

Summary
The growth of many cancers depends on self-renewing cells called cancer stem cells or tumor-propagating cells (TPCs). In human brain tumors, cells expressing the stem cell marker CD133 have been implicated as TPCs. Here we show that tumors from a model of medulloblastoma, the Patched mutant mouse, are not propagated by CD133+ cells but by cells expressing the progenitor markers Math1 and CD15/SSEA-1. These cells have a distinct expression profile that suggests increased proliferative capacity and decreased tendency to undergo apoptosis and differentiation. CD15 is also found in a subset of human medulloblastomas, and tumors expressing genes similar to those found in murine CD15+ cells have a poorer prognosis. Thus, CD15 may represent an important marker for TPCs in medulloblastoma.
Significance
Although tumor-propagating cells have been described in human brain tumors, such cells have not been identified in mouse models of the disease. Finding TPCs in mouse models is critical because it allows studies of their developmental origins, and experimental manipulation and targeting of these cells in a species-matched microenvironment. Here we identify a population of TPCs in a model of medulloblastoma, and show that these cells express CD15 (also known as SSEA-1 or LeX) and resemble neural progenitors. Our data challenge the notion that all brain tumors are propagated by stem-like cells, and raise the possibility that CD15 may be used to identify and target TPCs in human brain tumors.
Introduction
The growth of many tumors has been suggested to depend on a subset of tumor cells with an extensive capacity for self-renewal, termed cancer stem cells, tumor-initiating cells or tumor-propagating cells (TPCs) (Huntly and Gilliland, 2005; Reya et al., 2001). These cells are not necessarily abundant or highly proliferative, but because they are long-lived and often resistant to conventional therapies (Bao et al., 2006; Liu et al., 2006; Singh et al., 2004), they are believed to contribute to tumor resistance and recurrence. Therefore, identifying these cells and finding approaches to targeting them has become an important goal in cancer biology.
TPCs were originally described in leukemia, where it was shown that a rare population of cells resembling hematopoietic stem cells was uniquely capable of propagating tumors following transplantation (Bonnet and Dick, 1997). Cells with similar properties have been identified in breast cancer, prostate cancer and other solid tumors (Al-Hajj et al., 2003; O'Brien et al., 2007; Singh et al., 2004; Xin et al., 2005). In many cases, TPCs express markers associated with stem cells from the corresponding tissue, and are capable of generating multiple cell types from that tissue. But a stem-like phenotype is not a necessary feature of TPCs: even cells that do not express stem cell markers or exhibit multipotent differentiation can propagate tumors (Krivtsov et al., 2006; Peacock et al., 2007). Drawing the distinction between stem-like cancer cells and cancer stem cells (TPCs) is essential for interpreting studies in this field.
Evidence for TPCs in brain tumors first came from the observation that human medulloblastomas, astrocytomas and ependymomas contain cells that express the neural stem cell marker CD133 (Hemmati et al., 2003; Singh et al., 2003). Like normal stem cells, these cells can form “neurospheres” that can be passaged repeatedly and induced to differentiate into neurons and glia (Hemmati et al., 2003; Singh et al., 2003; Taylor et al., 2005). Most importantly, these cells are highly enriched for tumor-propagating capacity: CD133+ cells can generate tumors in immunocompromised mice, whereas CD133− cells cannot (Singh et al., 2004; Taylor et al., 2005). CD133+ cells from human gliomas have also been shown to be resistant to radiation and chemotherapy (Bao et al., 2006; Liu et al., 2006). These data suggest that CD133+ cells represent TPCs for human brain tumors.
Although TPCs have been studied in human brain tumors, such cells have not been identified in mouse models of these tumors. Identifying mouse counterparts of TPCs is important since it allows studies of their origin and development, and experimental manipulation and targeting of these cells in a species-matched (murine) microenvironment. This is critical in light of recent studies suggesting that the xenotransplantation assay used to identify human TPCs may select for cells that can survive in a foreign host, and may therefore lead to underestimation or incorrect identification of tumor-propagating cells (Kelly et al., 2007).
We sought to identify TPCs in a widely used model of medulloblastoma, the Patched (Ptc) mutant mouse (Goodrich et al., 1997). Ptc encodes an antagonist of the Hedgehog (Hh) signaling pathway (Rohatgi and Scott, 2007). Germline mutations in Ptc result in Gorlin syndrome, a hereditary disorder associated with skin tumors, craniofacial abnormalities and an increased incidence of medulloblastoma (Hahn et al., 1996; Johnson et al., 1996). Among sporadic medulloblastomas, 15-25% also contain mutations in Ptc or other elements of the Hh pathway (Pietsch et al., 1997; Taylor et al., 2002; Thompson et al., 2006). Although homozygous Ptc knockout mice die during embryogenesis, Ptc heterozygotes (Ptc+/- mice) survive to adulthood, and 15-20% develop tumors that resemble human medulloblastoma (Goodrich et al., 1997). These animals have provided insight into the early stages of tumorigenesis (Oliver et al., 2005), the genes that cooperate with loss of Ptc to promote tumor progression (Pogoriler et al., 2006; Uziel et al., 2005; Wetmore et al., 2001) and the efficacy of Hh antagonists in treating the disease (Berman et al., 2002; Romer et al., 2004). However, they have not been used to study the cells responsible for tumor propagation.
Here we show that tumors from Ptc+/- mice are not propagated by stem-like CD133+ cells, but by cells expressing the neuronal progenitor marker Math1 and the carbohydrate antigen CD15. CD15+ cells are also found in a subset of human medulloblastomas, and expression of genes similar to those found in murine CD15+ cells (a “CD15+ signature”) predicts patient survival. These studies identify CD15 as a marker for TPCs in Ptc+/- mice, and raise the possibility that a similar population of cells may be important for growth or maintenance of human medulloblastoma.
