

Abstract
Escherichia coli lipid A is a hexa-acylated disaccharide of glucosamine with secondary laurate and myristate chains on the distal unit. Hexa-acylated lipid A is a potent agonist of human Toll-like receptor 4, whereas its tetra- and penta-acylated precursors are antagonists. The inner membrane enzyme LpxL transfers laurate from lauroyl-acyl carrier protein to the 2′-R-3-hydroxymyristate moiety of the tetra-acylated lipid A precursor Kdo2-lipid IVA. LpxL has now been over-expressed, solubilized with n-dodecyl-β-d-maltopyranoside (DDM) and purified to homogeneity. LpxL migration on a gel-filtration column is consistent with a molecular weight of 80 kDa, suggestive of an LpxL monomer (36 kDa) embedded in a DDM micelle. Mass spectrometry showed that deformylated LpxL was the predominant species, non-covalently bound to as many as twelve DDM molecules. Purified LpxL not only catalyzed the formation in vitro of Kdo2-(lauroyl)-lipid IVA but also a slow second acylation, generating Kdo2-(dilauroyl)-lipid IVA. Consistent with the Kdo-dependence of crude LpxL in membranes, Kdo2-lipid IVA is preferred 6,000-fold over lipid IVA by the pure enzyme. Sequence comparisons suggest that LpxL shares distant homology with the glycerol-3-phosphate acyltransferase (GPAT) family, including a putative catalytic dyad located in a conserved H(X)4D/E motif. Mutation of H132 or E137 to alanine reduces specific activity by over three orders of magnitude. Like many GPATs, LpxL can also utilize acyl-CoA as an alternative acyl donor, albeit at a slower rate. Our results show that the acyltransferases that generate the secondary acyl chains of lipid A are members of the GPAT family and set the stage for structural studies.
The outer leaflet of the outer membrane of Gram-negative bacteria is composed of an unusual saccharolipid glycan, termed lipopolysaccharide (LPS). LPS is made up of a hydrophobic anchor (lipid A), a non-repeating oligosaccharide core, and a distal polysaccharide (O-antigen) (1). Escherichia coli lipid A is a hexa-acylated disaccharide of glucosamine that contains two or three phosphate residues (1, 2). The lipid A moiety of LPS is recognized with exquisite specificity and affinity by the innate immune system (3). Wild-type E. coli lipid A activates the Toll-like receptor-4/MD-2 (TLR-4) complex (2–4). The human receptor complex specifically senses hexa-acylated LPS to propagate the signals inside the cell that initiate the inflammatory cascade (5). For otherwise healthy animals, the LPS shed by Gram-negative bacteria contributes to the innate immune response, leading to clearance of the bacteria (3). However, during systemic infections the innate immune response may become over-stimulated, leading to Gram-negative septic shock (6, 7). In the United States there are approximately 750,000 cases of sepsis leading to over 200,000 deaths per year (6, 7). About 40% are caused by Gram-negative bacteria (7).
Mutants of Gram-negative bacteria modified to accumulate under-acylated lipid A precursors are up to 1,000-fold less immunogenic against human cells (8–10), and under-acylated lipid A actually antagonizes the human TLR-4/MD-2 response (5, 8, 10, 11). In addition, reduced virulence has been demonstrated for many such mutants of pathogens including Haemophilus influenzae, Salmonella typhimurium, Neisseria meningitidis and N. gonorrhoeae (9, 10, 12–14). Therefore, the ability to inhibit the full acylation of lipid A could help to ameliorate sepsis and/or inflammation associated with Gram-negative bacterial infection. Re-engineering of Gram-negative bacteria to produce under-acylated lipid A may also be an attractive option for live vaccine development (10, 14). Lipid IVA and other under-acylated lipid A species produced by these bacteria would not provoke an acute innate immune response via TLR-4/MD-2. Instead, the adaptive immune system might have time to respond to and eliminate the vaccine strain, in the process establishing immunity against the pathogenic wild-type strains (10, 14). Moreover, Gram-negative bacteria with under-acylated lipid A, while viable, possess compromised outer membranes and are more susceptible to antibiotics, detergents and cationic peptides (10, 13, 15–20).
The biosynthesis of lipid A begins with UDP-N-acetylglucosamine, and through eight steps, catalyzed by seven conserved enzymes (Scheme 1), yields tetra-acylated lipid IVA linked to two 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) moieties (2). Next, Kdo2-lipid IVA is the preferred substrate for LpxL (Scheme 1), which transfers laurate from lauroyl-acyl carrier protein (ACP) to the free OH group of the 2′-R-3-hydroxymyristoyl chain of Kdo2-lipid IVA (21). LpxL is a 35.5 kDa protein (306 amino acids) that contains one predicted transmembrane helix at its amino-terminus (15–17). Before its biochemistry was elucidated (21, 22), the lpxL gene had already been identified as a high-temperature requirement gene (termed htrB), because the null mutant is temperature-sensitive for growth above 33 °C on nutrient broth (15–17). Under non-permissive conditions, cell division ceases, the membranes bulge, and the cells slowly lyse (15–17).
Scheme 1. Incorporation of laurate into E. coli lipid A by LpxL.

The 2′ secondary laurate chain incorporated by LpxL is shown in red in the context of the entire constitutive lipid A pathway. Other enzyme designations are shown in magenta. The glucosamine disaccharide of lipid A is highlighted in blue, with the numbering system indicated in red. LpxM does not function efficiently without the laurate chain in its substrate (23, 24).
LpxL shares significant sequence homology and functional similarity to LpxM, the final enzyme in the lipid A biosynthetic pathway (Scheme 1) (18, 21, 23). E. coli LpxM works subsequent to LpxL to catalyze the transfer of myristate from myristoyl-ACP to the free hydroxy group of the 3′-R-3-hydroxymyristoyl chain of Kdo2-(lauroyl)-lipid IVA (23). LpxM strongly prefers the penta-acylated LpxL product as its substrate (24), and under normal conditions will not acylate Kdo2-lipid IVA very efficiently (18, 23). In addition, E. coli possesses a third LpxL ortholog, termed LpxP (25, 26), which is induced below 20 °C and competes with LpxL to transfer a palmitoleate chain to Kdo2-lipid IVA (25, 26). The shift from laurate to palmitoleate may serve to maintain the optimal outer membrane fluidity at low growth temperatures (26). There is at least one LpxL ortholog in nearly every species of Gram-negative bacteria. All of these orthologs are known or predicted to be involved in lipid A biosynthesis. The LpxL and LpxM acyltransferases lack homologyto LpxA and LpxD, the early acyltransferases of the lipid A pathway (Scheme 1) (2, 21, 27). Alignments of LpxL and LpxM proteins indicate the presence of a single conserved histidine residue, but no other obvious clues to the structure or catalytic mechanism (27).
Here, the purification and characterization of E. coli LpxL are reported. Our work provides the first unambiguous demonstration that LpxL by itself catalyzes the transfer of laurate from lauroyl-ACP to Kdo2-lipid IVA. The presence of the Kdo disaccharide in the substrate accelerates laurate transfer by at least three orders of magnitude. Pure LpxL was also found to catalyze a slow second acylation reaction leading to the formation of Kdo2-(dilauroyl)-lipid IVA, which is similar in structure to Kdo2-lipid A isolated from heptose-deficient mutants (28); however, this does not occur in cells of the lpxL- lpxM- and lpxP- deficient triple mutant MKV15b that over-express LpxL from a plasmid. Distant sequence homology between LpxL and the glycerol 3-phosphate acyltransferase (GPAT) family of enzymes was noted for the first time and confirmed by site-directed mutagenesis and analysis of substrate selectivity. These findings allow the assignment of an H(X)4E motif as the catalytic dyad of LpxL, wherein His 132 is proposed to interact with the R-3-hydroxyl group of the 2′-R-3-hydroxymyristoyl chain of Kdo2-lipid IVA, possibly activating it for nucleophilic attack on the thioester carbonyl group of lauroyl-ACP.
Experimental Procedures
Materials
Ammonium acetate, sodium acetate, chloroform and silica gel 60 (0.25 mm) thin layer chromatography (TLC) plates were obtained from EMD Chemicals Inc. (Gibbstown, NJ). [γ-32P]ATP was from PerkinElmer Life And Analytical Sciences, Inc. (Waltham, MA). The detergent n-dodecyl-β-d-maltopyranoside (DDM) was from Anatrace, Inc. (Maumee, OH). Yeast extract, agar, and tryptone were from Becton, Dickinson, and Company (Franklin Lakes, NJ). Sodium chloride and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were from VWR International (West Chester, PA). Triton X-100, NP-40, and the bicinchoninic acid (BCA) protein concentration determination kit (29) were from Thermo Fisher Scientific (Waltham, MA). Diethylaminoethyl (DEAE) cellulose (type DE52) was from Whatman Inc. (Florham Park, NJ). Tetrabutylammonium phosphate (TBAP) was from Regis Technologies, Inc. (Morton Grove, IL). Isopropyl-1-thio-β-d-galactopyranoside (IPTG) was from Invitrogen Corp. (Carlsbad, CA). R-3-hydroxylauroyl-methylphosphopantetheine was synthesized by Avanti Polar Lipids Inc. (Alabster, AL). All other chemicals were reagent grade and were purchased from either Sigma-Aldrich (St. Louis, MO) or Mallinckrodt Baker, Inc. (Phillipsburg, NJ).
Bacterial Strains
The bacterial strains used in this study are described in Table I. In general, the strains were grown at 37 °C in lysogeny broth (LB) (30). When required for selection of plasmids, cells were grown in the presence of 100 µg/mL ampicillin or 34 µg/mL chloramphenicol. In the absence of a plasmid, MKV15b (24) was selected on 12.5 µg/mL tetracycline.
Table 1.
