role of GSTA4-4 in regulation of 4-HNE-induced, p53-mediated apoptosis in RPE cells. For these studies, we have chosen RPE cells of human fetal origin and ARPE-19 cells developed from the retina of adult young male. Results of these studies indicate that in these cells 4-HNE causes activation, phosphorylation, and enhanced nuclear accumulation of p53, accompanied with activation of the signaling components involved in p53-mediated apoptosis. Over-expression of either human GSTA4-4 or the corresponding murine isozyme mGsta4-4 as well as the silencing of cellular p53 blocks these effects of 4-HNE.
Materials and methods
Chemicals
Dulbecco's modified Eagle's medium (DMEM), penicillin-streptomycin solution (P/S), phosphate buffered saline (PBS), fetal bovine serum (FBS), HEPES, trypsin, and MEM non-essential amino acid solution were purchased from Gibco (Grand Island, NY). 4-HNE was purchased from Cayman Chemical (Ann Arbor, MI). Reagents for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transblotting were purchased from Bio-Rad (Hercules, CA). RIPA lysis buffer was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The Western blot stripping buffer was from Pierce (Rockford, IL). The apoptosis detection system (CaspACE™ FITC-VAD-FMK in situ marker) was purchased from Promega Inc. (Madison, WI). All other chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Antibodies
Antibodies against p53 (DO-1, sc-126, mouse monoclonal), JNK1 (sc-571, polyclonal), p-JNK (G-7, sc-6254, monoclonal) p21 (sc-6246, monoclonal), Bax (N-20, sc-493, polyclonal) caspase3 (sc-7148, polyclonal) and MDM2 (sc-965, monoclonal), Histone H1 (sc-8030, monoclonal), GAPDH (sc-32233, monoclonal) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) while those against phosphorylated p53 (ser15, 9284S, polyclonal) were obtained from Cell Signaling Technology, Inc. (MA). Polyclonal antibodies developed against recombinant hGSTA4-4 in chicken and mGsta4-4 in rabbit have been characterized and described by us previously [33,34]. Horseradish peroxidase (HRP)-conjugated secondary antibodies as well those against GAPDH were purchased from Southern Biotech (Birmingham, AL).
Cell lines
The simian virus SV40-transformed human fetal male RPE 28 cells (Coriell Institute, Camden, NJ) which exhibit epitheloid morphology and retain physiological functions characteristic of the primary human RPE cells were chosen as a suitable model for investigating the effect of low levels of oxidative stress [31]. The ARPE-19 (ATCC CRL-2302) is a spontaneously arising retinal pigment epithelia cell line derived from the normal eyes of a young male. Cells were cultured in their standard medium containing 10% fetal bovine serum and antibiotics in a humidified incubator at 37 °C in 5% CO2 atmosphere. Both cell lines were trypsinized and passaged every 3-4 days.
Cell viability
The sensitivity of the RPE and ARPE-19 cells to 4-HNE was measured by the MTT assay as described by Mosmann [35] with slight modifications. After determining the number of viable cells in an aliquot of a log-phase culture by counting trypan-blue excluding cells in a hemocytometer, the cells were re-suspended in the regular complete medium. About 2 × 104 cells in an aliquot of 190 μl of full serum medium were seeded in 96-well flat bottomed microtiter plates for 24 h to allow attachment to the culture plates. After confirming cell attachment, the cells were incubated for 18 h in fresh serum-free medium to avoid any interaction between serum proteins and 4-HNE. No significant apoptosis was observed in serum-deprived conditions. The next day, 10 μl of PBS containing various amounts of 4-HNE was added. Eight replicate wells were used for each concentration of 4-HNE in these studies. After 24 h incubation of the plates at 37 °C, 10 μl of MTT solution (5 mg/ml in PBS) was added to each well, and the plates incubated for an additional 4 h at 37 °C. The plates were then centrifuged at 2000g for 10 min and the medium aspirated from each well. 100 μl of dimethylsulfoxide (DMSO) was added to each well and the intracellular formazan dye crystals were dissolved by shaking the plates at room temperature for at least 30 min. The absorbance of formazan at 562 nm was measured using a microplate reader (El × 808 BioTek Instruments, Inc). A dose-response curve was plotted and the concentration of 4-HNE resulting in a 50% decrease in formazan formation was calculated as the IC50 value of 4-HNE.
Exposure of cells to 4-HNE for signaling studies
RPE and ARPE-19 cells were plated at a density of 4 × 105 per 8.0 ml of complete medium in 100 mm dishes overnight. To investigate the effects of 4-HNE in p53-mediated cellular signaling, we cultured the cells overnight in serum-free conditions as above and then incubated them with different concentrations (0-75 μM) of 4-HNE prepared in phosphate buffered saline (PBS) for a period of 2 h.
Preparation of cell and tissue extracts
Cells treated as above were thereafter collected, washed with PBS and re-suspended in radio-immunoprecipitation assay (RIPA) lysis buffer, containing 1 × PBS (pH 7.4), 1% Nonidet-P-40 (NP-40), 0.5% sodium deoxycholate, 0.1% sodium dodecyl suphate, 1 mM phenylmethanesulfonyl fluoride (PMSF), and 2 μg/ml of pepstatin. After sonicating three times for 5 s, the lysates were centrifuged at 14,000 rpm for 15 min and the supernatants collected.
Brain, heart, lung, kidney, liver, and eye from wild-type and mGsta4 knockout mice were quickly removed after they had been sacrificed and immediately transferred to -80 °C until they were to be used. After washing with 20 mM potassium phosphate buffer (pH 7.0) containing 1.4 mM β-mercaptoethanol, the tissue samples were homogenized in ice-cold RIPA buffer containing PMSF and incubated on ice for at least 30 min. Homogenates were centrifuged at 14,000 rpm for 15 min and the extracts collected as supernatants were used for analyses. Protein concentrations were determined by the Bradford assay [36].