Results
Medulloblastomas from Ptc+/- mice can be propagated by orthotopic transplantation
To identify TPCs in Ptc+/- tumors, we established an orthotopic transplantation assay. Tumor cells were isolated from Ptc+/- mice and immediately stereotaxically implanted into the cerebellum of SCID-beige hosts. As shown in Figure 1A-B, implantation of 1 × 106 tumor cells resulted in secondary tumors that histologically resembled the primary tumors from which the cells were derived. In particular, both primary and secondary tumors consisted of small round cells with little cytoplasm and clearly demarcated borders, as is the case for many human medulloblastomas. To determine how many cells were required to generate tumors, we repeated these experiments using varying numbers of cells. When animals were transplanted with 50,000 or 100,000 cells, only a subset of recipients (17% and 42% respectively) developed tumors, and the latency of tumor formation was variable. In contrast, animals receiving ≥ 200,000 tumor cells showed 100% tumor take, with a median latency of 49 days (Figure 1C). These data demonstrate that Ptc+/- tumors can be propagated by transplantation into the cerebellum.
Figure 1. Propagation of Ptc+/- tumor cells by orthotopic transplantation.
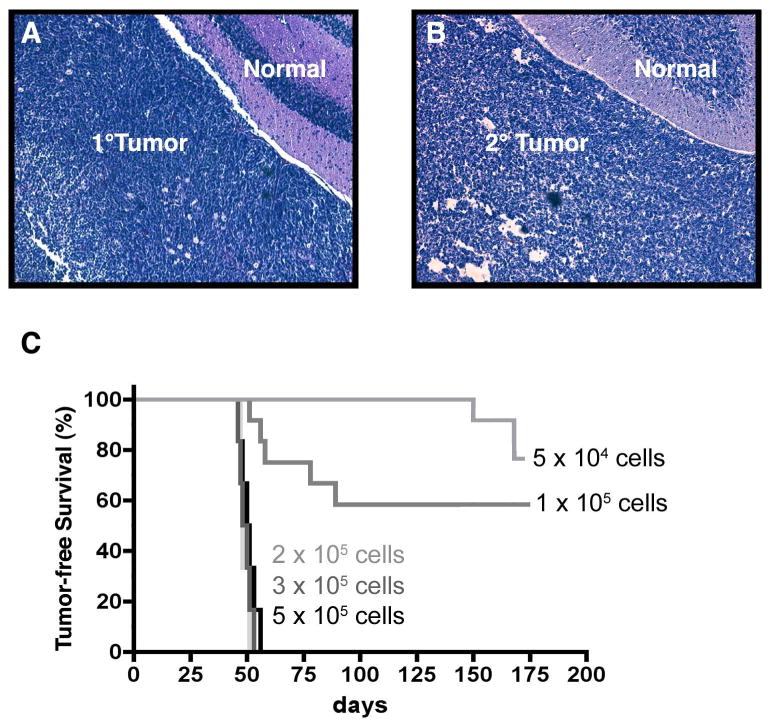
Tumor cells were implanted into the cerebellum of SCID-beige hosts. Animals were sacrificed when they developed symptoms, and tumor tissue was stained with H&E. (A) Primary tumor from a Ptc+/- mouse. (B) Secondary tumor from a SCID-beige host that received 5 × 105 Ptc+/- tumor cells. (C) Tumor-free survival of animals receiving indicated numbers of Ptc+/- tumor cells. Animals implanted with 2-5 × 105 cells developed tumors within 6-8 weeks (median latency = 49 days). Animals implanted with 0.5 × 105 or 1 × 105 cells developed tumors with a lower incidence (42% and 17% respectively) and a longer latency (58 days and 159 days respectively). Scale bars = 100 μm.
Ptc+/- medulloblastomas are not propagated by CD133+ cells
Since CD133+ cells have been shown to be critical for propagation of human brain tumors (Singh et al., 2004), we examined whether a similar population of cells was present in tumors from Ptc+/- mice. As shown in Figure 2A-B, CD133 expression varied greatly among Ptc+/- tumors, with most tumors containing <5% CD133+ cells but some containing as many as 56% CD133+ cells (median = 2.5%). In general, tumors in the posterior cerebellum (adjacent to the brainstem) had higher levels of CD133 than those in anterior regions.
Figure 2. CD133 does not mark tumor-propagating cells in Ptc+/- tumors.
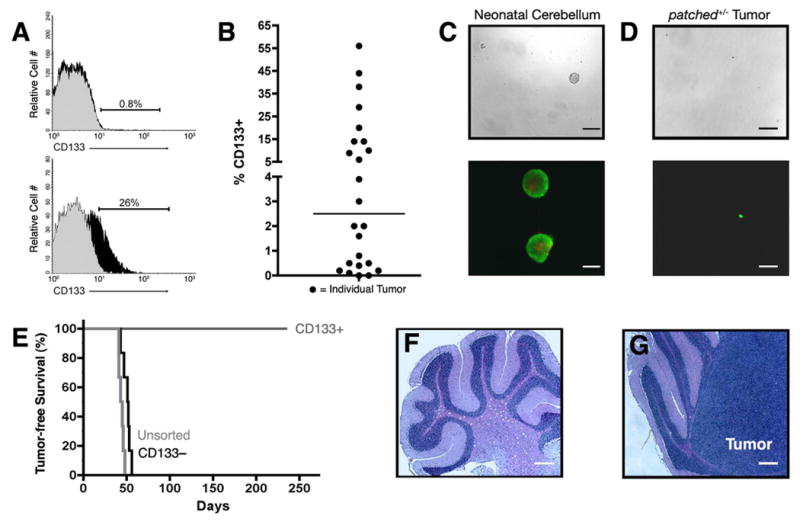
(A-B) Tumor cells from Ptc+/- or Math1-GFP/Ptc+/- mice were stained with anti-CD133 (black histograms) or isotype-matched control antibodies (gray histograms) and analyzed by FACS. (A) Representative histograms from CD133-low (top) and CD133-high (bottom) tumors. (B) Scatter-plot showing percentage of CD133+ cells in 24 tumors. Horizontal line indicates median percentage of CD133+ cells (2.5%). (C-D) CD133+ cells were sorted from neonatal cerebellum (C) or from Ptc+/- tumors (D) and cultured at clonal density in serum-free medium with 25 ng/ml EGF and bFGF for 10 days. Cells were photographed under bright field (top, scale bar = 200 μm) or stained using the Live-Dead Assay Kit, which labels live cells green and dead cells red (bottom, scale bar = 100 μm). Passageable neurospheres were consistently obtained from neonatal cerebellum but not from Ptc+/- tumors. (E) Survival of SCID-beige mice implanted with 3 × 105 unsorted, CD133+ or CD133− tumor cells. Tumors never developed in mice implanted with CD133+ cells (cerebellum shown in F), but developed in all mice implanted with CD133− cells (cerebellum with tumor shown in G). Scale bars for F and G = 200 μm.