Relevant strains and plasmids.
| Strain/Plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| BLR(DE3)pLysS | F− ompT hsdSB(rB − mB −) gal dcm (DE3) D(srl-recA)306::Tn10 pLysS (CamR, TetR) |
Novagen |
| W3110 | Wild-type, F−, λ− |
E. coli Genetic Stock Center (Yale) |
| MKV15b | W3110 lpxL::Tn10, lpxM::Ωcam, lpxP::kan, suppressor | Ref. (24) |
| XL1-Blue |
recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′proAB lacIqZDM15 Tn10 (TetR)] |
Stratagene |
| Plasmids | ||
| pET21a(+) | Expression vector containing a T7 promoter, AmpR | Novagen |
| pWSK29 | Low-copy expression vector, lac promoter, AmpR | Ref. (33) |
| pLpxR2 | pWSK29 containing lpxR | Ref. (40) |
| pLpxL | pET21a(+) containing lpxL | Ref. (32) |
| pWSK-LpxL | pWSK29 containing lpxL | This work |
| pWSK-LpxLΔN1 | pWSK29 containing lpxLΔN1 | This work |
| pWSK-LpxLΔN2 | pWSK29 containing lpxLΔN2 | This work |
| pH132A | pWSK29 containing lpxL-H132A | This work |
| pE137A | pWSK29 containing lpxL-E137A | This work |
| pR169A | pWSK29 containing lpxL-R169A | This work |
| pD200A | pWSK29 containing lpxL-D200A | This work |
| pP238A | pWSK29 containing lpxL-P238A | This work |
Molecular Biology Protocols
The plasmids used in this study are described in Table I. Plasmids were isolated using the Qiagen Spin Miniprep kit, and DNA fragments were isolated with the Qiaquick Spin Kits (Qiagen Valencia, CA). Pfu Turbo DNA polymerase (Stratagene La Jolla, CA), T4 DNA ligase (Invitrogen Corp. Carlsbad, CA), restriction endonucleases (New England Biolabs, Inc. Ipswich, MA), and shrimp alkaline phosphatase (USB Corp. Cleveland, OH) were used according to the manufacturers’ instructions. Chemically-competent cells were prepared as previously described (31).
Construction of Vectors
The cDNA for LpxL (accession number AAC74138) was previously cloned into the NdeI and BamHI sites of the pET21a(+) vector (EMD Chemicals Inc.) and named pLpxL (32). LpxL was subcloned out of the pET vector and into the pWSK29 vector using the XbaI and HindIII sites yielding a fragment which included the pET21a(+) ribosome binding site. The resulting plasmid is termed pWSK-LpxL. The pWSK29 plasmid is a relatively low-copy vector in which the inserted gene of interest is under control of the lac promoter. These plasmids (33) have been found to be particularly effective for the expression of membrane proteins in our laboratory.
Expression of LpxL
As determined by activity measurements and a visible band following sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), LpxL is significantly overexpressed after IPTG-induction of BLR(DE3)-pLysS harboring the pET construct, pLxpL (32). However, this strain contains intact, chromosomally-encoded lipid A secondary acyltransferase genes, potentially complicating purification and mutant analysis. Therefore, pWSK-LpxL was transformed into the E. coli strain MKV15b, which lacks all three lipid A secondary acyltransferase genes, lpxL (htrB), lpxM (msbB), and lpxP (ddg) (24). MKV15b is a spontaneous revertant of the parental triple-knockout strain MKV15, but unlike the parental strain, MKV15b grows well in LB broth at 37 °C and does not lyse during centrifugation (24). Because MKV15b lacks all the chromosomally-encoded secondary acyltransferases, the LPS from this strain contains predominantly the tetra-acylated lipid IVA (24) as its membrane anchor, and there is no background acyltransferase activity detected in vitro in cell extracts or membranes using acyl-ACP as the acyl-chain donor (24). MKV15b does still contain PagP, an outer membrane lipid A palmitoyltransferase that specifically employs phospholipids as the acyl-chain donors (24, 34). LpxL overexpression in MKV15b or BLR(DE3) pLysS was accomplished as described previously (32). Briefly, MKV15b with pWSK-LpxL was inoculated at an A600 of 0.02 in LB broth containing 1 mM IPTG and grown for 4 h before harvesting. In contrast, BLR(DE3) pLysS harboring pLpxL was inoculated at A600 of 0.02 and grown to 0.4 – 0.6. IPTG was added to 1 mM final concentration. The cells were grown for 3 h and harvested. Membranes were prepared from crude cell-free lysates by centrifugation as described previously (35), except that the buffer for washing the cell pellets and membranes contained 30 mM HEPES, pH 7.5, 1 mM ethylene glycol tetraacetic acid (EGTA), and 1 mM ethylenediaminetetraacetic acid (EDTA).
Kdo2-lipid IVA Preparation
The LpxL substrate, Kdo2-lipid IVA, was synthesized from lipid IVA. Lipid IVA was isolated and purified from MKV15b, which was grown at 37 °C in LB broth. The MKV15b cell pellet was washed and resuspended in phosphate-buffered saline (PBS) and extracted with a single-phase Bligh-Dyer mixture (consisting of chloroform:methanol:PBS, 1:2:0.8, v/v) (36) to remove phospholipids. Lipid IVA was liberated by resuspending the extracted pellet in 12.5 mM sodium acetate, pH 4.5. The suspension was subjected to probe sonic irradiation to homogenize the debris, and, if necessary, the pH was adjusted to 4.5 with acetic acid. The addition of 1% SDS, which is often employed in this procedure, was not required for efficient hydrolysis of MKV15b LPS. The suspension was boiled for 30 min, cooled to room temperature, acidified to pH 1 with HCl, and converted to a two-phase Bligh-Dyer mixture (chloroform:methanol:PBS, 2:2:1.8, v/v) (36). After centrifugation, the lower phase was retrieved, while the upper phase was washed once with pre-equilibrated acidic lower phase and centrifuged again. The second lower phase was retrieved, pooled with the first lower phase, neutralized with a few drops of pyridine, and dried with a rotary evaporator. The dried, crude lipid IVA was purified over DEAE cellulose, as described (37). After TLC in a chloroform:pyridine:formic acid:water system (50:50:16:5, v/v), the lipids were visualized by sulfuric acid charring. The small amounts of contaminating penta-acylated lipid IVB (10–20%) (2), generated by the outer membrane enzyme PagP (34), was removed by reverse-phase chromatography (octadecyl resin), employing TBAP counter-ions (37). The TBAP was removed by a second purification over DEAE cellulose, and the pure lipid IVA was retrieved by Bligh/Dyer partitioning (36, 37), yielding approximately 5 mg of lipid IVA from 9 g of MKV15b cell paste. The resulting pure lipid IVA was converted in vitro to Kdo2-lipid IVA (38). The reaction mixture was extracted by the Bligh/Dyer method (36), and the Kdo2-lipid IVA was purified over DEAE cellulose (37) to remove detergent and phospholipids from the in vitro reaction. The pure Kdo2-lipid IVA was weighed and resuspended to a stock concentration of approximately 1 mM in 25 mM Tris, pH 7.8, 0.1% Triton X-100, 1 mM EGTA, and 1 mM EDTA. The exact concentration was determined using the Kdo release assay (39). Radiolabeled Kdo2-[4′-32P]-lipid IVA was synthesized as described previously (38) The isolation proceeded as described for Kdo2-[4′-32P]-lipid A (38).
Lauroyl-ACP Synthesis
Lauroyl-ACP was synthesized from lauric acid and holo-ACP (accession number AAB27925) using acyl-ACP-synthase (accession number AAC75875) (21). The lauric acid was dissolved in ethanol and diluted into the synthesis reaction mixture. Lauroyl-ACP was purified and quantified as described previously (21).
LpxL In Vitro Activity Assay
LpxL activity was determined by the method previously described, with modification (21). Briefly, assays in 10 µL were conducted at 30 °C with 6.25 µM Kdo2-[4′-32P]-lipid IVA (1,000 cpm/µL) and 12.5 µM lauroyl-ACP. Final assay conditions included 0.1 mg/mL fatty acid-free bovine serum albumin (BSA), 5 mM MgCl2, 50 mM NaCl, 0.1% Triton X-100, and 50 mM HEPES, pH 7.5. BSA and Mg2+ were not essential but enhanced activity 2- to 3-fold at longer incubation times (data not shown). DDM did not substitute for Triton X-100 in the assay, although it did not interfere with the Triton X-100-containing assay system (data not shown). The assays were terminated by spotting 4-µL samples onto TLC plates, unless otherwise indicated. The plates were developed in tanks equilibrated with chloroform:pyridine:88% formic acid:water (30:70:16:10, v/v). The plates were dried, exposed to a PhosphorImager screen (Molecular Dynamics, Inc. Sunnyvale, CA), and quantified using a Storm PhosphorImager and ImageQuant software (GE Healthcare, Chalfont St. Giles, Buckinghamshire, United Kingdom). With membranes of MKV15b-pWSK29, no activity was detected (less than 0.15 nmol/min/mg protein, the limit of detection). Cell extracts and membranes expressing LpxL, as well as pure LpxL, exhibited linear substrate conversion when varying either time or enzyme concentration.
In Vitro Formation of a Second LpxL Product
The reaction conditions for the in vitro formation of a second LpxL product were modified from the standard LpxL in vitro activity assay (see above) with the following modifications. The substrate concentrations were increased to 300 µM lauroyl-ACP and 100 µM Kdo2-[4′-32P]-lipid IVA, and the LpxL concentration was increased to 17 µg/mL. A 2-µL sample of the 12-µL reaction mixture was spotted onto a TLC plate prior to the addition of 1 µL of the LpxL stock, and following the addition of LpxL, 2-µL samples were spotted at 10, 60, 120, and 180 min. The reaction progress was determined with TLC and PhosphorImager analysis. A larger scale, non-radiolabeled reaction mixture (270 µL) was incubated side-by-side with the radiolabeled reaction. After a 3-h incubation, this non-radiolabeled reaction was extracted by the two-phase Bligh/Dyer method and the lower phase isolated and dried under nitrogen.
In Vitro LpxR Treatment of Kdo2-(dilauroyl)-lipid IVA
Membranes from pWSK29-W3110 and pLpxR2-W3110 were prepared as described (40). A non-radiolabeled LpxL reaction (300 µL) was prepared as described above to form Kdo2-(dilauroyl)-lipid IVA. After a 3-h incubation, the reactions was split in half, and each half was diluted 3-fold into LpxR reaction conditions (40), consisting of 50 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.5, 1% Triton X-100, 5 mM CaCl2, and 0.033 mg/mL of the vector control or LpxR-expressing membranes. The reaction was incubated for 12 h at 30 °C. The reaction was extracted by the method of Bligh/Dyer (36), and the lower phase was dried under nitrogen. The resulting lipids were purified over DEAE cellulose, as described above (37), with the 30 mM ammonium acetate elution containing the fatty acid product of LpxR (40).
Solubilization of LpxL
Membranes containing over-expressed LpxL from pWSK-LpxL in MKV15b were solubilized in 2% detergent at 1 mg/mL final protein concentration for 2 h at 4 °C with gentle mixing. Triton X-100, DDM, and octyl glucoside efficiently solubilized LpxL based on SDS-PAGE and activity assays (data not shown). Because it is a single, chemically-defined compound, DDM was adopted for all the following steps.