Western blot analysis
Total cell extracts, containing 25-60 μg protein were separated by SDS-PAGE (4-20% gels) and transferred onto a nitrocellulose membrane (Bio-Rad). 16% gels were used for the detection of caspase3. After blocking the membrane with 5% non-fat dry milk in Tris-buffered saline (TBS) at room temperature for 1 h, it was incubated overnight at 4 °C with the appropriate primary antibody in blocking buffer. After washing with TBS, the membrane was incubated with the appropriate secondary antibody at room temperature for 1 h. The membrane was again washed with TBS, and immunoblots were developed with the ECL (chemiluminescence) reagents from Pierce according to the manufacturer's instructions. For JNK and p-JNK immunoblot detection, the procedure was slightly modified in that the membranes were blocked with 1% non-fat milk and 1% BSA instead of the blocking buffer containing 5% non-fat milk. Also, the primary JNK antibody was incubated in Tris-buffered saline (TBS) containing 1% milk, 1% BSA, 50 mM NaF and 0.05% Tween 20 (T-TBS).
Transient transfection with GSTA4
We carried out transient transfection in RPE cells using 10 μg of either empty pTarget-T vector (VT) or the pTarget vector with the open reading frame (ORF) of the restored Kozak hGSTA4 sequence (hGSTA4-Tr), using Lipofectamine PLUS reagents (Invitrogen, Carlsbad, CA) as per the manufacturer's instructions. Similarly, ARPE-19 cells were transiently transfected with the mouse pRC/CMV mGsta4 [37] and with the vector alone. For 4-HNE exposure experiments, GSTA4 transiently transfected RPE and ARPE-19 cells were treated for 1 h with 20 μM 4-HNE in serum-free medium and cell lysates prepared again as described above.
Silencing of p53expression using siRNA
The p53 siRNA transfection was essentially carried out according to manufacturer's instructions. P53 si RNA (h) (sc-29435) and control si RNA (sc-37007) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) to transfect RPE cells. Briefly, RPE cells (2 × 105 cells per well) were plated in a six-well tissue culture plate, in 2 ml of antibiotic free growth medium supplemented with FBS. Cells were cultured at 37 °C until 60-80% confluency. For each transfection, 50 pmol of siRNA was diluted with 100 μl siRNA transfection medium (solution A) and 8 μl of siRNA transfection reagent was diluted with 100 μl siRNA transfection medium (solution B). Solution A was directly added to solution B, mixed gently and the mixture was incubated for 30 min at room temperature. Cells were washed with 2 ml of siRNA transfection medium and 0.8 ml of siRNA transfection medium was added to the mixture of the solutions A and B, gently mixed and overlaid onto the washed cells and incubated for 24 h at 37 °C. After 24 h, 2 ml of fresh normal medium was added to each well and cells were further incubated for 48 h. Control cells were treated in a similar manner with a mixture of scrambled siRNA. Cells were harvested at appropriate time points and the silencing of p53 was examined.
In situ detection of apoptosis
ARPE-19 (2 × 104) cells were plated in complete medium onto chamber slides a day before the experiment. On the day of the experiment, the cells were assessed for their confluency (60-80%) and starved of serum for at least 2 h before treatment with different concentrations (0-50 μM) of 4-HNE in serum-free medium for 2 h at 37 °C. About 1 μ 105 RPE cells (VT and hGSTA4-Tr) were treated with 20 μM 4-HNE in serum-free medium for 1 h at 37 °C. Apoptotic cells were detected by staining with 10 μM CaspACE™ FITC-VAD-FMK in situ marker for 30 min in the dark. After rinsing twice with PBS, the slides were fixed with 4% paraformaldehyde for 1 h, mounted in a medium containing DAPI (1.5 μg/ml), and observed under the fluorescence microscope (Olympus, Japan).
Determination of 4-HNE levels
Intracellular 4-HNE levels in RPE cells were measured spectro-photometrically by using the Biotech LPO-586 228 kit (Oxis International, Portland, OR) as per the manufacturer's instructions as well as by the HPLC method described below. Empty vector- and hGSTA4-transfected RPE cells were sus pended in ice and sonicated (3×, 10 s, 30 W) on ice. Cellular proteins were precipitated with ice-cold 70% perchloric acid (1 ml). After centrifuging at 10000g for 10 min, the supernatants were extracted with 2 ml of dichloromethane (HPLC grade) by gentle vortexing. The extracts were once again centrifuged at 1500 rpm for 10 min. The organic layer was collected, dried under nitrogen, re-suspended in 100 μl ethanol and filtered through nylon 66 filters (Micron Separations Inc. NY). The Beckman Coulter System Gold HPLC equipment connected to a Beckman 168 Photo Diode Array (PDA) detector and the Beckman Ultrasphere (5 μm, 4.6 × 25 cm) column was used for HPLC analysis of the filtrate, using 70% sodium phosphate, pH 2.6, 30% acetonitrile as the mobile phase. The column eluate was monitored for 4-HNE at 202 and 224 nm [38] and peaks were analyzed by Beckman 32 Karat software. 4-HNE concentrations of the samples were quantified by using a calibration graph prepared by HPLC analysis of standard 4-HNE concentrations (0-1000 pmol) plotted against peak areas.
Results
4-HNE is toxic to RPE/ARPE-19 cells
Because retina is rich in polyunsaturated fatty acids and LPO is believed to be involved in the etiology of retinopathy [39], we first determined the cytotoxicity of 4-HNE to RPE and ARPE-19 cells in cultures which was estimated from the percentage of cells surviving the exposure of varying concentrations of 4-HNE for 2 h. Results of these experiments showed decrease in the cell viability with increasing concentrations of 4-HNE (Fig. 1). The IC50 values for 4-HNE for the RPE and ARPE-19 cells were found to be 52 ± 1.4, and 46 ± 3.2 μM (n = 8), respectively.
Fig. 1.