CD133+ cells from human brain tumors, like normal neural stem cells, have been reported to generate neurospheres when cultured at low density in the presence of growth factors (Hemmati et al., 2003; Singh et al., 2003). To test whether this was true for CD133+ cells from Ptc+/- tumors, we cultured these cells at clonal density (200 cells/ml) in serum-free medium containing epidermal and fibroblast growth factors. Under these conditions, CD133+ cells from neonatal mouse cerebellum reproducibly generated neurospheres (Figure 2C), and these spheres could be passaged repeatedly in vitro ((Lee et al., 2005) and data not shown). In contrast, CD133+ cells from Ptc+/- tumors never generated neurospheres at clonal density (Figure 2D). At higher density (40,000-80,000 cells/ml), CD133+ cells formed multicellular aggregates within 24 hours. However, the viability of these cells declined dramatically over the next 5-7 days, and cells could not be maintained or expanded by passaging. Similar results were seen with unsorted Ptc+/- tumor cells. These results suggest that CD133+ cells from Ptc+/- tumors do not form neurospheres.
To determine whether CD133+ cells were capable of tumor propagation, we used the orthotopic transplantation assay. Ptc+/- tumor cells were sorted into CD133+ and CD133− fractions, and 300,000 cells from each fraction were transplanted into the cerebellum of SCID-beige mice. As shown in Figure 2E, 100% of the mice that received unsorted cells or CD133− cells developed tumors between 6 and 8 weeks (Figure 2G). In contrast, animals that received CD133+ cells did not develop tumors, and no remnants of these cells could be detected by H&E staining > 8 months after transplantation (Figure 2F). Since Ptc+/- tumor cells contain a beta-galactosidase (β-gal) transgene (Goodrich et al., 1997), we also stained cerebella from animals transplanted with CD133+ cells with anti-β-gal antibodies; this approach also failed to detect CD133+ cells at the time of sacrifice (data not shown). These results suggest that Ptc+/- tumors are propagated by CD133− rather than CD133+ cells.
Ptc+/- tumors are propagated by cells expressing the progenitor marker Math-1
Since CD133+ cells could not generate tumors following transplantation, we looked for other possible markers of tumor-propagating cells in Ptc+/- tumors. We and others (Lee et al., 2003; Oliver et al., 2005) have shown that many Ptc+/- tumor cells express the transcription factor Math1, a marker of immature granule neuron precursors (GNPs). To determine whether tumors are propagated by Math1+ cells, we used Math1-GFP/Ptc+/- mice, in which Math1-expressing cells express green fluorescent protein (Lumpkin et al., 2003). Consistent with previous findings (Oliver et al., 2005), 80-95% of tumor cells from these animals expressed GFP (Figure 3A). To assess the tumorigenicity of these cells, we sorted GFP+ and GFP− cells and transplanted them into the cerebellum of SCID-beige mice. GFP+ cells gave rise to tumors in all recipients (Figure 3B), and these tumors resembled donor tumors in terms of GFP expression (Figure 3C-D) and proliferative index (Ki67, Figure 3E-F). In contrast, none of the recipients of GFP− cells developed tumors. Thus, the tumor-propagating cell in Ptc+/- medulloblastoma is contained within the GNP-like Math1-GFP+ population.
Figure 3. Ptc+/- tumors are propagated by Math1+ cells.
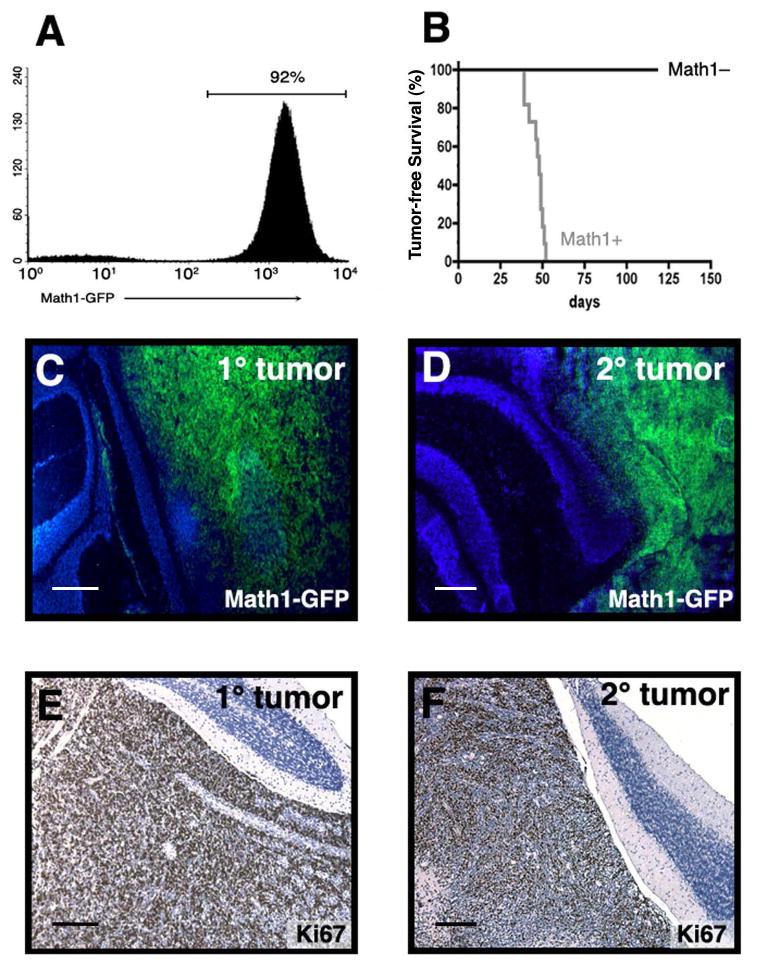
(A) FACS analysis of tumors from Math1-GFP/Ptc+/- mouse shows that the majority of tumor cells are Math1-GFP+. (B) Survival of SCID-beige mice implanted with 3 × 105 GFP+ or GFP− cells. Tumors developed in 100% of animals implanted with GFP+ cells (median latency: 48 days). (C-F) Tumors from Math1-GFP/Ptc+/- mice (1° tumors in C, E) and from SCID-beige mice implanted with Math1-GFP/Ptc+/- tumor cells (2° tumors in D, F) were cryosectioned and stained with DAPI (blue) to label nuclei and with anti-GFP antibodies to label Math1-expressing cells (green, C-D). Additional tumors were paraffin embedded, sectioned and stained with hematoxylin (blue) to label nuclei and with anti-Ki67 to label proliferating cells (brown, panels E-F). Scale bars = 200 μm.