Purification of LpxL by Cation-Exchange Chromatography
Cellulose phosphate resin (10 g, Sigma C3145) was prepared according to the manufacturer’s instructions and transferred into a XK 50/20 column (5 × 20 cm, 200 mL bed volume, GE Healthcare). The column was washed at 5 mL/min. Solubilized membranes contained approximately 25% LpxL at 0.5 – 0.75 mg/mL protein. The solubilized membranes were diluted to a final protein concentration of 0.032 mg/mL in 10 mM phosphate buffer, pH 7.4, 0.1% DDM, 1 mM EGTA, and 1 mM EDTA. The dilute detergent and protein concentrations were essential for complete LpxL binding to the cellulose phosphate resin (data not shown). Typically 100 – 200 mg of solubilized protein were flowed over the column with a peristaltic pump, with essentially complete binding of LpxL (data not shown). Once loaded, the column was attached to an ÄKTAFPLC controlled by the UNICORN system (GE Healthcare). Elution of LpxL was accomplished by flowing a linear gradient of NaCl from 0 to 1.5 M over three column volumes, followed by two column volumes of 1.5 M NaCl. LpxL begins eluting in a single peak after 0.5 M NaCl. The other elution buffer components were essentially the same as the load buffer, except that the DDM concentration was dropped to 0.01%. The fractions containing pure LpxL were pooled and concentrated about 100-fold with the Centricon Plus-70 Ultracel PL-10 and Amicon Ultra-15 Ultracel 10k (Millipore Corp. Billerica, MA). To reach a defined buffer composition, the LpxL concentrate was diluted 10-fold into 10 mM phosphate buffer, pH 7.4, 1 mM EGTA, 1 mM EDTA, 750 mM NaCl followed by another concentration step. The dilution and concentration was repeated a second time. In the presence of 750 mM NaCl, LpxL could be concentrated to 15 mg/mL with no precipitation. Below 500 mM NaCl, LpxL precipitated when it was concentrated. The final concentration of DDM was calculated to be no higher than 1%.
Size-exclusion Chromatography
Purified, concentrated LpxL was run over a Superdex 200 10/300 GL column (10 × 300 mm, 24 mL bed volume, GE Healthcare) using an ÄKTAFPLC controlled by the UNICORN system, according to the manufacturer’s recommendations. Typically, the flow rate was 0.4 – 0.5 mL/min. The column was pre-equilibrated in 0.1% DDM, 500 mM NaCl, 10 mM phosphate buffer, pH 7.4, 1 mM EGTA, and 1 mM EDTA. The high level of NaCl was required to maintain LpxL solubility. The concentrated eluate from the cellulose phosphate column was loaded onto the Superdex column after centrifugation at 13,000 × g for 10 min. The column was calibrated with blue dextran, BSA, and the High Molecular Weight Gel Filtration Calibration Kit (GE Healthcare) according to the manufacturer’s protocol. The LpxL buffer conditions were used during the calibration, allowing for comparison to LpxL.
Intact Protein Liquid Chromatography-Mass Spectrometry (LC/MS)
Purified LpxL (15 mg/mL, ~1% DDM, 10 mM phosphate buffer, pH 7.4, 1 mM EGTA, 1 mM EDTA, and 750 mM NaCl was diluted in water to 0.5 mg/mL and subjected to LC/MS in the positive ion mode. A Shimadzu Scientific Instruments (Columbia, MD) LC system (comprising a solvent degasser, two LC-10A pumps and a SCL-10A system controller) was coupled to a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (ABI/MDS-Sciex, Foster City, CA) equipped with an electrospray source. LC was operated at a flow rate of 200 µL/min with a linear gradient as follows: 100% A was held isocratically for 2 min and then linearly increased to 60% B over 18 min and then increased to 100% B over 5 min. Mobile phase A consists of water:acetonitrile (98:2, v/v) with 0.1% acetic acid. Mobile phase B consists of acetonitrile:water (90:10, v/v) with 0.1% acetic acid. A Vydac C4 reverse phase column (2.1 × 50 mm) was used for LC/MS analysis. MS data acquisition and analysis were performed using the Analyst QS software with the BioAnalyst extension installed (ABI). The Bayesian Protein Reconstruction tool was employed for the determination of the average molecular weights from the ESI-generated multiply-charged ions.
LpxL Mutagenesis
Site-directed mutants and truncations of lpxL were synthesized according the QuikChange system (Stratagene) using either pLpxL or pWSK-LpxL as the template for PCR. Supporting Table 1 lists the primers that yielded LpxL truncations and site-directed mutants. LpxL has a single predicted transmembrane helix between Tyr 16 and Tyr 38 (2). LpxL ΔN1 lacks the first 38 amino acids, replacing them with Met, Gly, Gly followed by LpxL amino acids 39–306 (270 amino acids total). The spacer between the ribosome binding site and the translation start site in pLpxL and pWSK-LpxL is 8 bp but increased to 10 bp for LpxL ΔN1 as a consequence of the deletion. LpxL truncation ΔN2 lacks amino acids 16–38 and replaces them with Pro Trp followed by LpxL amino acids 39–306 (286 amino acids total). The sequences of the resulting constructs were confirmed using an Applied Biosystems 3730 DNA Analyzer at the Duke DNA Sequencing Facility.
Negative Ion Electrospray Ionization (ESI) of Lipid A Species
Lipid A species extracted from cells or aqueous enzymatic reactions were re-dissolved in chloroform:methanol (2:1, v/v), supplemented with 1% piperidine (v/v), and analyzed by ESI/MS and ESI/MS/MS as described previously (41).
Results
Overexpression, Purification, and Activity Determination
E. coli LpxL has previously been over-expressed in BLR(DE3) from a pET vector, resulting in very high lauroyltransferase specific activity in the membrane fraction (32). Rather than purifying LpxL from this BLR strain, however, LpxL was over-expressed in MKV15b, a triple null mutant lacking the chromosomal copies of lpxL, lpxM, and lpxP (24). Because it lacks all acyl-ACP-dependent secondary lipid A acyltransferases, this strain allows for the unambiguous quantification of LpxL acyltransferase activity from various plasmid-borne constructs. The LPS lipid anchor of MKV15b is predominantly lipid IVA (Figure 1A). The expression of wild-type LpxL in this mutant causes a complete shift from lipid IVA to lauroyl-lipid IVA, as determined by TLC (Figure 1A). The identities of these lipid A species were confirmed by ESI/MS and tandem MS (MS/MS). Figure 1B highlights the prominent lipid IVA [M-2H]2− ion at a mass-to-charge ratio (m/z) of 701.42 atomic mass units (amu) characteristic of MKV15b harboring the empty vector (blue tracing). This peak is absent when LpxL is over-expressed in this strain (red tracing). Figure 1C shows the concomitant appearance of the lauroyl-lipid IVA [M-2H]2− ion at m/z 792.50 in MKV15b expressing LpxL (red tracing), which is absent in the vector control strain (blue tracing). Collision-induced tandem mass spectrometry of the [M-2H]2− ion at m/z 792.50 confirmed the proposed site of lauroyl chain attachment. The MS/MS spectrum (Supporting Figure 1A) reveals several peaks corresponding to laurate-containing fragments that are diagnostic of a laurate moiety attached to the 2′ position of lipid IVA (Supporting Figure 1B).
Figure 1. An assay for LpxL in living cells of E. coli.

A. The lipid A species released from the LPS of IPTG-induced mutant MKV15b (lacking lpxL, LpxM and LpxP), harboring either the vector control pWSK29 (left lane) or pWSK-LpxL (right lane), were subjected to TLC in the solvent chloroform:pyridine:88% formic acid:water (50:50:16:5, v/v) and visualized by charring. B. The lipid A species isolated from MKV15b cells harboring either the vector control pWSK29 or pWSK-LpxL were subjected to ESI/MS in the negative ion mode, and the spectra were normalized and overlayed. The major peak for the vector control sample (blue) was seen at m/z 701.42 and is interpreted as the [M-2H]2− ion of lipid IVA. The minor peak at m/z 712.41 is the corresponding sodium adduct [M-3H+Na]2−. The spectrum of lipid A species from MKV15b cells harboring pWSK-LpxL (red) shows no peaks in this range, consistent with the TLC analysis. C. The major peak for the lipid A species from MKV15b cells harboring the pWSK-LpxL (red) is seen at m/z 792.50, corresponding to the [M-2H]2− ion of (lauroyl)-lipid IVA. The minor peak at m/z 806.52 may correspond to (myristoyl)-lipid IVA, reflecting slightly relaxed LpxL substrate specificity. The spectrum of the vector control sample (blue) contains no peaks in this range.
After induction with IPTG, membranes of MKV15b cells harboring pWSK-LpxL contained up to 25% LpxL protein, as judged by SDS-PAGE analysis (Figure 2, lane 1). These membranes possessed very high lauroyltransferase specific activity in vitro (Table II), qualitatively consistent with the extent of protein over-production. In order to purify LpxL from these membranes, it was necessary to solubilize them with a non-ionic detergent. LpxL was efficiently solubilized from the membranes by DDM (Figure 2 and Table II), although some LpxL protein and activity remained behind in the pellet (not shown). DDM-solubilized LpxL was then purified to near homogeneity over a cellulose phosphate column (Figure 2 and Table II). The specific activity increased about 4-fold, which again qualitatively matches the SDS-PAGE analysis (Figure 2). A typical activity assay is shown in Figure 3, demonstrating the differential TLC migration of the Kdo2-[4′-32P]-lipid IVA substrate versus the Kdo2-[4′-32P]-(lauroyl)-lipid IVA product. The reaction progress is linear with the concentration of LpxL and with time at low percent conversion (not shown), and nearly all of the substrate can be converted to product at higher concentrations of LpxL.
Figure 2. Purification of LpxL to near homogeneity.

This Coomassie-Blue-stained SDS polyacrylamide gel shows the degree of protein purity for each fraction assayed in Table II. Approximately 20 µg of protein was loaded in each lane. The molecular weight standards are present in the outside lanes, as indicated. Lane 1, membranes from induced MKV15b/pWSK-LpxL; Lane 2, 2% DDM-treated membranes before centrifugation; Lane 3, DDM-solubilized membranes (the high-speed supernatant of the DDM-treated membranes); and Lane 4, LpxL purified over a cellulose phosphate column, pooled, and concentrated. The molecular weight of LpxL was confirmed to be 35.5 kDa.
Table 2.
Purification of LpxL over-expressed in MKV15b
| Enzyme source | Protein (mg) |
Specific Activity (µmol/min/mg) |
Units (µmol/min) |
Overall Yield (%) |
|---|---|---|---|---|
| Membranes | 127 | 19 | 2405 | 100 |
| DDM-treated membranesa | 118 | 19 | 2235 | 93 |
| DDM-solubilized membranesb | 82 | 16 | 1315 | 55 |
| Purified LpxL | 14 | 80 | 1158 | 48 |
Membranes treated with DDM before high-speed centrifugation.