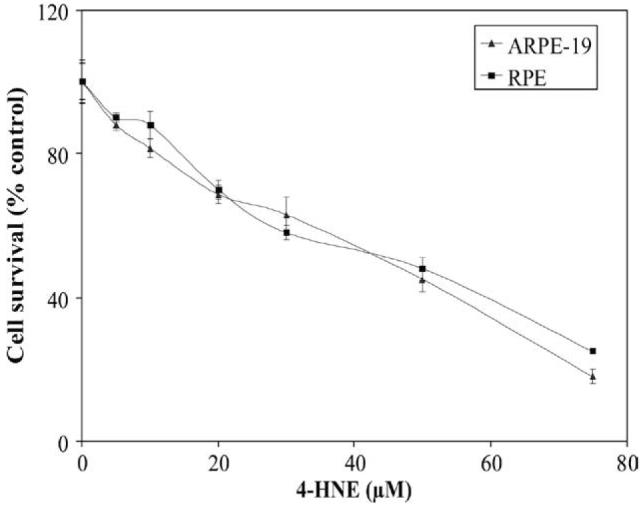
Cytotoxicity of 4-HNE to RPE and ARPE-19 cells: Cells (2 × 104) were plated in 96-well plates in complete growth medium for 24 h to allow for complete attachment to the culture plates. Next day, cells were incubated for 18 h in fresh serumfree medium to avoid any interaction between serum proteins and 4-HNE and solutions containing various amounts of 4-HNE prepared in PBS were added to achieve the final concentrations of 0-75 μM 4-HNE. Eight replicate wells were used for each concentration of 4-HNE used in these studies and the plates were incubated at 37 °C for 2 h. The MTT assay was performed as described in Materials and methods section. The OD562 values of samples subtracted from those of respective blanks (no cells) were normalized with control values. Results presented are percent cell survital in 4-HNE treated groups with respect to control cells (mean ± SD; n = 8).
4-HNE enhances p53 levels in RPE/ARPE-19 cells
It has been shown that 4-HNE causes apoptosis in a variety of cells including RPE cells [19,20,24,25]. P53 is known to be involved in the intrinsic pathway for apoptosis [4,5,40,41]. Therefore, to examine any possible role of p53 in the mechanisms of 4-HNE-induced apoptosis, we first determined the effect of 4-HNE on the levels of p53 in RPE and ARPE-19 cells. Upon 4-HNE treatment, a dose dependent increase in the intracellular levels of p53 was observed which peaked at 30 μM 4-HNE (Fig. 2A). In cells treated with 50 lM 4-HNE the level of p53 was slightly less than the optimally induced levels at 30 μM 4-HNE. This was probably because as seen in Fig. 1, close to 50% cell death was observed at this concentration of 4-HNE. There was also a time dependent induction of p53 at least up to 2 h in RPE cells exposed to 20 lM 4-HNE (Fig. 2B).
Fig. 2.

Effect of 4-HNE on induction and phosphorylation of p53 in RPE and ARPE-19 cells: Western blot analyses of 4-HNE treated RPE and ARPE-19 cells showing the activation of p53 with dose (A) and time (B), and p53 phosphorylation (C). For (A) and (C) cells were treated with 0-50 μM 4-HNE in the medium for 2 h. To assess the time dependent activation of p53 (B), RPE cells (4 × 105) were treated with 20 μM 4-HNE and incubated for 0, 15, 30, 60, and 120 min. Cells were harvested after completion of incubation and extracted in RIPA lysis buffer as described in the Methods section. Cell extracts (50 μg protein) were resolved on 4-20% SDS-PAGE and immunoblotted using anti-p53 (A), (B) and anti-phospho p53 (serine-15) (C) antibodies, respectively as the primary antibodies. GAPDH was used as the loading control. Blots were developed with West Pico-chemiluminescence reagent (Pierce). Representative blot from three different experiments yielding similar results has been presented.
4-HNE induces phosphorylation of p53
One of the potential mechanisms through which the activity of p53 protein may be regulated is through its phosphorylation on Ser and Thr residues within its N and C-terminal regions [12,13,42]. Therefore, we examined the phosphorylation status of p53 in 4-HNE-treated RPE cells using the anti-phosphoserine-15 antibody which specifically detects p53 phosphorylated at serine 15. The results of these studies showed that within 0-30 μM range, 4-HNE caused a dose-dependent increase in phosphorylated p53 in RPE and ARPE-19 cells which correlated with the increased intracellular levels of total p53 protein (Fig. 2C). At 50 μM 4-HNE, the level of phosphorylated p53 was slightly less than the maximally induced level by 30 μM 4-HNE, because of the toxicity of 50 μM 4-HNE to the cells as discussed above.
4-HNE-induces accumulation of p53 innucleus
In order to investigate whether or not 4-HNE facilitates nuclear accumulation of p53, we examined p53 expression in the cytoplasmic and nuclear fractions of RPE cell extracts by Western blot analysis. The results presented in Fig. 3A indicated a time dependent increase in p53 levels in the nuclear fraction of cells treated with 20 μM of 4-HNE. These results were consistent with the results of immunocytochemical localization experiments (Fig. 3B) which clearly indicated an increase in the concentration of p53 in the nuclear compartment of cells treated with 4-HNE. In previous studies [4,6], it has been shown that oxidative stress causes enhanced nuclear accumulation of p53. It is possible that this effect of oxidative stress may in fact be mediated by 4-HNE.
Fig. 3.

4-HNE-induced nuclear accumulation of p53 and degradation of MDM2 in RPE cells: RPE cells (2 × 106 cells in 100 mm petri dishes and 1 × 104 cells/chamber) were treated with 20 μM 4-HNE for different time intervals (0, 30, and 60 min). (A) Cells were harvested from the petri dishes and their cytoplasmic and nuclear extracts were prepared by using Imgenex cell processing kit according to the manufacturer's instructions. Western blot analyses for the expression of p53 in cytoplasmic and nuclear extracts (50 μg protein) were carried out using anti-p53 antibodies. Western blot presented show a time dependent decrease of p53 in cytoplasmic fractions with corresponding increase of p53 in the nuclear fractions. (B) 4-HNE treated cells in chamber slides were fixed in 4% paraformaldehyde, blocked with 1% goat serum and incubated with anti-p53 antibodies overnight at 4 °C. The cells were washed with PBS, incubated with FITC-conjugated secondary antibodies for 2 h, mounted with Vectashield mounting medium containing DAPI nuclear stain and the slides were viewed under Olympus fluorescence microscope. Photographs were taken with 40× objective. Photomicrographs show an enhanced nuclear accumulation of p53 with time as judged by the increase in green fluorescent stain in the nucleus. (C) Western blot analysis showing a gradual decrease of MDM2 levels in RPE cells treated with 0-50 μM 4-HNE for 2 h. Blots were probed with anti-MDM2 polyclonal antibodies. (D) Bar chart showing the percentage of cells with 4-HNE (20 μM) induced nuclear accumulation of p53 at different time points (mean ± SD, n = 3). (For interpretation of color mentioned in this figure the reader is referred to the web version of the article.)