The tumor propagating cell in Ptc+/- medulloblastomas expresses CD15
Since the majority of Ptc+/- tumor cells express Math1-GFP, sorting for these cells does not significantly enrich for tumor-propagating cells. To identify other possible markers of tumor-propagating cells within the Math1-GFP+ population, we stained Ptc+/- and Math1-GFP/Ptc+/- tumors with antibodies previously shown to mark neural progenitors and stem cells. Figure S1A shows the markers tested and the expression range in the tumors we screened. Among these markers, several (A2B5, PSA-NCAM, CD24, CD44, Sca-1, CD117/c-kit) were not consistently expressed and were excluded from further analysis. Three markers – HNK1, CD15 and GD3 − were found in all tumors examined, and were tested to determine if they could enrich for tumor-propagating cells. Tumor cells lacking or expressing these markers were isolated by FACS and transplanted into the cerebellum of SCID-beige mice. As shown in Figure S1B, tumors developed with similar frequency in animals that received HNK1+ and HNK1− cells, indicating that this marker could not be used for enrichment. Animals transplanted with GD3+ cells did not develop tumors, whereas those transplanted with GD3− cells did (Figure S1C). However, since 92-99% of tumor cells lack GD3, selecting for GD3− cells did not result in substantial enrichment of the tumor-propagating population.
The only marker we tested that consistently enriched for tumor propagation was CD15. Also known as stage-specific embryonic antigen (SSEA-1), Lewis X (LeX) or Forse-1 (Allendoerfer et al., 1995; Capela and Temple, 2002; Solter and Knowles, 1978), CD15 is a carbohydrate epitope expressed on normal neutrophils (Skubitz and Snook, 1987), Hodgkin's lymphoma cells (Hall and D'Ardenne, 1987) and cells from a variety of solid cancers (Fox et al., 1983; McCarthy et al., 1985). In addition, several studies suggest that it is expressed on stem cells and progenitors in the embryonic and adult nervous system (Allendoerfer et al., 1995; Capela and Temple, 2002; Capela and Temple, 2006). As shown in Figure 4A, CD15+ cells consistently gave rise to tumors, whereas CD15− cells never did. These studies suggested that CD15 marks TPCs in tumors from Ptc mutant mice.
Figure 4. CD15 is expressed on a subset of medulloblastoma cells and enriches for tumor propagation.
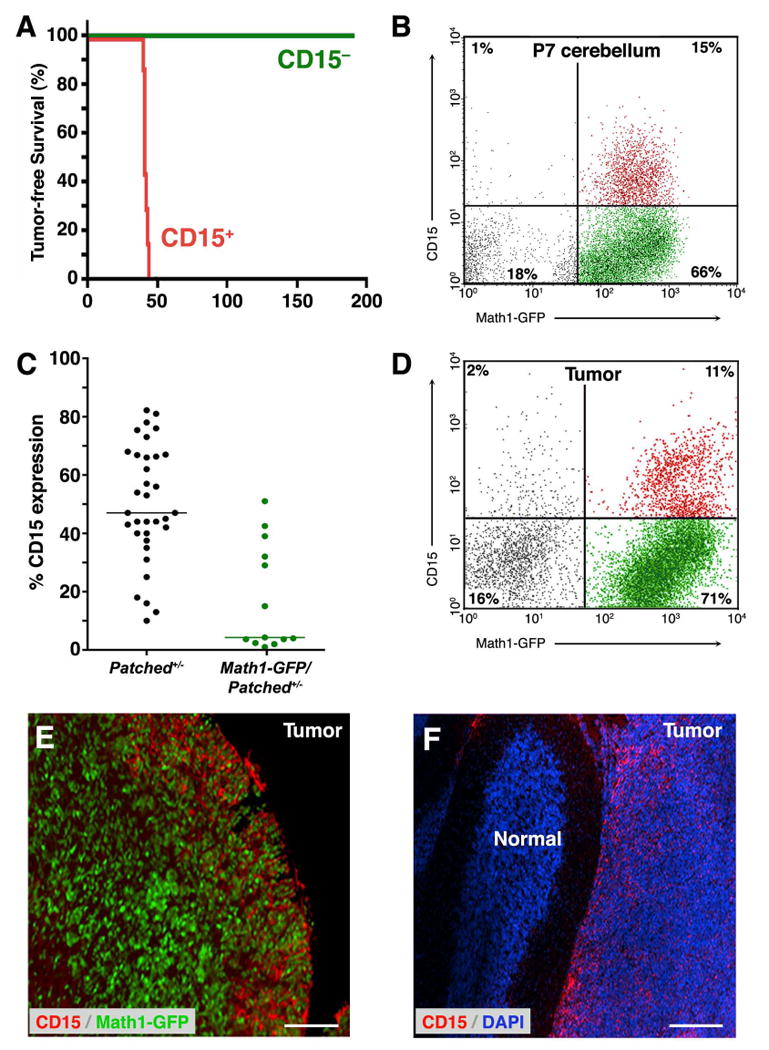
(A) Tumor cells from Ptc+/- or Math1-GFP-Ptc+/- mice were stained with anti-CD15 antibodies and sorted into CD15+ and CD15− fractions. 3 × 105 cells from each fraction were implanted into SCID-beige mice, and animals were monitored for tumor formation. Animals transplanted with CD15+ cells all developed tumors, whereas those transplanted with CD15− cells did not. (B) Expression of CD15 in neonatal cerebellum. Cerebellar cells from P7 Math1-GFP mice were stained with anti-CD15 antibodies and analyzed by FACS. Among cells that expressed Math1-GFP (right quadrants), a subset (15% of all cells, 18.5% of Math1-GFP+ cells) expressed CD15 (red dots). (D-F) Expression of CD15 in Ptc+/- tumor cells. (C) Scatter-plot showing the percentage of CD15+ cells in a panel of 34 Ptc+/- tumors (black dots) and 13 Math1-GFP/Ptc+/- tumors (green dots). Horizontal lines indicate median percentage of CD15+ cells: 47% for Ptc+/- and 4.3% for Math1-GFP/Ptc+/- tumors. (D) FACS analysis of CD15 expression in a representative Math1-GFP/Ptc+/- tumor. Among tumor cells expressing Math1-GFP, 13.4% (11% of all tumor cells) expressed CD15 (red dots). (E-F) Tumor-bearing Math1-GFP/Ptc+/- mice (E) and Ptc+/- mice (F) were injected intracranially with anti-CD15-secreting hybridomas to label CD15+ cells in vivo (see Supplementary Methods). Cerebella were sectioned and stained with secondary antibodies to detect cells that had bound anti-CD15 antibodies (red) and with anti-GFP antibodies to label Math1-expressing cells (green) or with DAPI (blue) to label all nuclei. Note the predominance of CD15 staining on the edge of the tumor (E) and at the boundary between tumor and normal tissue (F). Scale bars = 100 μm.