The supernatant after high-speed centrifugation.
Figure 3. LpxL in vitro activity assay.

Serial dilutions of pure LpxL (from 220 ng/mL to 0.1 ng/mL) were assayed in vitro with 6.25 µM Kdo2-[4′-32P]-lipid IVA (1,000 cpm/µL) and 12.5 µM lauroyl-ACP at 30 °C for 10 min in a final volume of 10 µL. The assay mixture also included 0.1 mg/mL BSA, 5 mM MgCl2, 50 mM NaCl, 0.1% Triton X-100, and 50 mM HEPES, pH 7.5. Portions of 4 µL were spotted on a silica TLC plate and developed with the solvent chloroform:pyridine:88% formic acid:water (30:70:16:5, v/v). The plates were dried and analyzed with a PhosphorImager system.
LC/MS of Pure, Intact LpxL
The LC/MS elution profile for purified LpxL on a C4 reverse phase column is complicated by overlapping elution of DDM (Supporting Figure 2A–C). However, some LpxL elutes without DDM between 7 and 7.7 min (Supporting Figure 2B versus 2C). The LpxL spectrum for this time window is shown in Figure 4A. The series of multiply-charged (protonated) species of LpxL extends over an m/z range of 670 – 1700 amu, carrying from 53 to 21 positive charges. Analysis with the reconstruction tool yielded an average molecular weight for LpxL of 35,536 Da (data not shown), which is within experimental error of deformylated LpxL (calculated average molecular weight 35,537.85 Da). This result indicates that LpxL is present as a single species that was deformylated at its N-terminal Met.
Figure 4. LC/ESI/MS of purified LpxL.

Pure LpxL was subjected to LC/ESI/MS in the positive ion mode, as described in the Materials and Methods. A. LpxL elutes without DDM between 7 and 7.7 min. The peaks for the m/z range of 650 – 1550 amu are shown, corresponding to various charge states (H+) of LpxL ranging from +23 to +53. B. LpxL elutes with DDM between 7.72 and 8.10 min. The protein identification program parameters were limited to proteins in the molecular weight range of 33 – 43 kDa using the m/z range of 640 – 1020. Although protein ion peaks were identified above 1020, a cutoff of 1020 was selected because of an interfering protonated DDM dimer at m/z 1021.64. The mass reconstruction of the deconvoluted masses for LpxL and LpxL-DDM adducts is shown.
LpxL co-eluted with DDM from 7.7 to 8.1 min (Supporting Figure 2B versus 2C). The average molecular weights for the species eluting in this time window were reconstructed from the ion peaks. The results, shown in Figure 4B, indicate that authentic LpxL was present and ionized as a series of LpxL-DDM non-covalent adducts. The identified adducts contained from 1 to 12 DDM molecules per LpxL monomer (Figure 4B). The observed and predicted molecular weights for the LpxL-DDM adducts differ by less than 0.01% in each case. A numerical comparison is shown in Supporting Table II.
Size-Exclusion Chromatography of LpxL
Although LpxL was purified to homogeneity and its identity confirmed by ESI/MS, the native aggregation state of LpxL and its behavior in solution were not known. Therefore, pure LpxL was subjected to size-exclusion chromatography. LpxL eluted in a sharp, symmetric peak under the conditions described in the Materials and Methods (data not shown). Using the equation derived from the calibration curve, the peak elution volume for LpxL corresponds to 80 kDa. Using the available range of aggregation numbers for DDM in water (Anatrace), the calculated micelle size of DDM in water ranges from 40–80 kDa. Therefore, the observed size of LpxL in the presence of DDM, 80 kDa, is consistent with one LpxL monomer (36 kDa) embedded in one DDM micelle (44 kDa). However, it remains a possibility that LpxL is present as a dimer with minimal DDM.
In vitro activity characterization
Because LpxL is an integral membrane protein and its substrate Kdo2-lipid IVA fully partitions into membrane interfaces, the kinetics of LpxL must be treated differently than that of soluble enzymes with soluble substrates (42). The apparent KM for each substrate was individually determined at a saturating concentration of the other substrate and at a fixed detergent concentration (Figure 5A,B). The apparent KM and Vmax for lauroyl-ACP are 7 µM and 95 µmol/min/mg, respectively. The apparent KM and Vmax for Kdo2-lipid IVA are 15 µM and 221 µmol/min/mg (kcat of 131 s−1), respectively. In each case, the concentration of Triton X-100 was held constant at 0.1% (v/v). The detergent is critical to form a surface that contains the substrate Kdo2-lipid IVA and LpxL, both of which should fully partition into the mixed micelle surface. Surface dilution kinetics predict that LpxL activity should decrease as detergent concentration increases (42). As is shown in Figure 5C, Triton X-100 greatly enhances the activity of LpxL at lower concentrations, but the activity declines only gradually as the concentration of detergent increases.
Figure 5. LpxL kinetics and detergent dependency.

To determine the apparent Km and Vmax for each of its two substrates, LpxL was assayed at a fixed, saturating concentration of one substrate while the other was varied. The buffer included 0.1 mg/mL BSA, 5 mM MgCl2, 50 mM NaCl, and 50 mM HEPES, pH 7.5. Portions of 4 µL were spotted onto a silica TLC plate, which was developed in the solvent chloroform:pyridine:88% formic acid:water (30:70:16:5, v/v) and quantified with a PhosphorImager. A. LpxL at 4.7 ng/mL was assayed at 30 min with 75 µM Kdo2-[4′-32P]-lipid IVA (5,000 cpm/µL), 0.1% Triton X-100, and varying concentrations of lauroyl-ACP (3 to 180 µM) in a total volume of 10 µL for each lauroyl-ACP concentration. The specific activity was determined at each lauroyl-ACP concentration. The Michaelis-Menten equation was fit to the data to provide an apparent KM and Vmax. For lauroyl-ACP, the apparent KM is 7 ± 2 µM, and the apparent Vmax is 95 ± 5 µmol/min/mg. B. LpxL at a concentration of 0.18 ng/mL was assayed from 10 to 30 min with 90 µM lauroyl-ACP, 0.1% Triton X-100 and varying concentrations of Kdo2-[4′-32P]-lipid IVA (700 cpm/µL, 3 µM to 180 µM) in a total volume of 10 µL for each Kdo2-lipid IVA concentration. The specific activity was determined at each Kdo2-[4′-32P]-lipid IVA concentration. For Kdo2-lipid IVA the apparent KM is 15 ± 3 µM and the apparent Vmax is 221 ± 7 µmol/min/mg. C. The effects of varying Triton X-100 on LpxL activity were determined. LpxL was assayed for 10 min at 1.6 ng/mL at 15 µM for Kdo2-[4′-32P]-lipid IVA (1,000 cpm/µL) and 90 µM lauroyl-ACP in a total volume of 10 µL at each Triton X-100 concentration. The Triton X-100 concentration was varied from 0.0155 to 2%, and the fraction of substrate conversion to product was determined. The data points are connected for ease of visualization only.
Second Acylation by LpxL
At longer reaction times or at higher concentrations of LpxL, where Kdo2-lipid IVA was nearly fully converted to the penta-acylated Kdo2 (lauroyl)-lipid IVA, a second reaction product was identified by TLC analysis. The faster migration of the new product was consistent with Kdo2-(dilauroyl)-lipid IVA, given that lauroyl-ACP was the only acyl donor provided to pure LpxL in the assay. Additionally, because LpxL was expressed in a strain that lacked LpxM and LpxP and was purified to homogeneity, the second acylation reaction is attributable strictly to LpxL. To identify this novel product, Kdo2-[4′-32P]-lipid IVA and excess lauroyl-ACP were incubated with LpxL. A sample was spotted onto a TLC plate prior to the addition of LpxL, and following the addition of LpxL, samples were spotted at various times. The reaction progress was determined with TLC and PhosphorImager analysis (Figure 6). Under these conditions, the first acylation reaction was complete by 10 min, and the second reaction was complete by 180 min.
Figure 6. Formation of Kdo2-[4′-32P]-(dilauroyl)-lipid IVA at high substrate and LpxL concentrations.

A large excess of LpxL (17 µg/mL) was incubated with 100 µM Kdo2-[4′-32P]-lipid IVA (1,000 cpm/µL) and 300 µM lauroyl-ACP for 10 to 180 min in a total volume of 11 µL. The buffer included 0.1% Triton X-100, 0.1 mg/mL BSA, 5 mM MgCl2, 50 mM NaCl, and 50 mM HEPES, pH 7.5. At the given time points, a 2-µL portion was spotted onto a silica TLC plate, which was developed with chloroform:pyridine:88% formic acid:water (30:70:16:5, v/v) and analyzed with a PhosphorImager system.
A non-radiolabeled reaction mixture was incubated side-by-side with the radiolabeled reaction. After a 3-h incubation, this non-radiolabeled reaction was extracted, and the lipid species analyzed by ESI/MS and MS/MS. The results of the MS analysis confirmed the synthesis of Kdo2-(dilauroyl)-lipid IVA. Although the MS was complicated by the presence of Triton X-100 and DDM from the assay, peaks were readily identified at m/z values of 1103.63 (Supporting Figure 3A), corresponding to the [M-2H]2− ion (predicted m/z 1103.645), and at 735.42 (data not shown), corresponding to the [M-3H]3− ion (predicted m/z 735.428). No ions were observed for Kdo2-lipid IVA or Kdo2-(lauroyl)-lipid IVA. The MS/MS spectrum was obtained for the [M-2H]2− at m/z 1103.6 (Supporting Figure 3B). The major fragment ion peaks are listed in Table III. The MS/MS spectrum of the closely-related compound Kdo2-lipid A, purified from heptose-deficient E. coli, has previously been determined (28). This Kdo2-lipid A contains one lauroyl and one myristoyl chain (28). For each MS/MS fragment peak of the LpxL-derived Kdo2-(dilauroyl)-lipid IVA there is an analogous peak in the MS/MS spectrum of naturally-occuring Kdo2-lipid A (Table III). However, the myristate anion peak at m/z 227.20 seen in the MS/MS of Kdo2-lipid A isolated from heptose-deficient E. coli (28) is missing and is replaced by the accentuated laurate anion peak at m/z 199.16 in the MS/MS spectrum of the LpxL-derived Kdo2-(dilauroyl)-lipid IVA (Supporting Figure 3B).
Table 3.