4-HNE causes degradation of MDM2
Mouse double minute 2 (MDM2), a ubiquitin ligase, is a negative regulator of the p53 protein that blocks p53 transcriptional activity directly and mediates the degradation of p53 protein [43]. Under the conditions of oxidative stress, MDM2 polyubiquitylates itself which leads to the degradation of MDM2 resulting in an increase in the half-life of p53 [43]. During present studies, we also examined the effect of 4-HNE on MDM2 in ARPE-19 cells exposed to different concentrations of 4-HNE. Results of Western blot analyses presented in Fig. 3C indicated that treatment with increasing concentrations of 4-HNE caused a gradual decrease in the intracellular levels of MDM2 which was consistent with the increased levels of p53 in 4-HNE treated cells.
4-HNE causes induction of p21 and Bax in RPE/ARPE-19 cells
Since induction of p53 in response to cellular stress has been linked to apoptosis by the transcriptional activation of its target genes, including p21WAFI and Bax [15-18], we examined the effect of 4-HNE on the expression of these p53 associated pro-apoptotic genes. Bax has been identified as the first p53 target gene to encode an effector protein for p53-mediated apoptosis [17]. An increase in level of Bax was observed in RPE and ARPE-19 cells treated with the increasing concentrations of 4-HNE (Fig. 4a left and right panels). A dose-dependent up-regulation in the expression of the p21WAFI was also observed in these cells (Fig. 4b, left and right panels). Together, these results indicated that exposure of 4-HNE leads to an increased nuclear accumulation and phosphorylation of p53 as well as an up-regulation of some of the pro-apoptotic genes associated with p53.
Fig. 4.

4-HNE-induced activation of Bax, p21, JNK, and p-JNK in RPE and ARPE-19 cells: The expression of Bax (a) p21 (b) p-JNK (c), and JNK (d) in RPE and ARPE-19 cells exposed to different concentrations of 4-HNE. RPE and ARPE-19 cells were treated with 4-HNE (0-50 lM) for 2 h in serum-free medium. Cell extracts (50 μg protein) were resolved on 4-20% SDS-PAGE and immunoblotted. The blots were probed with anti-Bax, anti-p21, anti-JNK, and anti-p-JNK antibodies, respectively. GAPDH (e) was used as the loading control. Blots were developed with West Pico-chemi luminescence reagent (Pierce).
4-HNE activates JNK in RPE cells
Enhanced LPO and rise in the intracellular levels of 4-HNE is a common occurrence when cells are exposed to stressors such as heat shock, oxidant chemicals, and UV radiation and a sustained activation of JNK is observed during stress-induced apoptosis in many different cell types [19,20,44,45]. Therefore, we examined the effect of 4-HNE on the expression and phosphorylation of JNK. 4-HNE caused a dose-dependent increase in the phosphorylation of JNK in both RPE and ARPE-19 cells (Fig. 4c). These results showed that a sustained activation of JNK occurred in RPE cells exposed to 4-HNE.
Exposure of ARPE-19 cells to increasing concentrations of 4-HNE resulted in an increase in JNK protein in these cells as indicated by significant differences in the intensities of the two bands (JNK1/2 46 and 54 kDa) in 4-HNE treated and the control cells (Fig. 4d right panel). However, in RPE cells (Fig. 4d left panel) such differences in JNK1/2 of control and 4-HNE treated cells were not observed.
4-HNE activates caspase3 in RPE/ARPE-19 cells
Over-expression of wild-type p53 has been shown to elicit a rapid loss of cell viability and trigger apoptosis in a wide array of cell types [46]. In the effector stage of apoptosis, caspase3 (CPP32) is activated by its proteolytic cleavage into 17- and 12-kDa fragments [47]. Results presented in Fig. 5(panel A) showed that 4-HNE caused a dose dependent cleavage of a 17-kDa fragment from the caspase3 zymogen, CPP32. These results were also supported by in situ immunofluorescence studies with ARPE-19 cells which also showed a dose-dependent increase in the activation of caspase3.
Fig. 5.

Effect of 4-HNE on caspase3 in RPE and ARPE-19 cells: (A) Cell extracts (50 μg protein) from RPE cells treated with 4-HNE (0-50 μM) for 2 h were resolved on 4-20% SDS-PAGE and immunoblotted using the anti-caspase3 antibody as the primary antibody. Activation of caspase3 was monitored by the appearance of the 20/17 kDa bands. The blot was developed using West Pico-chemiluminescence reagent (Pierce). (B) In situ analysis of activated caspase3 in ARPE-19 cells. About 2 × 104 cells were grown in chamber slides and treated with 0, 10, 30, 50 μM 4-HNE for 2h. The activation of caspase3 in these cells was examined by staining with 10 μM CaspACE™ FITC-VAD-FMKin situ marker following the manufacturer's instructions. The slides were mounted with Vectashield DAPI mounting medium and observed with a fluorescence microscope (Olympus) using the standard filter sets for DAPI and FITC. Appropriately marked different panels show blue DAPI-stained and green FITC-stained cells in the figure. The photographs were taken at 400× magnification. (For interpretation of color mentioned in this figure the reader is referred to the web version of the article.)
Silencing of p53 expression in RPE cells attenuates 4-HNE-induced cell death
In order to further establish the role of p53 in 4-HNE induced apoptosis in RPE cells, the cells were transfected with p53 siRNA to silence the expression of p53. Silencing of p53 expression in these cells was achieved in RPE cells after 48 h of p53 siRNA transfection as judged by the Western blot analyses (Fig. 6A) which showed almost complete depletion of p53 expression when compared with control and scrambled siRNA transfected RPE cells. P53 depleted (-/-) RPE cells were found to be significantly resistant to the 4-HNE induced cell death as compared to p53 expressing (+/+) cells as analyzed by the MTT assay (Fig. 6B). These results support the involvement of p53 in the mechanisms of 4-HNE-induced cell death in RPE cells.
Fig. 6.