CD15 is expressed on subset of granule neuron precursors and tumor cells
CD15 is expressed on a stem cells and tumor cells from a variety of tissues. Although expression has been reported in the cerebellum (Ashwell and Mai, 1997; Marani and Tetteroo, 1983), its localization and function during cerebellar development are unknown. To determine whether CD15 is expressed on GNPs, we isolated cerebellar cells from neonatal Math1-GFP mice, stained them with anti-CD15 antibodies, and analyzed them by FACS. As we have shown previously (Lee et al., 2005), the majority of cells that can be isolated from the cerebellum at postnatal day 7 (P7) are Math1-GFP+ (Figure 4B); of these, 15-20% co-express CD15. Staining of sections of neonatal cerebellum revealed CD15 expression in the white matter as well as on proportion of GNPs in the external germinal layer (Figure S2). Thus, a subset of GNPs expresses CD15.
To characterize expression of CD15 in Ptc+/- tumors, we stained tumor cells from a large cohort of Ptc+/- and Math1-GFP/Ptc+/- mice with anti-CD15 antibodies. FACS analysis revealed CD15 expression in all tumors (Figure 4D), although there was marked tumor-to-tumor variation in the percentage of CD15+ cells (Figure 4C). In general, tumors from Math1-GFP/Ptc+/- mice contained fewer CD15+ cells (median = 4.3%) than those from Ptc+/- mice (median = 47%); this may reflect differences in the strain backgrounds of these mice (see Experimental Procedures). Importantly, the CD15+ population in Math1-GFP/Ptc+/- tumors was predominantly GFP+, consistent with the notion that tumor-propagating cells express Math1 (Figure 4C-D and Figure S3). Staining of tumor sections showed CD15+ cells scattered throughout the tumor, but with a higher density near the surface of the cerebellum and at the boundary between tumor and normal tissue (Figure 4E-F). The fact that a subset of cells within every tumor expressed CD15, and the fact that only these cells were capable of forming tumors following transplantation, suggested that CD15 marks TPCs in Ptc+/- medulloblastoma.
CD15+ cells recapitulate the heterogeneity of Ptc+/- medulloblastoma
A cardinal feature of TPCs is their ability to regenerate all the cell types in the original tumor. To determine whether CD15+ cells could recapitulate the heterogeneity of the tumor from which they were isolated, we analyzed tumors formed following transplantation of CD15+ cells. As shown in Figure S4A-B, tumors resulting from transplantation of CD15+ cells histologically resembled the tumors from which they were derived. Secondary tumors from Math1-GFP/Ptc+/- mice consisted largely of Math1-GFP+ cells (Figure S4C-D), indicating that the overall phenotype of the tumor did not change following transplantation. Moreover, secondary tumors contained CD15+ and CD15− cells in proportions similar to those found in the primary tumor (Figure S4E-F). These studies indicate that upon transplantation CD15+ cells not only generate more of themselves but also give rise to the other cell types that make up a histologically complex tumor.
CD15+ cells have a distinct gene expression profile
Having seen a marked difference in the ability of CD15+ and CD15− cells to generate tumors, we sought to determine the molecular basis for this difference. To this end, we isolated RNA from CD15+ and CD15− cells and performed microarray analysis. Principal components analysis, a statistical method that facilitates global comparison of gene expression between multiple samples, indicated that CD15+ and CD15− cells were readily distinguishable from one another (Figure 5A). Differences between CD15+ and CD15− cells were also apparent from hierarchical clustering (Figure 5B), which revealed groups of genes whose expression was increased or decreased in CD15+ vs. CD15− cells. Among the 45,101 probe sets on the arrays, 438 (approximately 1%) showed significant differences in expression (increased or decreased by ≥ 1.8-fold, FDR < 0.05 using paired t-test) between these populations. Of these, 70 were higher in CD15+ cells, and 368 were lower (see Table S1).
Figure 5. CD15+ cells have a distinct gene expression profile.