MS/MS product ions for the [M-2H]2− ion of LpxL-generated Kdo2-(dilauroyl)-lipid IVA versus Kdo2-lipid A.
| MS/MS Ions Kdo2-(dilauroyl)-lipid IVA | Designation | MS/MS Ions kdo2-lipid Aa | Designation | Difference (amu) |
|---|---|---|---|---|
| 78.95 | PO3− | 78.96 | PO3− | 0.01 |
| 199.16 | [12:0]− | 227.20 | [14:0]− | 28.04 |
| 219.04 | [Kdo-H2O-H]− | 219.04 | [Kdo-H2O-H]− | 0.00 |
| 439.08 | [Kdo2-H2O-H]− | 439.09 | [Kdo2-H2O-H]− | 0.01 |
| 679.38 | [M-Kdo2-12:0-βOH14:0]2− | 679.40 | [M-Kdo2-14:0-βOH14:0]2− | 0.02 |
| 783.46 | [M-2H-Kdo2-12:0]2− | 783.46 | [M-2H-Kdo2-14:0]2− | 0.00 |
| 883.55 | [M-2H-Kdo2]2− | 897.59 | [M-2H-Kdo2]2− | 28.08 |
| 993.59 | [M-2H-Kdo]2− | 1007.65 | [M-2H-Kdo]2− | 28.12 |
| 1567.89 | [M-H-Kdo2-12:0-H2O]− | 1567.95 | [M-H-Kdo2-14:0-H2O]− | 0.06 |
| 1688.15 | [M-Kdo2-PO3]− | 1716.19 | [M-Kdo2-PO3]− | 28.04 |
| 1768.06 | [M-H-kdo2]− | 1796.16 | [M-H-kdo2]− | 28.10 |
Product ion values and designations were taken from Reference (28). Kdo2-lipid A was purified from the heptose-deficient mutant WBB06.
To confirm the attachment site of the second lauroyl chain, the in vitro product was digested with the specific lipid A 3′-O-deacylase, LpxR of Salmonella (40). Treatment with LpxR-expressing membranes, but not with those of the vector control, released a compound with a diagnostic peak at m/z 425.37 that matches the predicted m/z of 425.364 for the anion of the lauroyl ester of R-3-hydroxymyristate (Figure 7). This finding confirms that LpxL can catalyze laurate transfer from lauroyl-ACP to the 3′-R-3-hydroxymyristoyl moiety of Kdo2-(lauroyl)-lipid IVA. No major increase in the amount of unmodified R-3-hydroxymyristate was detected in either spectrum (m/z 243.197, data not shown), consistent with LpxL-dependent formation of a single molecular species of Kdo2-(dilauroyl)-lipid IVA.
Figure 7. Confirmation of a 3′-secondary lauroyl chain in LpxL-generated Kdo2-(dilauroyl)-lipid IVA.

The Kdo2-(dilauroyl)-lipid IVA generated by LpxL was treated with membranes from either vector control cells or LpxR over-expressing cells. The lipids were extracted from the reaction mixture and fractionated on DEAE cellulose. The compounds that eluted with 30 mM ammonium acetate (42) were isolated and subjected to ESI/MS, and the spectra were normalized and overlayed. The LpxR-treated Kdo2-(dilauroyl)-lipid IVA released a lipid characterized by m/z 425.365 (red), which is interpreted as the [M-H]− ion of the lauroyl ester of R-3-hydroxymyristic acid. The spectrum from the vector control-treated Kdo2-(dilauroyl)-lipid IVA (blue) does not contain this species.
Substrate Specificity
LpxL and many other lipid A secondary acyltransferases have a strong preference for Kdo-containing substrates (9, 21, 23, 25, 27, 43, 44). For E. coli LpxL, this specificity was most clearly demonstrated in an E. coli mutant lacking Kdo (with a second site suppressor that allows viability), which produced only lipid IVA, despite containing intact lpxL and lpxM (19). Lipid IVA is generated in the inner leaflet of the inner membrane (presumably in the vicinity of LpxL) in nearly all Gram-negative bacteria. In order to confirm the intrinsic substrate specificity of pure LpxL, its ability to acylate lipid IVA was assayed. The results, shown in Figure 8A, indicate that LpxL is capable of acylating lipid IVA in vitro. However, under these assay conditions the specific activity with lipid IVA as the substrate is 6,000-fold lower than for Kdo2-lipid IVA.
Figure 8. Dependence of LpxL on the Kdo and 1-phosphate moieties.

In vitro assays with these alternative substrates were performed at 30 °C in reaction mixtures containing 0.1 mg/mL BSA, 5 mM MgCl2, 50 mM NaCl, 0.1% Triton X-100, and 50 mM HEPES, pH 7.5. A. Serial dilutions of pure LpxL (from 18 µg/mL to 0.22 µg/mL) were assayed with 6.25 µM [4′-32P]-lipid IVA (1,000 cpm/µL) and 25 µM lauroyl-ACP at for 60 min in a total volume of 10 µL at each LpxL concentration. Portions of 4 µL were spotted on a silica TLC plate, which was developed with chloroform:pyridine:88% formic acid:water (50:50:16:10, v/v). B. Serial dilutions of pure LpxL (from 25 ng/mL to 0.8 ng/mL) were assayed with 1.25 µM 1-dephospho-Kdo2-[4′-32P]-lipid IVA (800 cpm/µL) and 12.5 µM lauroyl-ACP at for 30 min in a total volume of 10 µL at each LpxL concentration. Portions of 4 µL were spotted onto a silica TLC plate, which was developed with chloroform:pyridine:88% formic acid:water (30:70:16:5, v/v) and quantified with a PhosphorImager.
Although the specificity of LpxL for Kdo sugars on the distal glucosamine is well established, it is not known whether the 1-phosphate group on the proximal glucosamine is equally important for recognition by LpxL. With the identification and cloning of the lipid A 1-phosphatase LpxE (45), the lipid A analogue 1-dephospho-Kdo2-lipid IVA was synthesized (45). Pure LpxL is readily able to acylate this substrate (Figure 8B), and the specific activity is within 10-fold of Kdo2-lipid IVA.
Acyltransferase Homology
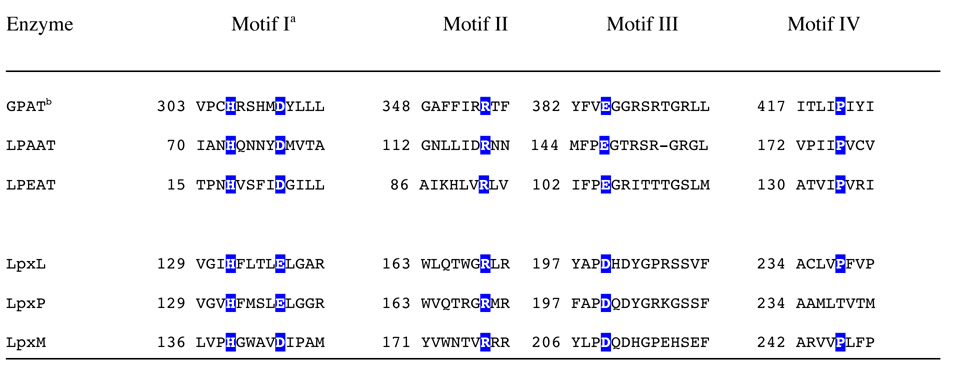
LpxL and the other secondary acyltransferases have no detectable homology to any other known proteins (27) using basic bioinformatics tools such as BLAST. In order to characterize LpxL more fully, in-depth bioinformatics approaches were taken. The Clusters of Orthologous Groups (COG) database classifies LpxL and the other lipid A secondary acyltransferases into COG1560, with a corresponding entry in the Conserved Domain Database (46). This entry contains a multiple sequence alignment of 47 diverse secondary acyltransferases (including E. coli LpxL, LpxM, and LpxP). The alignment shows that there are only two absolutely conserved residues, corresponding to His 132 and Arg 169 of LpxL. In addition, the position corresponding to E. coli LpxL Glu 137 is either an Asp or Glu in all 47 sequences, forming a conserved His and Asp/Glu pair.
Rock and co-workers demonstrated that a conserved catalytic His/Asp pair is essential for glycerolipid acyltransferase catalysis and that this pair is present in an absolutely conserved motif of H(X)4D, where X is any amino acid (47). E. coli enzymes that contain conserved H(X)4D motifs include glycerol-3-phosphate acyltransferase (GPAT or PlsB), lysophosphatidic acid acyltransferase (LPAAT or PlsC), and lysophosphatidylethanolamine acyltransferase (LPEAT or Aas). Research by Coleman and co-workers expanded this observation to three other conserved motifs containing key residues (48). Motif II contains a conserved Arg, and Motif III contains a conserved Glu (48). The Arg and Glu were found to be involved in substrate binding (48). Motif IV contains a conserved Pro that is important for full GPAT activity (48). The acyltransferases with these motifs form the GPAT family of acyltransferases.
Analysis by iterative PSI-BLAST (49) for E. coli LpxL shows that orthologs of E. coli LPAAT (PlsC) share statistically significant homology with the lipid A secondary acyltransferase family. This homology includes the H(X)4D/E motif (data not shown). This distant homology is also predicted by HHpred (50), which models a 99 % probability of homology between LpxL and COG0204 (LPAATs) over approximately 150 amino acids, including the H(X)4D/E motif (data not shown). An alignment of the four conserved GPAT motifs from the known E. coli GPAT homologs is shown in Table IV along with the putative GPAT motifs for the E. coli lipid A secondary acyltransferases.
Table 4.
Sequence alignment of the E. coli lipid A secondary acyltransferases and various GPAT family members.
 |
LpxL GPAT Point Mutants
Bioinformatic evidence suggests that putative active site residues of LpxL (His 132, Glu 137, Arg 169, Asp 200, and Pro 238) may be homologous to the GPAT family. To test this hypothesis, the following LpxL mutants were constructed: H132A, E137A, R169A, D200A, and P238A. Wild-type LpxL and the mutants were expressed in MKV15b, the membranes harvested, and the samples analyzed by SDS-PAGE (data not shown). None of the mutants formed inclusion bodies, and each mutant overexpressed to a similar level as wild-type LpxL.
Membranes containing each LpxL mutant were assayed to determine their specific activities under the same conditions in which wild-type LpxL and its vector control were tested. The vector control membranes did not demonstrate any activity, even at the highest membrane concentration tested. Like wild-type LpxL, each mutant’s activity was linear with time and amount of protein, except for H132A, which had an initial linear time-course followed by a gradual reduction in activity (data not shown). Of the mutants tested, H132A and E137A appeared to be most impaired, having >1,000-fold and >3,000-fold reduced activity relative to wild-type LpxL, respectively (Table V). R169A and D200A were also significantly impaired under these conditions (170-fold and 15-fold, respectively, Table V). The P238A mutation had little or no effect on LpxL activity (<2-fold, Table V).
Table 5.