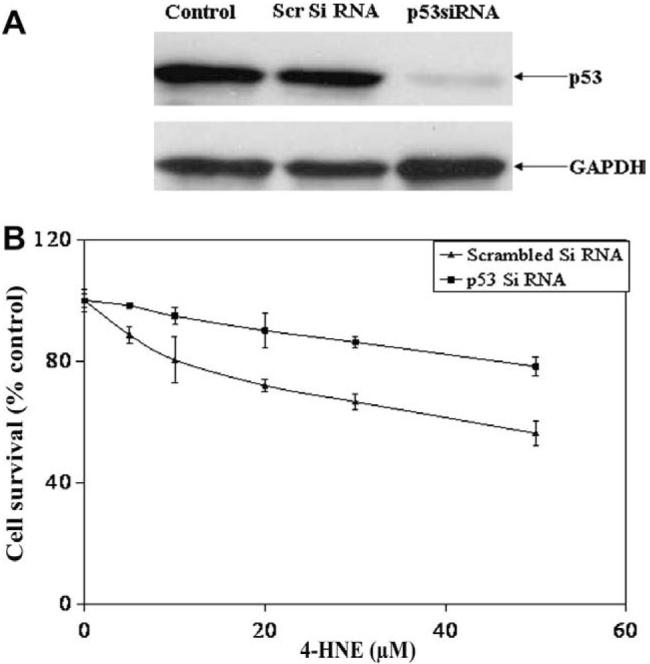
Silencing of p53 protects RPE cells from 4-HNE induced cytotoxicity: (A) RPE cells (1 × 105) were transfected with si RNA of p53 and scrambled siRNA as described in Methods section. The depletion of p53 in cells was confirmed by Western blot analysis. (B) P53 depleted and p53 expressing control cells were treated with 0-50 μM of 4-HNE for 2 h and assayed for cytotoxicity by MTT assay. The plot shows the percent cell survival (mean ± SD, n = 4) at different concentrations of 4-HNE.
Over-expression of GSTA4 inhibits 4-HNE induced apoptosis
During stress conditions causing LPO, the induction of GST isozymes with high catalytic efficiency to conjugate 4-HNE to GSH has been shown [19,20]. These cells with induced GSTs acquire an enhanced capability to dispose off 4-HNE and are resistant to apoptosis caused by various oxidants [19,20].We therefore, examined whether or not the over expression of GSTA4-4 in RPE/ARPE-19 cells had any effect on 4-HNE-induced and p53-mediated apoptosis. RPE cells were transiently transfected with the hGSTA4 isozyme and in parallel experiments; ARPE-19 cells were transfected with the murine isozyme mGsta4. Both these enzymes have high catalytic efficiency towards 4-HNE. Expression of hGSTA4-4 and mGsta4-4 in respective cells was confirmed by Western blot analysis as indicated by the results presented in Fig. 7(panels A and B) showing a strong expression of these isozymes in the transfected cells. As reported previously, there was a marked reduction in the intracellular levels of 4-HNE in GSTA4-transfected cells which was about 30% of that observed in the empty vector-transfected cells.
Fig. 7.

Effect of GSTA4 transfection on 4-HNE-induced caspase3 activation and apoptosis: (A) Expression of hGSTA4-4 and (B) mGsta4-4 in the transfected RPE and ARPE-19 cells, respectively. Aliquots (50 μg) of 28,000 g supernatant fraction of homogenates of the vector (VT) and hGSTA4-transfected (hGSTA4-Tr) RPE cells and mGsta4-transfected ARPE-19 cells were subjected to SDS-PAGE in 4-20% gels. Western blot analyses were performed using the polyclonal primary antibody against recombinant hGSTA4-4 and mGsta4-4. The blot was developed using chemiluminescence (Supersignal West Pico, Pierce) reagents. (C) Western blot analysis for caspase3 activation in the VT and hGSTA4-Tr RPE cells treated with 20 μM 4-HNE for 1 h. Cell extracts (50 μg protein) were resolved on 4-20% SDS-PAGE and immunoblotted using the anti-caspase3 antibody as the primary antibody. Activation of caspase3 was monitored by the appearance of 20/17 kDa band in the VT transfected, 20 μM 4-HNE-treated RPE cells only. The blots were developed using chemiluminescence (Supersignal West Pico, Pierce) reagents. (D) In situ analysis of activated caspase3 in VT and mGsta4-Tr RPE cells. 1 × 105 RPE cells were treated with 20 μM 4-HNE for 1 h. The activation of caspase3 in these cells was examined by staining with 10 μM CaspACE™ FITC-VAD-FMK in situ marker following the manufacturer's instructions. The slides were mounted with Vectashield DAPI mounting medium and observed with a fluorescence microscope (Olympus) using the standard filter sets for DAPI and FITC. Different panels show blue DAPI-stained and green FITC-stained cells as marked in the figure. Green FITC-labeled VT cells show a significant increase in the level of caspase3 activation after 4-HNE treatment. (For interpretation of color mentioned in this figure the reader is referred to the web version of the article.)
The extent of 4-HNE-induced apoptosis was compared in the vector and GSTA4-transfected RPE cells by assessing the activation of caspase3. Results of these experiments showed that both, hGSTA4-transfected RPE and mGsta4-transfected ARPE-19 cells acquired significant resistance to apoptosis as judged by the lack of caspase3 activation in 4-HNE treated transfected cells examined by Western blots (Fig. 7; panel C), and in situ immunofluorescence studies (Fig. 7; panel D). Together, these results suggested that GSTA4-transfected RPE cells acquired resistance to 4-HNE-induced p53-mediated apoptosis due to an accelerated metabolism of 4-HNE in these cells.
GSTA4-4 over-expression attenuates P53 expression and activation of associated pro-apoptotic genes
We have also compared the effect of 4-HNE on activation of p53 in empty vector and hGSTA4-transfected RPE cells. Results in Fig. 8A indicated that transfection of RPE cells either with empty vector or the hGSTA4 vector did not affect the expression of p53. However, exposure of empty vector-transfected RPE cells to 4-HNE resulted in a significant up-regulation of p53 expression. On the other hand, 4-HNE-treatment did not up-regulate p53 expression in hGSTA4-transfected cells. In fact, expression of p53 was suppressed in these cells. Since phosphorylation of p53 has been shown to influence its activity under conditions of stress, we examined the effect of 4-HNE on the levels of phospho p53 in these cells. Results presented in Fig. 8B indicated that levels of phospho p53 increased in 4-HNE treated empty vector-transfected cells but were significantly suppressed in hGSTA4-transfected cells. Interestingly, the treatment of mGsta4-4 over-expressing ARPE-19 cells with 4-HNE (20 μM) also resulted in the significant suppression of p53 expression (Fig. 8C). The effect of hGSTA4-transfection on the components of p53-mediated pathway for apoptosis in RPE cells exposed to 4-HNE was also examined. Transfection of RPE cells with hGSTA4 resulted in a significant down-regulation of the basal expression of Bax (Fig. 8A and B) in these cells. Treatment with 4-HNE resulted in a pronounced up-regulation of Bax in empty vector-transfected cells which was not seen in the hGSTA4-transfected cells. Cells transfected with empty vector also showed an increased expression of pJNK upon 4-HNE treatment which was not seen in hGSTA4-transfected cells (data not shown). Taken together, these results suggest a regulatoryrole of GSTA4-4 in p53-mediated, 4-HNE-induced apoptosis.