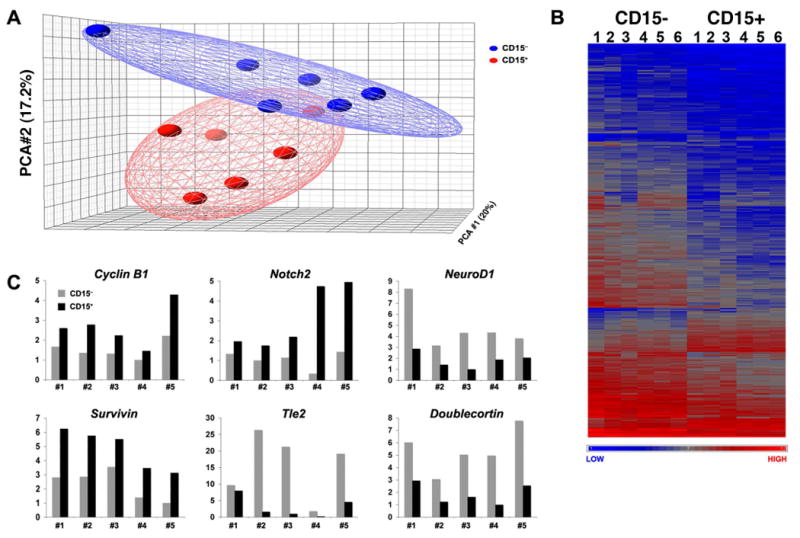
Cells from 6 Ptc+/- tumors were sorted into CD15+ and CD15− samples, and RNA from these samples was labeled and hybridized to Affymetrix expression microarrays. (A) Principle components analysis. Each sample is represented by a spot (red = CD15+, blue = CD15−) whose position in the grid reflects its overall expression profile. The distance between spots is proportional to the difference in gene expression. Note that CD15+ samples are clustered together (red ellipse) and occupy a region of the grid distinct from that occupied by CD15− samples (blue ellipse). (B) Unsupervised hierarchical clustering highlights groups of genes whose expression is significantly increased or decreased in CD15+ vs. CD15− cells. Each column represents a distinct sample (CD15− or CD15+) and each row represents an individual gene. The normalized relative level of gene expression is denoted by color (blue = low, gray = intermediate, red = high), as indicated in the gradient at the bottom. (C) Validation of differential gene expression by RT-PCR. CD15+ and CD15− samples from five independent Ptc+/- tumors (distinct from those used for microarray analysis) were analyzed by quantitative RT-PCR using primers for the indicated genes. Gene expression was normalized based on Actin levels.
To gain insight into the properties that distinguished CD15+ and CD15− cells, we performed Gene Ontology analysis (Table S2) and carried out literature searches for each of the differentially expressed genes, focusing in particular on their functions in the nervous system. Based on this analysis, we identified 5 major categories of genes that were differentially expressed between CD15+ and CD15− cells (see Tables 1 and S3). Regulators of proliferation and self-renewal (e.g. Cyclins B1 and B2, Aurora kinase, and Notch2) accounted for the majority of genes upregulated in CD15+ cells. Expression of pro-apoptotic genes (including those encoding the ubiquitin ligase Fbxo7, the Bcl2-interacting protein Harakiri and the apoptosis-inducing factor Aifm3) were markedly lower in CD15+ cells compared to CD15− cells, while the anti-apoptotic protein Birc5/Survivin was higher. Genes implicated in cell migration and axon growth (including L1CAM, Contactin2/Tag-1, Semaphorin 6A and Doublecortin) were also significantly decreased in CD15+ cells, consistent with the notion that these cells have a decreased propensity to migrate away from the surface of the cerebellum and elaborate axons, processes associated with normal granule neuron differentiation. Finally, the list of differentially expressed genes included a significant number of RNA binding proteins (which regulate mRNA processing and stability), transcription factors and regulators of chromatin structure. While some of these (NeuroD1, Nhlh1/Nscl, Elavl4/HuD, Actin-like 6b/Baf53b) have been shown to regulate differentiation in GNPs and other types of neurons, the function of most remains to be determined. Differential expression of selected genes from each of these categories was validated by quantitative RT-PCR (Figure 5C). Overall, these data indicate that CD15+ and CD15− cells have distinct gene expression profiles, and that the tumor-propagating capacity of CD15+ cells is associated with an increased capacity to proliferate and a decreased tendency to undergo apoptosis and differentiation.
Table 1. Categories of Genes Differentially Expressed in CD15+ vs. CD15− Cells.
For the 438 genes whose expression was significantly different between CD15+ and CD15− cells (see Table S1), we used Gene Ontology (Table S2) and literature searches to gain insight into gene function. Based on this analysis, many genes could be grouped into 5 functional categories. Representative genes from each category are listed. Gene names and symbols are derived from the Entrez-Gene database. Fold change represents the difference in expression between CD15+ and CD15− cells; genes increased in CD15+ cells are preceded by an up arrow (↑), those decreased in CD15+ cells are preceded by a down arrow (↓). Gene functions are based on published literature, with emphasis placed on functions in the nervous system whenever possible. For references supporting the functions of each gene, see Supplementary Table 3 and Supplementary References.