LpxL mutant activities
| Construct | % of WT LpxL Activity (± S.D.)a |
|---|---|
| Wild-type LpxL | 100 |
| H132A | 0.08 ± 0.01 |
| E137A | 0.03 ± 0.01 |
| R169A | 0.59 ± 0.05 |
| D200A | 7.4 ± 0.9 |
| P238A | 66 ± 14 |
The specific activities of wild-type (WT) and mutant LpxL constructs were determined by separately varying time and enzyme amount. These values were then averaged, and the standard deviation determined. The enzyme source was membranes from MKV15b cells overexpressing each construct. Standard assay conditions were used. The activities were normalized to values for wild-type LpxL.
The expression of LpxL or the LpxL point mutants in MKV15b led to different levels of laurate incorporation in the lipid A species (data not shown). The expression of H132A had no effect on the lipid IVA produced in MKV15b, while the expression of E137A, R169A, and D200A resulted in a mixture of lipid IVA and lauroyl-lipid IVA. The expression of P238A yielded only lauroyl-lipid IVA, matching the results for wild-type LpxL expression. These levels of laurate incorporation by LpxL mutants in MKV15b are consistent with the in vitro activity results.
Two other constructs were also examined for their in vivo activity. LpxL ΔN1 lacks the first 38 amino acids, while LpxL ΔN2 lacks amino acids 16–38, encompassing the predicted N-terminal transmembrane helix. Neither of these constructs expressed to the same levels as wild-type LpxL in MKV15b, although a band of the correct size was visible for LpxL ΔN2 by SDS-PAGE analysis (data not shown). The band corresponding to LpxL ΔN2 was also readily apparent in membrane preparations from the overexpressing strain, indicating it retains significant membrane affinity. The expression of the truncated LpxL constructs had no effect on the lipid A species synthesized by MKV15b (data not shown), demonstrating the importance of the membrane anchor for proper LpxL activity.
Promiscuity in Acyl Donor Selectivity
A preliminary study employing E. coli membranes reported that lauroyl-ACP was strongly prefered as the acyl chain donor for incorporation of laurate into Kdo2-lipid A, with only traces of incorporation observed when lauroyl-CoA was present as the sole acyl chain donor (44). In Pseudomonas aeuruginosa extracts, lauroyl-CoA appeared to inhibit the incorporation of laurate into lipid A (51). Using pure LpxL, lauroyl-CoA was found to serve as an alternative acyl donor for LpxL. When the concentration of lauroyl-CoA was varied, it appeared to function as a substrate up to 50 µM (albeit at only 5% the specific activity of lauroyl-ACP), but as the concentration of lauroyl-CoA increased beyond 50 µM, product formation and specific activity actually decreased (Figure 9). Moreover, lauroyl-CoA concentrations above 50 µM were found to inhibit product formation even in the presence of lauroyl-ACP.
Figure 9. Lauroyl-CoA is both a slow substrate and an inhibitor of LpxL.

Pure LpxL (40 ng/mL) was assayed with 6.25 µM Kdo2-[4′-32P]-lipid IVA (1,000 cpm/µL) and varying concentrations of lauroyl-CoA (from 1 µM to 500 µM) at 30 °C for 30 min in a total volume of 10 µL at each lauroyl-CoA concentration. The buffer included 0.1 mg/mL BSA, 0.5 mM LiCl, 5 mM MgCl2, 50 mM NaCl, 0.1% Triton X-100, and 50 mM HEPES, pH 7.5. Portions of 4 µL were spotted on a silica TLC plate, which was developed with chloroform:pyridine:88% formic acid:water (30:70:16:5, v/v) and analyzed with a PhosphorImager.
Although LpxL is specific for lauroyl-donors in vivo and in vitro, it can utilize acyl donors of other chains lengths (21). Of the commercially-available acyl-CoA species, n-decanoyl-CoA, and to a lesser extent myristoyl-CoA, also functioned as acyl donors at low concentrations with inhibition of acylation at higher concentrations (Supporting Figure 4). Palmitoyl-CoA was inefficiently utilized as an acyl donor, but some acylation was observed (Supporting Figure 4). Under the same conditions, no acylation was observed in the presence of stearoyl-CoA, palmitoleoyl-CoA, or oleoyl-CoA (not shown). Two other acyl donors were tested. R-3-hydroxymyristoyl-ACP (the substrate of LpxA and LpxD) was an extremely poor acyl chain donor for LpxL. Synthetic R-3-hydroxylauroyl-methylphosphopantetheine was also a poor acyl chain donor for LpxL (not shown).
Discussion
LpxL is the inner membrane acyltransferase responsible for the incorporation of the 2′ secondary lauroyl chain into E. coli lipid A (Scheme 1). This conclusion is based on structural studies of lipid A isolated from lpxL mutants, and on in vitro enzymatic studies with membranes from lpxL mutants and lpxL over-expressing strains (21–24). E. coli contains two closely related LpxL orthologues, designated LpxP and LpxM. LpxP incorporates palmitoleate in place of laurate when E. coli cells are grown at 15 °C, whereas LpxM efficiently incorporates the 3′ secondary myristoyl group of lipid A (1, 2, 24), provided the 2′ secondary acyl chain is already in place. The mutant MKV15b contains insertion mutations in all three of these genes (24), providing an ideal chromosomal background for evaluating the functions of diverse LpxL orthologues and LpxL point mutants. Related LpxL orthologs are found in virtually all Gram-negative bacteria and are likewise responsible for the attachment of the secondary acyl chains in their respective organisms. In the case of E. coli lipid A, both secondary acyl chains are needed for agonist activity against human TLR-4/MD2. Tetra- or penta-acylated E. coli lipid A species are antagonists of human TLR-4/MD2 (1, 2).
We have now devised the first purification of a lipid A secondary acyltransferase to near homogeneity, facilitating the unambiguous characterization of its structure and function. LpxL and its orthologues are inner membrane proteins with a predicted single N-terminal transmembrane helix (2). Despite the inherent difficulties of working with integral membrane proteins, wild-type E. coli LpxL is well-behaved in terms of retention of activity and stability following solubilization. It therefore proved amenable to purification and kinetic characterization. The isoelectric point of LpxL is approximately 9.8. This indicates that LpxL is positively charged near a neutral pH, possibly contributing to its interaction with its acidic lipid A precursor substrate, Kdo2-lipid IVA. The presence of two phosphate groups in its natural substrate furthermore suggested that LpxL might bind to cation-exchange resins, like cellulose phosphate. Indeed, overexpressed LpxL was easily purified to near homogeneity from solubilized membranes over a single cellulose phosphate column with an appropriately chosen protein dilution and salt gradient (Figure 2).
The identity of pure LpxL was confirmed by LC/ESI/MS analysis of the intact protein (Figure 4). The single species that was detected matched the predicted average molecular weight for the deformylated version of LpxL. Successful ESI/MS of intact membrane proteins is not trivial, as the hydrophobic nature of integral membrane proteins and the high levels of detergent needed to solubilize them can interfere with MS. In addition, the most common detergent used for solubilization of catalytically active membrane enzymes, Triton X-100, is incompatible with MS analysis because Triton X-100 is a mixture of related species with different molecular weights, which interferes over a wide m/z range. In the present study, LpxL was purified at a minimal DDM-to-protein ratio, and as noted above, it is highly charged (up to +53) (Figure 4). The additional identification of LpxL-DDM adducts by MS (Figure 4) is consistent with the behavior of LpxL as an integral membrane protein and the predicted membrane-spanning helix (2).
LpxL displays robust in vitro lauroyltransferase activity and is particularly well-behaved for a detergent-solubilized membrane protein. The modest inhibitory effects of increasing detergent concentrations on LpxL activity (Figure 5C) are not entirely consistent with simple surface dilution kinetics (42). Instead, LpxL may directly interact with its substrate Kdo2-lipid IVA in a manner controlled by bulk kinetics. Such a high affinity direct interaction was reported for the pleckstrin homology domain of phospholipase Cd1 interacting with phosphatidylinositol 4,5-bisphosphate or its soluble head group, inositol 1,4,5-trisphosphate (52). This idea is supported by the extraordinarily high affinity of LpxL for cellulose phosphate (Figure 3) and its remarkable specificity for Kdo-containing substrates (Figure 8). It is conceivable that O-deacylated Kdo2-lipid IVA or even fully O- and N-deacylated Kdo2-lipid IVA, which could be prepared by hydrazinolysis of Kdo2-lipid IVA or Kdo2-lipid A, might still bind to LpxL. The former might be an alternative substrate, whereas the latter might be an inhibitor. If so, this would greatly simplify the analysis of LpxL kinetics and investigation into its mechanism, because the co-substrate lauroyl-ACP is also fully soluble at concentrations below 100 µM.
While LpxL was confirmed to catalyze the acylation of the free hydroxyl group of the 2′-R-3-hydroxymyristoyl chain of Kdo2-lipid IVA, the discovery that it could catalyze a second acylation was unexpected because this does not happen in living cells (Figure 1). The two acylations of Kdo2-lipid IVA by LpxL produce a dilauroyl derivative of Kdo2-lipid IVA, which is almost the same in structure as natural Kdo2-lipid A isolated from heptose-deficient mutants (28). The structure of Kdo2-(dilauroyl)-lipid IVA was confirmed by comparing its MS/MS spectrum with that of natural Kdo2-lipid A, which contains lauroyl and myristoyl groups at 2′ and 3′ secondary positions respectively (28). While fragmentation of Kdo2-lipid A gives rise to a myristate anion at m/z 227.3 (predicted m/z 227.20), which is released from the 3′ secondary position, no such myristate anion is seen with the Kdo2-(dilauroyl)-lipid IVA. Instead, the laurate anion at m/z 199.2 (predicted m/z 199.17) is detected after fragmentation of Kdo2-(dilauroyl)-lipid IVA. The lack of the myristate anion combined with the prominent laurate anion in the Kdo2-(dilauroyl)-lipid IVA MS/MS spectrum (Supporting Figure 3B) is strongly suggestive of laurate attachment to the 3′ secondary position.
To establish conclusively that Kdo2-(dilauroyl)-lipid IVA contains a secondary laurate moiety at the 3′ position, the compound was treated with the recently characterized 3′-O-deacylase of Salmonella (40). The enzyme, LpxR, cleaves the ester-linked R-3-hydroxymyristoyl group at the 3′ position of lipid A, regardless of the presence or absence of a secondary acyl chain (40). Figure 7 shows that LpxR liberates a compound from the 3′ position of Kdo2-(dilauroyl)-lipid IVA with the mass expected for the lauroyl ester of R-3-hydroxymyristate.