Fig. 8.

Effect of 4-HNE on the expression of p53, phospho p53, and Bax in GSTA4-4 over-expressing RPE cells: (A) Expression of p53, phospho p53, and Bax, in VT and hGSTA4-Tr RPE cells treated with 4-HNE (20 μM) for 1 h was monitored by Western blot analysis. Cell extracts (50 μg protein) were resolved on 4-20% SDS-PAGE and immunoblotted using the anti-p53, anti-phospho p53, and anti-Bax, respectively as the primary antibodies. GAPDH was used as the loading control. The blot was developed using chemiluminescence (Supersignal West Pico, Pierce) reagents. (B) A bar graph showing densitometric analysis of bands (using Kodak 1D image analysis software) of p53, phospho p53, and Bax, respectively. (C) Effect of 4-HNE on p53 expression in mGsta4-4 overexpressing ARPE-19 cells.
Increased levels of 4-HNE in tissues of mGsta4 null mice lead to the induction of p53
Earlier studies in our laboratory have shown that 4-HNE levels in the tissues of mGsta4 (-/-) null mice are about 3-fold higher than that observed in the tissues of wild-type (+/+) mice [48]. Therefore, we compared the levels of p53 in the brain, heart, lung, kidney, liver, and eye tissues of mGsta4-4 (+/+) and (-/-) mice. The results of these studies presented in Fig. 9 indicated enhanced p53 levels in all these tissues of mGsta4 -/- mice as compared to the tissues of +/+ mice. These results show for the first time that in vivo expression of p53 is determined by the intracellular or ambient levels of 4-HNE in tissues and it is regulated by GSTs.
Fig. 9.
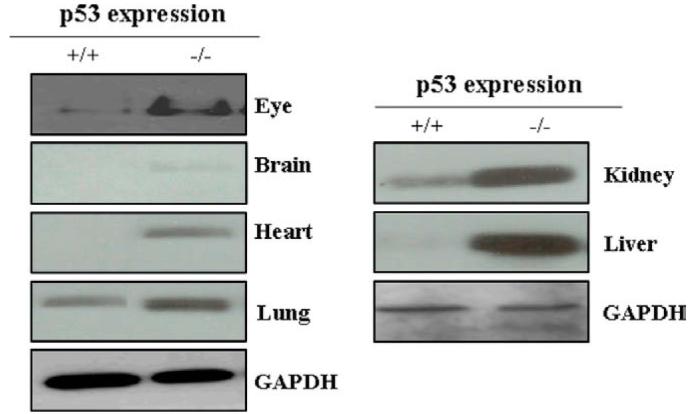
Expression of p53 in different tissues of mGSTA4 (+/+) and (-/-) mice: Mice eye, brain, heart, lung, kidney, and liver were homogenized in lysis buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM sodium orthovanadate, 50 mM NaF, PMSF (20 μg/ml), 4 mM EDTA, 1 mM EGTA, 1% NP-40, 150 mM sodium chloride, 100 μM leupeptin, 0.07 μg/ml pepstatin, 10 μg/ml soybean trypsin inhibitor, 1:100 protease inhibitor cocktail and centrifuged at 14000 rpm. Supernatants containing 50 μg tissue proteins were loaded on 4-20% SDS-PAGE and p53 was assayed by Western blot analysis using anti-p53 antibody as the primary antibody. GAPDH was used as a loading control. Blots were developed with West Pico-chemiluminescence reagent (Pierce).
Discussion
The role of 4-HNE as a small signaling molecule has been advocated during the past decade and numerous studies have shown that it can modulate the expression of many genes, affect cell cycle signaling, and activate membrane tyrosine kinase receptors to influence the activi ties of key regulatory proteins such as protein kinase C isoenzymes [6,49-51]. Present studies demonstrating that 4-HNE can modulate the intracellular levels and functions of p53 further substantiate the role of 4-HNE as a molecule which can modulate cell cycle events. Here we demonstrate that exogenous addition of 4-HNE to the media of RPE cells in culture leads to an induction of p53. Earlier studies have shown a similar 4-HNE-induced dose-dependent increase in the expression of p53 in the cytoplasmic and nuclear compartments of an unrelated cell line, CRL2571 [52]. This would suggest that this effect of 4-HNE is not limited to a specific cell line and is perhaps a generalized phenomenon. A dose-dependent induction of the p53 in various cells by ionizing radiations [53] and hydrogen peroxide has been reported [54,55]. Since 4-HNE levels are increased in cells exposed to ionizing radiations or oxidants it is more than likely that 4-HNE is involved in the induction of p53 in ARPE-19 cells by radiation/oxidants.
Earlier studies have also shown that lowering of the intracellular levels of 4-HNE can lead to the suppression of p53 expression [26,27]. The results of present studies when taken together with these earlier studies suggest that the intracellular concentration of 4-HNE is one of the major determinants for the cellular levels of p53 which is widely accepted as a major cell cycle regulatory protein [20].
The induction of p53 by 4-HNE can be inhibited by the over expression of either hGSTA4 or mGsta4 which accelerates disposition of 4-HNE. This would suggest that the effect of 4-HNE on p53 is either a direct effect of 4-HNE or any of its metabolites or degradation products of 4-HNE that can also trigger p53-mediated apoptosis. Present studies show that not only do the p53 levels increase in 4-HNE treated cells, the levels of phosphorylated p53 are also elevated. Since phosphorylation of p53 has been suggested to contribute to its stabilization, it is possible that the overall increase in p53 levels in 4-HNE-treated cells may be the consequence of a two-pronged effect of 4-HNE where it down-regulates the degradation of p53 by MDM2 and also stabilizes the gene product through its phosphorylation.