| PROLIFERATION/SELF-RENEWAL | |||
|---|---|---|---|
| Gene Name | Gene Symbol/Aliases | Fold Change | Gene Function |
| cyclin B1 | Ccnb1 | ↑ 2.4 | G2/mitotic cyclin |
| ubiquitin-conjugating enzyme E2C | Ube2c, UBCH10 | ↑ 2.1 | ubiquitination, cell cycle regulation |
| cell division cycle 25 homolog C | Cdc25c | ↑ 2.1 | phosphatase, regulates mitosis |
| cyclin B2 | Ccnb2 | ↑ 2.0 | G2/mitotic cyclin |
| polo-like kinase 1 | Plk1 | ↑ 2.0 | kinase, regulates cell cycle checkpoints |
| aurora kinase A | Aurka, Ark1, Stk6 | ↑ 1.9 | kinase, regulates spindle formation, cytokinesis |
| cyclin A2 | Ccna2 | ↑ 1.9 | G1/S cyclin |
| discs large homolog 7 | Dlgap5, Dlg7, HURP | ↑ 1.8 | regulates mitotic spindle formation |
| Notch2 | Notch2 | ↑ 1.8 | receptor, reguates proliferation/self-renewal |
| APOPTOSIS | |||
| Gene Name | Gene Symbol/Aliases | Fold Change | Gene Function |
| F-box protein 7 | Fbxo7, Fbx7 | ↓ 4.5 | ubiquitin ligase, regulates apoptosis and cell cycle |
| cytoplasmic FMR1 interacting protein 2 | Cyfip2, Pir121 | ↓ 3.1 | p53 target, promotes apoptosis |
| mitogen activated protein kinase 10 (JNK3) | Mapk10, JNK3 | ↓ 2.7 | kinase, promotes apoptosis |
| harakiri, BCL2 interacting protein | Hrk, Bid3, DP5 | ↓ 2.7 | BH3-only protein, promotes apoptosis |
| SH3-domain kinase binding protein 1 | Sh3kbp1, Ruk, CIN85 | ↓ 2.5 | adaptor, negative regulator of PI3 kinase |
| apoptosis-inducing factor | Aifm3, Aifl | ↓ 1.8 | mitochondrial protein, inducer of apoptosis |
| baculoviral IAP repeat-containing 5 | Birc5, Survivin, Api4 | ↑ 1.9 | prevents apoptosome assembly, inhibits apoptosis |
| MIGRATION and AXON GROWTH | |||
| Gene Name | Gene Symbol/Aliases | Fold Change | Gene Function |
| L1 cell adhesion molecule | L1cam, CD171 | ↓ 6.4 | surface protein, regulates migration, neurite growth |
| G prot-regulated inducer of neurite outgrowth 3 | Gprin3, Grin3 | ↓ 5.3 | cytoplasmic protein, binds G alpha subunits |
| dihydropyrimidinase-like 3 | Dpysl3, Crmp4, ulip | ↓ 4.9 | cytoplasmic protein, involved in growth cone signaling |
| contactin 2 (TAG-1) | Cntn2, Tag1 | ↓ 3.3 | surface protein, reguates neurite growth |
| semaphorin 6A | Sema6a, Semaq | ↓ 3.0 | surface protein, regulates migration, neurite growth |
| doublecortin | Dcx, Lisx | ↓ 2.5 | microtubule binding protein, regulates migration |
| neuron-glia-CAM-related cell adhesion molecule | Nrcam, Bravo | ↓ 2.5 | surface protein, regulates neurite growth |
| cyclin-dependent kinase 5, regulatory subunit | Cdk5r1, p35 | ↓ 2.2 | kinase, regulates migration, axon growth, apoptosis |
| p21 (CDKN1A)-activated kinase 1 | Pak1 | ↓ 2.1 | regulates cell motility and morphology |
| ephrin B2 | Efnb2, Elf-2, Lerk5 | ↓ 1.9 | transmembrane protein, regulates migration |
| hyaluronan mediated motility receptor | Hmmr, Rhamm | ↑ 2.3 | surface/cytoplasmic protein, regulates motility |
| RNA PROCESSING/TRANSLATION | |||
| Gene Name | Gene Symbol/Aliases | Fold Change | Gene Function |
| bruno-like 5 | Brunol5, Celf5 | ↓ 3.0 | RNA binding protein, regulates differentiation |
| ELAV-like 4 (Hu antigen D) | Elavl4, Hud | ↓ 2.4 | RNA binding protein, regulates differentiation |
| ataxin 2 binding protein 1 | A2bp1, Hrnbp1, Fox-1 | ↓ 2.3 | RNA binding protein, regulates splicing in neurons |
| cytoplasmic polyA binding protein 4 | Cpeb4 | ↓ 2.3 | RNA binding protein, regulates splicing in neurons |
| bruno-like 4 | Brunol4, Celf4 | ↓ 2.1 | RNA binding protein, regulates neural differentiation |
| trinucleotide repeat containing 4 | Tnrc4, Brunol1, CAGH4 | ↓ 1.9 | RNA binding protein, regulates splicing in neurons |
| TRANSCRIPTION/DIFFERENTIATION | |||
| Gene Name | Gene Symbol/Aliases | Fold Change | Gene Function |
| zinc finger protein, multitype 2 | Zfpm2, Fog2 | ↓ 3.3 | binds GATA transcription factors, regulates differentiation |
| nescient helix loop helix 1 | Nhlh1, Nscl, Tal2 | ↓ 3.1 | bHLH transcription factor, regulates differentiation |
| transducin-like enhancer of split 2 | Tle2, Grg2, Esg2 | ↓ 2.7 | co-repressor, regulates Wnt, Notch signaling |
| neurogenic differentiation 1 | Neurod1 | ↓ 2.7 | bHLH transcription factor, regulates differentiation |
| actin-like 6B | Actl6b, Baf53b, ArpNa | ↓ 2.5 | chromatin remodeling protein regulates differentiation |
| scratch homolog 2, zinc finger protein | Scrt2 | ↓ 2.2 | Snail family transcriptional repressor |
| Kruppel-like factor 7 | Klf7, Uklf | ↓ 2.0 | Zn-finger transcription factor, regulates differentiation |
| Kruppel-like factor 12 | Klf12, AP-2rep | ↓ 1.9 | Zn-finger transcriptional repressor |
CD15+ cells exhibit increased Hh pathway activation and proliferation
TPCs in human leukemia are more quiescent than the “blasts” that make up the majority of the tumor (Bonnet and Dick, 1997; Holyoake et al., 1999). In contrast, brain tumor stem cells have been reported to exhibit higher proliferation than non-stem cells from the same tumor (Singh et al., 2003; Singh et al., 2004). The fact that CD15+ cells from Ptc+/- tumors expressed elevated levels of cell cycle regulators suggested that these cells might also be more proliferative than CD15− cells. To examine this, we sorted CD15+ and CD15− cells from Ptc+/- tumors, cultured them for 48 hours, and measured incorporation of tritiated thymidine. As shown in Figure 6A, CD15+ cells consistently showed a higher level of thymidine incorporation than CD15− cells from the same tumor. (These studies involved culturing cells at high density in the absence of added growth factors; when cells were cultured at clonal density in the presence of EGF and bFGF, they survived poorly, did not form neurospheres, and showed no evidence of multi-lineage differentiation). CD15+ cells also showed increased proliferation in vivo, as measured by incorporation of bromodeoxyuridine (BrdU, Figure S5). Together these data suggest that CD15+ cells have an increased capacity for proliferation in vitro and in vivo.