While LpxL prefers lauroyl-ACP in vitro, LpxL was found to utilize the alternative substrate, lauroyl-CoA at about 5% the rate under matched, optimized conditions. In E. coli, it is likely that lauroyl-ACP is the preferred substrate for LpxL, but the possibility that lauroyl-CoA is utilized as the donor under circumstance when the lauroyl-ACP pool is limiting cannot be completely excluded. Assays with varying concentrations of lauroyl-CoA further show that it is both a substrate and an inhibitor of LpxL, suggesting that it might play a regulatory role. In vitro, at least one lauroyl-CoA molecule must be able to bind productively to the active site to be utilized as a donor substrate. A second lauroyl-CoA molecule likely binds in a different mode, thereby inhibiting LpxL. Lauroyl-CoA can also inhibit the ability of LpxL to utilize lauroyl-ACP (data not shown). It is likely that the LpxL-catalyzed acylation of Kdo2-lipid IVA with lauroyl-CoA as the donor was overlooked previously because of its inhibitory effect at concentrations above 50 µM.
The ability of LpxL to utilize alternative acyl chain donors in vitro and to catalyze a second LpxM-like reaction make LpxL an attractive enzyme for the semi-synthesis of unnatural lipid A analogues. In addition to its ability to catalyze the formation of Kdo2-(dilauroyl)-lipid IVA, LpxL is also able to catalyze the formation of (dilauroyl)-lipid IVA (Figure 8A) and Kdo2-(dimyristoyl)-lipid IVA (data not shown).
A multiple sequence alignment of LpxL and homologous lipid A secondary acyltransferases had identified only a few strongly conserved residues (27). The absolute conservation of the H(X)4D/E motif in the lipid A secondary acyltransferase family (COG1560) provided the first clue to their evolutionary relationship to the GPAT family. GPAT enzymes, though extremely diverse, have a well-conserved set of key active site residues, including the catalytic dyad, H(X)4D (53). When the His or Asp of the H(X)4D motif was mutated to alanine in E. coli GPAT and LPEAT, there was no detectible transferase activity from crude membranes expressing the mutant enzymes (47). The Asp to Glu mutant of GPAT maintains significant activity (48). The only structural information for the GPAT family comes from a highly divergent, soluble chloroplast GPAT (54). There is only 19% sequence identity between the chloroplast and bacterial GPATs, which occurs over the four GPAT motifs and corresponds to about 150 amino acids. Reflecting their divergence, the GPAT motifs for the chloroplast homologs are somewhat different (53). The crystal structure of the chloroplast GPAT (54) suggests that the His and Asp from the GPAT H(X)4D motif orient and activate a substrate hydroxyl group for nucleophilic attack on its acyl donor substrate. This proposed mechanism is reminiscent of the serine protease catalytic triad in the sense that the serine hydroxyl group of the protease is replaced with the substrate (acceptor) hydroxyl group. This hypothetical model is consistent with mutational data from many GPAT family members (47, 48, 53).
Although LpxL has the variant H(X)4E motif, LpxM and many other homologs of LpxL possess the H(X)4D motif. Indeed, an acidic Asp or Glu is present five amino acid positions away from the invariant His in every sequence in COG1560. Moreover, within the GPAT family, a few members also have the variant H(X)4E motif. A Neisseria meningitidis LPAAT ortholog, NlaB, contains the H(X)4E motif and was confirmed to have LPAAT activity (55). In addition, the Asp to Glu point mutant of E. coli LPAAT retained significant acyltransferase activity (47). GPAT motifs II and III, containing a conserved Arg or Glu, respectively, were found to be important for substrate binding in GPAT family members (48). Their putative counterparts in LpxL are Arg 169 (the second absolutely conserved residue) and Asp 200. The role for the conserved Pro of Motif IV is unclear, but it is important for GPAT activity (48). While no Pro in COG1560 is absolutely conserved, the HHpred alignment and spacing to Asp 200 suggested that LpxL Pro 238 (66% identity within COG1560) might be homologous to the GPAT Motif IV Pro. However, the P238A mutant retained full wild-type LpxL activity.
While the sequence homology is not definitive, other indirect evidence also supports an evolutionary relationship between the GPAT family and LpxL. In contrast to LpxA and LpxD, many GPAT enzymes utilize either acyl-ACP or acyl-CoA as their acyl donors (56). LpxL has now been demonstrated to utilize either acyl-ACP or acyl-CoA as its acyl donor. Perhaps most intriguing is the discovery of LPAAT homologs from Sinorhizobium meliloti that are responsible for the acyl-ACP-dependent acylation of ornithine (by OlsB) and lyso-ornithine (by OlsA) to form the unusual ornithine lipids (57). These lipids, like lipid A, each have an amide-linked R-3-hydroxy fatty acid moiety that is further acylated to give an acyloxyacyl unit (57). The acylation reaction performed by OlsA is therefore very similar to that performed by LpxL.
Another clue to the distant homology between the lipid A secondary acyltransferases and the GPAT family comes from COG1560, which contains putative lipid A lauroyl/myristoyltransferase homologs from Gram-positive bacteria, such as Corynebacteria and Mycobacteria. Because Gram-positive bacteria lack lipid A and the first seven enzymes in its biosynthesis, it is likely that these putative acyltransferase genes are annotated incorrectly, despite their readily apparent homology to the lipid A secondary acyltransferases. A recent report indicates that the Mycobacterium tuberculosis LpxL homolog, Rv2611c, functions as an acyltransferase in the biosynthesis of phosphatidylinositol mannosides (58). Utilizing the default search parameters, a PSI-BLAST search of the non-redundant database with LpxL picks up Rv2611c in iteration 2 with an E value of 7e-12, 17% sequence identity, 30% sequence similarity, and 5% gaps over 237 amino acids.
The recognition that LpxL is likely to be a member of the GPAT family, thus sharing a common mechanism, may enhance its value as a target for antibiotic development, or perhaps, intervention in Gram-negative sepsis. In nearly all reports, the under-acylation of LPS reduces the virulence of human pathogens and renders lipid A ineffective as an activator of human TLR-4/MD2 (9, 12, 13, 20). Bacteria with under-acylated lipid A are usually viable, but they often have compromised growth phenotypes with increased sensitivity to temperature, antibiotics, and surfactants such as bile salts (10, 13, 15, 17, 18, 20, 24). These strains are promising vaccine candidates (9, 10, 12, 14). Pharmacologic intervention leading to the disruption of lipid A secondary acylation would enhance the susceptibilty of Gram-negative pathogens to killing by the cationic peptides of the innate immune system and to many existing antibiotics.
Supplementary Material
Supplementary figures and tables provide the details of the mutagenesis procedures, mass spectrometry and substrate selectivity assays using acyl-coenzyme A variants as donors. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by NIH Grant GM-51310 to C. R. H. Raetz. The mass spectrometry facility in the Department of Biochemistry at the Duke University Medical Center and Z.G. were supported by the LIPID MAPS Large Scale Collaborative Grant number GM-069338 from the National Institutes of Health.
The authors thank all members of the Raetz Laboratory, past and present, for their assistance, especially Drs. Suparna Kanjilal-Kolar and Allison Williams. Brian Ingram isolated the 1-dephospho-Kdo2-lipid IVA. Craig Bartling supplied R-3-hydroxymyristoyl-ACP and myristoyl-ACP. Dr. C. Michael Reynolds provided lipid IVA as well as helpful discussions. Louis Metzger and Adam Barb offered insightful comments to improve the manuscript. Reza Kordestani provided assistance with mass spectrometry.
List of Abbreviations
- ACP
acyl carrier protein
- amu
atomic mass units
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- COG
Clusters of Orthologous Groups
- DDM
n-dodecyl-β-d-maltopyranoside
- DEAE
diethylaminoethyl cellulose
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- ESI
electrospray ionization
- GPAT
glycerol 3-phosphate acyltransferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IPTG
isopropyl-1-thio-β-d-galactopyranoside
- Kdo
3-deoxy-d-manno-oct-2-ulosonic acid
- LB
lysogeny broth
- LC
liquid chromatography
- LPS
lipopolysaccharide
- MS
mass spectrometry
- m/z
mass-to-charge ratio
- PBS
phosphate-buffered saline
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TBAP
tetrabutylammonium phosphate
- TLC
thin layer chromatography
- TLR-4
Toll-like receptor-4
References
- 1.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 4.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Coats SR, Do CT, Karimi-Naser LM, Braham PH, Darveau RP. Antagonistic lipopolysaccharides block E. coli lipopolysaccharide function at human TLR4 via interaction with the human MD-2 lipopolysaccharide binding site. Cell Microbiol. 2007;9:1191–1202. doi: 10.1111/j.1462-5822.2006.00859.x. [DOI] [PubMed] [Google Scholar]
- 6.Opal SM. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol. 2007;297:365–377. doi: 10.1016/j.ijmm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Leaver SK, Finney SJ, Burke-Gaffney A, Evans TW. Sepsis since the discovery of Toll-like receptors: disease concepts and therapeutic opportunities. Crit Care Med. 2007;35:1404–1410. doi: 10.1097/01.CCM.0000261883.16943.4B. [DOI] [PubMed] [Google Scholar]
- 8.Somerville JE, Jr, Cassiano L, Bainbridge B, Cunningham MD, Darveau RP. A novel Escherichia coli lipid A mutant that produces an antiinflammatory lipopolysaccharide. J Clin Invest. 1996;97:359–365. doi: 10.1172/JCI118423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols WA, Raetz CRH, Clementz T, Smith AL, Hanson JA, Ketterer MR, Sunshine M, Apicella MA. htrB of Haemophilus influenzae: determination of biochemical activity and effects on virulence and lipooligosaccharide toxicity. J Endotoxin Res. 1997;4:163–172. [Google Scholar]
- 10.Fisseha M, Chen P, Brandt B, Kijek T, Moran E, Zollinger W. Characterization of native outer membrane vesicles from lpxL mutant strains of Neisseria meningitidis for use in parenteral vaccination. Infect Immun. 2005;73:4070–4080. doi: 10.1128/IAI.73.7.4070-4080.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuneyoshi N, Kohara J, Bahrun U, Saitoh S, Akashi S, Gauchat JF, Kimoto M, Fukudome K. Penta-acylated lipopolisaccharide binds to murine MD-2 but does not induce the oligomerization of TLR4 required for signal transduction. Cell Immunol. 2006;244:57–64. doi: 10.1016/j.cellimm.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Geurtsen J, Angevaare E, Janssen M, Hamstra HJ, Hove JT, de Haan A, Kuipers B, Tommassen J, van der Ley P. A Novel Secondary Acyl Chain in the Lipopolysaccharide of Bordetella pertussis Required for Efficient Infection of Human Macrophages. J Biol Chem. 2007;282:37875–37884. doi: 10.1074/jbc.M706391200. [DOI] [PubMed] [Google Scholar]