The effect of 4-HNE on the expression of various genes has been extensively studied in cell cultures [7,26,27,56]. However, the in vivo relevance, if any, of these studies is not clear. Present studies demonstrate for the first time that the rise in p53 levels in tissues of these mice positively correlates with increased 4-HNE levels in these tissues. We have previously demonstrated that in the tissues of mGsta4 null mice the levels of 4-HNE increase by about three-fold [48]. This would suggest that the levels of p53 in mice in vivo, are regulated by the intracellular and/or ambient levels of 4-HNE. The levels of p53 were found to be increased in most of the tissues examined in these studies suggesting that the observed effects of 4-HNE on expression and stabilization of p53 may have physiological significance in vivo. Furthermore, these results provide support to the idea that 4-HNE mediated induction of p53 is not limited to any particular cell type. It is important to realize that within the cells, 4-HNE has a short half-life and it is difficult to assess its concentrations within the cells at a given time point. However, our studies clearly demonstrate that p53 accumulation within cells increases up to a certain threshold with any rise in the ambient 4-HNE levels. MDM2 was found to be down regulated in RPE cells exposed to different concentrations of 4-HNE. Under the influence of stress stimuli such as radiations and chemicals, MDM2 polyubiquitylates itself to allow enhanced accumulation of p53 with a concomitant increase in the half-life of p53 from minutes to hours and therefore acts as a negative regulator of p53 expression. The intracellular levels of 4-HNE correlate with oxidative stress [19,20] and therefore it is possible that the promotion of polyubiquitylation and degradation of MDM2 [43] in UV or oxidant treated cells is mediated via 4-HNE.
Our results also demonstrate that 4-HNE modulates not only the intracellular levels of p53 but also its functions. It is well-established that oxidative stress-induced apoptosis is mediated through an intrinsic pathway involving p53 [27,56]. The involvement of p53 in the mechanisms of 4-HNE induced cell death was further confirmed by silencing the expression of p53 which lead to enhanced resistance of the RPE cells to 4-HNE-induced cytotoxicity. Increased resistance in p53 depleted ARPE-19 cells to apoptosis caused by gamma radiations and other genotoxic agents also support these findings [57].
We demonstrate that along with the induction and phosphorylation of p53, 4-HNE also activates the signaling components of the p53-mediated pathway of apoptosis. Bax, p21, JNK, and caspase3 are all activated in response to exposure to 4-HNE. Since these effects can be inhibited by accelerating GSTA4-4-mediated conjugation of 4-HNE to GSH, it is further indicated that 4-HNE directly modulates these components of p53-linked apoptotic machinery. 4-HNE has also been shown to inhibit thioredoxin reductase (TrxR), which is a component of defense mechanisms against oxidative stress [58-60]. It will be of interest to see if the effects of 4-HNE on p53 are mediated by TrxR enzyme system in RPE cells. Present findings showing that 4-HNE can mimic the effects of stress or stress-induced DNA damage in signaling for apoptosis suggest that 4-HNE may be an initiator of chemical signals in stress-induced apoptosis via the p53-mediated intrinsic pathway.
Acknowledgments
Supported in part by NIH Grants ES012171, EY 04396 (Y.C.A.), CA77495 (S.A.).
References
- [1].Esterbauer H, Schaur RJ, Zollner H. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- [2].Awasthi YC, Sharma R, Cheng JZ, Yang Y, Sharma A, Singhal SS, Awasthi S. Mol. Aspects Med. 2003;24:219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- [3].Dianzani MU. Mol. Aspects Med. 2003;24:263–272. doi: 10.1016/s0098-2997(03)00021-9. [DOI] [PubMed] [Google Scholar]
- [4].Levine AJ. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- [5].Gottlieb TM, Oren M. Semin. Cancer Biol. 1998;8:359–368. doi: 10.1006/scbi.1998.0098. [DOI] [PubMed] [Google Scholar]
- [6].Harris SL, Levine AJ. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- [7].Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- [8].Prives C. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- [9].Kohli M, Jorgensen TJ. Biochem. Biophys. Res. Commun. 1999;257:168–176. doi: 10.1006/bbrc.1999.0389. [DOI] [PubMed] [Google Scholar]
- [10].Lin JY, Simmons DT. J. Virol. 1991;65:6447–6453. doi: 10.1128/jvi.65.12.6447-6453.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hess RD, Brandner G. Oncogene. 1997;15:2501–2504. doi: 10.1038/sj.onc.1201404. [DOI] [PubMed] [Google Scholar]
- [12].Shieh SY, Ikeda M, Taya Y, Prives C. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- [13].Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. Genes Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- [15].Hunter T, Pines J. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- [16].El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- [17].Miyashita T, Reed JC. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- [18].Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- [19].Cheng JZ, Sharma R, Yang Y, Singhal SS, Sharma A, Saini MK, Singh SV, Zimniak P, Awasthi S, Awasthi YC. J. Biol. Chem. 2001;276:41213–41223. doi: 10.1074/jbc.M106838200. [DOI] [PubMed] [Google Scholar]
- [20].Yang Y, Sharma A, Sharma R, Patrick B, Singhal SS, Zimniak P, Awasthi S, Awasthi YC. J. Biol. Chem. 2003;278:41380–41388. doi: 10.1074/jbc.M305766200. [DOI] [PubMed] [Google Scholar]
- [21].Sharma R, Brown D, Awasthi S, Yang Y, Sharma A, Patrick B, Saini MK, Singh SP, Zimniak P, Singh SV, Awasthi YC. Eur. J. Biochem. 2004;271:1690–1701. doi: 10.1111/j.1432-1033.2004.04067.x. [DOI] [PubMed] [Google Scholar]
- [22].Patrick B, Li J, Jeyabal PV, Reddy PM, Yang Y, Sharma R, Sinha M, Luxon B, Zimniak P, Awasthi S, Awasthi YC. Biochem. Biophys. Res. Commun. 2005;334:425–432. doi: 10.1016/j.bbrc.2005.06.099. [DOI] [PubMed] [Google Scholar]
- [23].Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Free Radic. Biol. Med. 2004;37:607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- [24].Liu W, Kato M, Akhand AA, Hayakawa A, Suzuki H, Miyata T, Kurokawa K, Hotta Y, Ishikawa N, Nakashima I. J. Cell Sci. 2000;113:635–641. doi: 10.1242/jcs.113.4.635. [DOI] [PubMed] [Google Scholar]
- [25].Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. J. Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li L, Hamilton RF, Kirichenko A, Holian A. Toxicol. Appl. Pharmacol. 1996;139:135–143. doi: 10.1006/taap.1996.0152. [DOI] [PubMed] [Google Scholar]
- [27].Laurora S, Tamagno E, Briatore F, Bearding P, Pizzimenti S, Toaldo C, Reffo P, Costelli P, Dianzani MU, Danni O, Barrera G. Free Radic. Biol. Med. 2005;38:215–225. doi: 10.1016/j.freeradbiomed.2004.10.014. [DOI] [PubMed] [Google Scholar]
- [28].Sickel W. In: Handbook of Sensory Physiology. Fuortes MGF, editor. Springer-Verlag; 1972. pp. 667–727. [Google Scholar]
- [29].Young RW. J. Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tate DJ, Newsome DA, Oliver PD. Invest. Ophthalmol. Vis. Sci. 1993;34:2348–2351. [PubMed] [Google Scholar]
- [31].Liang FQ, Godley BF. Exp. Eye Res. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- [32].Klein R, Wang Q, Klein BE, Moss SE, Meuer SM. Invest. Ophthalmol. Vis. Sci. 1995;36:182–191. [PubMed] [Google Scholar]
- [33].Cheng JZ, Yang Y, Singh SP, Singhal SS, Awasthi S, Pan SS, Singh SV, Zimniak P, Awasthi YC. Biochem. Biophys. Res. Commun. 2001;282:1268–1274. doi: 10.1006/bbrc.2001.4707. [DOI] [PubMed] [Google Scholar]
- [34].Zimniak L, Awasthi S, Srivastava SK, Zimniak P. Toxicol. Appl. Pharmacol. 1997;143:221–229. doi: 10.1006/taap.1996.8070. [DOI] [PubMed] [Google Scholar]
- [35].Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- [36].Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [37].Guo J, Zimniak L, Zimniak P, Orchard JL, Singh SV. Biochem. J. 2002;366:817–824. doi: 10.1042/BJ20020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hartley DP, Ruth JA, Petersen DR. Arch. Biochem. Biophys. 1995;316:197–205. doi: 10.1006/abbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- [39].Bazan NG. Prog. Clin. Biol. Res. 1989;312:95–112. [PubMed] [Google Scholar]
- [40].Thompson CB. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- [41].Strasser A, O'Connor L, Dixit VM. Annu. Rev. Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- [42].Meek DW. Semin. Cancer Biol. 1994;5:203–210. [PubMed] [Google Scholar]
- [43].Riley T, Sontag E, Chen P, Levine A. Nature Rev. Mol. Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- [44].Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. J. Biol. Chem. 1999;274:2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- [45].Biasi F, Vizio B, Mascia C, Gaia E, Zarkovic N, Chiarpotto E, Leonarduzzi G, Poli G. Free Radic. Biol. Med. 2006;41:443–454. doi: 10.1016/j.freeradbiomed.2006.04.005. [DOI] [PubMed] [Google Scholar]
- [46].Oren M. Semin. Cancer Biol. 1994;5:221–227. [PubMed] [Google Scholar]
- [47].Fernandes-Alnemri T, Litwack G, Alnemri ES. J. Biol. Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- [48].Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, Yang Y, Awasthi S, Awasthi YC, Zimniak P. Toxicol. Appl. Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- [49].Negre-Salvayre A, Vieira O, Escargueil-Blanc I, Salvayre R. Mol. Aspects Med. 2003;24:251–261. doi: 10.1016/s0098-2997(03)00020-7. [DOI] [PubMed] [Google Scholar]
- [50].Leonarduzzi G, Robbesyn F, Poli G. Free Radic. Biol. Med. 2004;37:1694–1702. doi: 10.1016/j.freeradbiomed.2004.08.027. [DOI] [PubMed] [Google Scholar]
- [51].Chiarpotto E, Domenicotti C, Paola D, Vitali A, Nitti M, Pronzato MA, Biasi F, Cottalasso D, Marinari UM, Dragonetti A, Cesaro P, Isidoro C, Poli G. Hepatology. 1999;29:1565–1572. doi: 10.1002/hep.510290510. [DOI] [PubMed] [Google Scholar]
- [52].Sharma R, Sharma A, Dwivedi S, Zimniak P, Awasthi S, Awasthi YC. Biochemistry. 2008;47:143–156. doi: 10.1021/bi701559f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jiang YL, Escano MFT, Sasaki R, Fujii S, Kusuhara S, Matsumoto A, Sugimura K, Negi A. Jpn. J. Ophthalmol. 2004;48:106–114. doi: 10.1007/s10384-003-0043-x. [DOI] [PubMed] [Google Scholar]
- [54].Jin GF, Hurst JS, Godley BF. Curr. Eye Res. 2001;22:165–173. doi: 10.1076/ceyr.22.3.165.5517. [DOI] [PubMed] [Google Scholar]
- [55].Uberti D, Yavin E, Gil S, Ayasola K-R, Goldfinger N, Rotter V. Mol. Brain Res. 1999;65:167–175. doi: 10.1016/s0169-328x(98)00339-8. [DOI] [PubMed] [Google Scholar]
- [56].Shibata T, Iio K, Kawai Y, Shibata N, Kawaguchi M, Toi S, Kobayashi M, Kobayashi M, Yamamoto K, Uchida K. J. Biol. Chem. 2006;281:1196–1204. doi: 10.1074/jbc.M509065200. [DOI] [PubMed] [Google Scholar]
- [57].Nair AR, Schliekelman M, Thomas MB, Wakefield J, Jurgensen S, Ramabhadran R. Cell Cycle. 2005;4:697–703. doi: 10.4161/cc.4.5.1672. [DOI] [PubMed] [Google Scholar]
- [58].Moos PJ, Edes K, Cassidy P, Massuda E, Fitzpatrick FA. J. Biol. Chem. 2003;278:745–750. doi: 10.1074/jbc.M211134200. [DOI] [PubMed] [Google Scholar]
- [59].Cassidy PB, Kornelia E, Nelson CC, Parsawar K, Fitzpatrick FA, Moos PJ. Carcinogeneisis. 2006;27:2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- [60].Fang J, Holmgren A. J. Am. Chem. Soc. 2006;128:1879–1885. doi: 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]