- 13.Clements A, Tull D, Jenney AW, Farn JL, Kim SH, Bishop RE, McPhee JB, Hancock RE, Hartland EL, Pearse MJ, Wijburg OL, Jackson DC, McConville MJ, Strugnell RA. Secondary acylation of Klebsiella pneumoniae lipopolysaccharide contributes to sensitivity to antibacterial peptides. J Biol Chem. 2007;282:15569–15577. doi: 10.1074/jbc.M701454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu T, Konig R, Sha J, Agar SL, Tseng CT, Klimpel GR, Chopra AK. Immunological responses against Salmonella enterica serovar Typhimurium Braun lipoprotein and lipid A mutant strains in Swiss-Webster mice: Potential use as live-attenuated vaccines. Microb Pathog. 2007;44:224–237. doi: 10.1016/j.micpath.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karow M, Fayet O, Cegielska A, Ziegelhoffer T, Georgopoulos C. Isolation and characterization of the Escherichia coli htrB gene, whose product is essential for bacterial viability above 33 degrees C in rich media. J Bacteriol. 1991;173:741–750. doi: 10.1128/jb.173.2.741-750.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karow M, Georgopoulos C. Sequencing, mutational analysis, and transcriptional regulation of the Escherichia coli htrB gene. Mol Microbiol. 1991;5:2285–2292. doi: 10.1111/j.1365-2958.1991.tb02159.x. [DOI] [PubMed] [Google Scholar]
- 17.Karow M, Raina S, Georgopoulos C, Fayet O. Complex phenotypesof null mutations in the htr genes, whose products are essential for Escherichia coli growthat elevated temperatures. Res Microbiol. 1991;142:289–294. doi: 10.1016/0923-2508(91)90043-a. [DOI] [PubMed] [Google Scholar]
- 18.Karow M, Georgopoulos C. Isolation and characterization of the Escherichia coli msbB gene, a multicopy suppressor of null mutations in the high-temperature requirement gene htrB. J Bacteriol. 1992;174:702–710. doi: 10.1128/jb.174.3.702-710.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meredith TC, Aggarwal P, Mamat U, Lindner B, Woodard RW. Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem Biol. 2006;1:33–42. doi: 10.1021/cb0500015. [DOI] [PubMed] [Google Scholar]
- 20.Phongsisay V, Perera VN, Fry BN. Expression of the htrB gene is essential for responsiveness of Salmonella typhimurium and Campylobacter jejuni to harsh environments. Microbiology. 2007;153:254–262. doi: 10.1099/mic.0.29230-0. [DOI] [PubMed] [Google Scholar]
- 21.Clementz T, Bednarski JJ, Raetz CRH. Function of the htrB high temperature requirement gene of Escherchia coli in the acylation of lipid A: HtrB catalyzed incorporation of laurate. J Biol Chem. 1996;271:12095–12102. doi: 10.1074/jbc.271.20.12095. [DOI] [PubMed] [Google Scholar]
- 22.Clementz T, Bednarski J, Raetz CRH. Escherichia-Coli Genes Encoding Kdo Dependent Acyltransferases That Incorporate Laurate and Myristate into Lipid-A. Faseb Journal. 1995;9:A1311–A1311. [Google Scholar]
- 23.Clementz T, Zhou Z, Raetz CRH. Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. Acylation by MsbB follows laurate incorporation by HtrB. J Biol Chem. 1997;272:10353–10360. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 24.Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CRH. A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. J Biol Chem. 2002;277:14194–14205. doi: 10.1074/jbc.M200409200. [DOI] [PubMed] [Google Scholar]
- 25.Carty SM, Sreekumar KR, Raetz CRH. Effect of cold shock on lipid A biosynthesis in Escherichia coli. Induction At 12 degrees C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. J Biol Chem. 1999;274:9677–9685. doi: 10.1074/jbc.274.14.9677. [DOI] [PubMed] [Google Scholar]
- 26.Vorachek-Warren MK, Carty SM, Lin S, Cotter RJ, Raetz CRH. An Escherichia coli mutant lacking the cold shock-induced palmitoleoyltransferase of lipid A biosynthesis: absence of unsaturated acyl chains and antibiotic hypersensitivity at 12 degrees C. J Biol Chem. 2002;277:14186–14193. doi: 10.1074/jbc.M200408200. [DOI] [PubMed] [Google Scholar]
- 27.Basu SS, Karbarz MJ, Raetz CRH. Expression cloning and characterization of the C28 acyltransferase of lipid A biosynthesis in Rhizobium leguminosarum. J Biol Chem. 2002;277:28959–28971. doi: 10.1074/jbc.M204525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raetz CRH, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Jr, Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, Dennis EA. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 30.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 31.Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- 32.Tran AX, Karbarz MJ, Wang X, Raetz CRH, McGrath SC, Cotter RJ, Trent MS. Periplasmic cleavage and modification of the 1-phosphate group of Helicobacter pylori lipid A. J Biol Chem. 2004;279:55780–55791. doi: 10.1074/jbc.M406480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- 34.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. Embo J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trent MS, Pabich W, Raetz CRH, Miller SI. A PhoP/PhoQ-induced Lipase (PagL) that catalyzes 3-O-deacylation of lipid A precursors in membranes of Salmonella typhimurium. J Biol Chem. 2001;276:9083–9092. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 36.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 37.Kanipes MI, Lin S, Cotter RJ, Raetz CRH. Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-D-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide. A novel membrane enzyme dependent upon phosphatidylethanolamine. J Biol Chem. 2001;276:1156–1163. doi: 10.1074/jbc.M009019200. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds CM, Kalb SR, Cotter RJ, Raetz CRH. A phosphoethanolamine transferase specific for the outer 3-deoxy-D-manno-octulosonic acid residue of Escherichia coli lipopolysaccharide. Identification of the eptB gene and Ca2+ hypersensitivity of an eptB deletion mutant. J Biol Chem. 2005;280:21202–21211. doi: 10.1074/jbc.M500964200. [DOI] [PubMed] [Google Scholar]
- 39.Lee CH, Tsai CM. Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate andheptose at the inner core by periodate oxidation. Anal Biochem. 1999;267:161–168. doi: 10.1006/abio.1998.2961. [DOI] [PubMed] [Google Scholar]
- 40.Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, Raetz CRH, Trent MS. An outer membrane enzyme encoded by Salmonella typhimurium lpxR that removes the 3′-acyloxyacyl moiety of lipid A. J Biol Chem. 2006;281:21974–21987. doi: 10.1074/jbc.M603527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanjilal-Kolar S, Basu SS, Kanipes MI, Guan Z, Garrett TA, Raetz CRH. Expression cloning of three Rhizobium leguminosarum lipopolysaccharide core galacturonosyltransferases. J Biol Chem. 2006;281:12865–12878. doi: 10.1074/jbc.M513864200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deems RA. Interfacial enzyme kinetics at the phospholipid/water interface: practical considerations. Anal Biochem. 2000;287:1–16. doi: 10.1006/abio.2000.4766. [DOI] [PubMed] [Google Scholar]
- 43.Goldman RC, Doran CC, Capobianco JO. Analysis of lipopolysaccharide biosynthesis in Salmonella typhimurium and Escherichia coli by using agents which specifically block incorporation of 3-deoxy-D-manno-octulosonate. J Bacteriol. 1988;170:2185–2191. doi: 10.1128/jb.170.5.2185-2191.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brozek KA, Raetz CRH. Biosynthesis of lipid A in Escherichia coli. Acyl carrier protein-dependent incorporation of laurate and myristate. J Biol Chem. 1990;265:15410–15417. [PubMed] [Google Scholar]
- 45.Karbarz MJ, Kalb SR, Cotter RJ, Raetz CRH. Expression cloning and biochemical characterization of a Rhizobium leguminosarum lipid A 1-phosphatase. J Biol Chem. 2003;278:39269–39279. doi: 10.1074/jbc.M305830200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heath RJ, Rock CO. A conserved histidine is essential for glycerolipid acyltransferase catalysis. J Bacteriol. 1998;180:1425–1430. doi: 10.1128/jb.180.6.1425-1430.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewin TM, Wang P, Coleman RA. Analysis of amino acid motifs diagnostic for the sn-glycerol-3-phosphate acyltransferase reaction. Biochemistry. 1999;38:5764–5771. doi: 10.1021/bi982805d. [DOI] [PubMed] [Google Scholar]
- 49.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 51.Mohan S, Raetz CRH. Endotoxin biosynthesis in Pseudomonas aeruginosa: enzymatic incorporation of laurate before 3-deoxy-D-manno-octulosonate. J Bacteriol. 1994;176:6944–6951. doi: 10.1128/jb.176.22.6944-6951.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V, McLaughlin S, Rebecchi MJ. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry. 1995;34:16228–16234. doi: 10.1021/bi00049a039. [DOI] [PubMed] [Google Scholar]
- 53.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 54.Turnbull AP, Rafferty JB, Sedelnikova SE, Slabas AR, Schierer TP, Kroon JT, Simon JW, Fawcett T, Nishida I, Murata N, Rice DW. Analysis of the structure, substrate specificity, and mechanism of squash glycerol-3-phosphate (1)-acyltransferase. Struure. 2001;9:347–353. doi: 10.1016/s0969-2126(01)00595-0. [DOI] [PubMed] [Google Scholar]
- 55.Shih GC, Kahler CM, Swartley JS, Rahman MM, Coleman J, Carlson RW, Stephens DS. Multiple lysophosphatidic acid acyltransferases in Neisseria meningitidis. Mol Microbiol. 1999;32:942–952. doi: 10.1046/j.1365-2958.1999.01404.x. [DOI] [PubMed] [Google Scholar]
- 56.Cronan JE. Bacterial membrane lipids: where do we stand? Annu Rev Microbiol. 2003;57:203–224. doi: 10.1146/annurev.micro.57.030502.090851. [DOI] [PubMed] [Google Scholar]
- 57.Gao JL, Weissenmayer B, Taylor AM, Thomas-Oates J, Lopez-Lara IM, Geiger O. Identification of a gene required for the formation of lyso-ornithine lipid, an intermediate in the biosynthesis of ornithine-containing lipids. Mol Microbiol. 2004;53:1757–1770. doi: 10.1111/j.1365-2958.2004.04240.x. [DOI] [PubMed] [Google Scholar]
- 58.Kordulakova J, Gilleron M, Puzo G, Brennan PJ, Gicquel B, Mikusova K, Jackson M. Identification of the required acyltransferase step in the biosynthesis of the phosphatidylinositol mannosides of mycobacterium species. J Biol Chem. 2003;278:36285–36295. doi: 10.1074/jbc.M303639200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures and tables provide the details of the mutagenesis procedures, mass spectrometry and substrate selectivity assays using acyl-coenzyme A variants as donors. This material is available free of charge via the Internet at http://pubs.acs.org.