Figure 6. CD15+ tumor cells exhibit increased proliferation and Hh pathway activation.
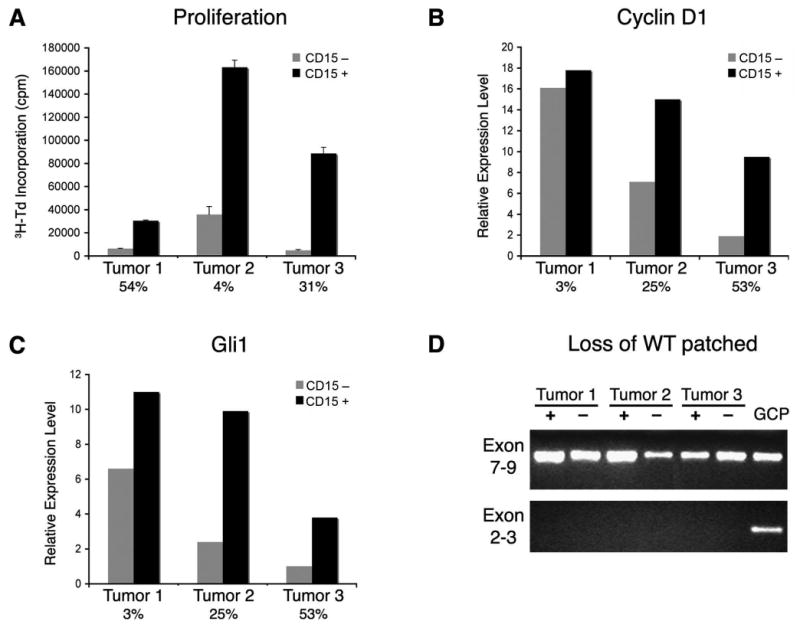
(A) CD15+ and CD15− tumor cells were cultured for 48 hours in serum-free medium, and then pulsed with tritiated thymidine (3H-Td) for 14 hours before assaying thymidine incorporation. Data represent means of triplicate samples ± SEM. (B-C) RNA from CD15+ and CD15− tumor cells was analyzed by real-time RT-PCR using primers specific for the Hh target genes cyclin D1 and Gli1. Expression levels are normalized to actin. In A-C, percentages of CD15+ cells are shown below bars for each tumor. CD15+ cells consistently exhibited higher proliferation and Hh target gene expression than CD15− cells from the same tumor. (D) Math1+CD15− cells have lost the remaining copy of Ptc. RNA from Math1+/CD15+ and Math1+/CD15− tumor cells, and from WT GNPs (as a positive control) were analyzed by RT-PCR using primers specific for Ptc exons 7-9 (found in both the mutant and WT allele) and Ptc exons 2-3 (found only in the WT allele). Transcripts containing exons 7-9 are found in all cell populations, but only GNPs express transcripts containing the WT allele of Ptc. These data suggest that both CD15+ and CD15− cells are tumor cells that have lost the WT allele.
Since proliferation of medulloblastoma cells from Ptc+/- mice is dependent on Hh pathway activation (Berman et al., 2002; Romer et al., 2004) we also examined expression of Hh target genes in CD15+ and CD15− tumor cells. As shown in Figure 6D-E, CD15+ cells expressed elevated levels of the Hh target genes Gli1 and Cyclin D1 (Kenney and Rowitch, 2000; Oliver et al., 2003) compared to CD15− cells. Interestingly, CD15+ cells isolated from the neonatal cerebellum also showed greater target gene induction and a stronger proliferative response to Sonic hedgehog than CD15− cells (data not shown). These data suggest that increased proliferative capacity of CD15+ cells is associated with increased Hh pathway activation.
One explanation for the low proliferative capacity of CD15− cells and for the inability of these cells to propagate tumors could be that they do not represent tumor cells at all. Among the features that have been used to define tumor cells in the Ptc mutant mouse are expression of Math1 (a marker of GNPs that is not expressed in the normal adult cerebellum) and loss of the remaining (wild-type, WT) copy of Ptc (Lee et al., 2003; Oliver et al., 2005). The fact that a substantial proportion of CD15− cells express Math1 (see Figures 5C, E) suggests that these cells represent tumor cells, rather than normal cells that have become trapped within the tumor. To determine whether these cells have also lost expression of WT Ptc, we analyzed RNA from Math1+/CD15+ and Math1+/CD15− tumor cells by real-time RT-PCR. To distinguish between WT and mutant Ptc transcripts, we used two sets of primers: one derived from exons 7-9, which are present in both WT and mutant alleles, and another derived from exons 2-3, which can only amplify sequences present in the WT allele (Oliver et al., 2005). As shown in Figure 6D, transcripts containing exons 7-9 were found in CD15+ and CD15− tumor cells, as well as in normal GNPs (a positive control for Ptc expression). In contrast, transcripts containing exons 2-3 (specific for the WT allele) were observed in GNPs, but were absent from both CD15+ and CD15− tumor cells. These results suggest that CD15− cells are tumor cells, but simply lack tumor-propagating capacity.
CD15 is expressed in a subset of human medulloblastoma
The fact that CD15 identified tumor-propagating cells in murine medulloblastoma raised the possibility that it might be important in human tumors as well. To determine if CD15 is expressed in human medulloblastoma, we performed immunohistochemical analysis on a panel of archival medulloblastoma samples. Immunoreactivity for CD15 in granulocytes and histiocytes was detected in all cases, serving as an internal control. CD15+ tumor cells were present in approximately one-third of cases (10/28 samples), including classic and large-cell tumors and medulloblastomas with extensive nodularity (Figure 7A-C). CD15 staining was also detected in patient samples analyzed by flow cytometry (data not shown), including one nodular medulloblastoma, one tumor with focal nodularity and one large-cell tumor. These data indicate that CD15 is expressed in a subset of human medulloblastomas.
Figure 7. CD15 is expressed in human medulloblastoma and expression of a CD15-associated gene signature predicts survival.
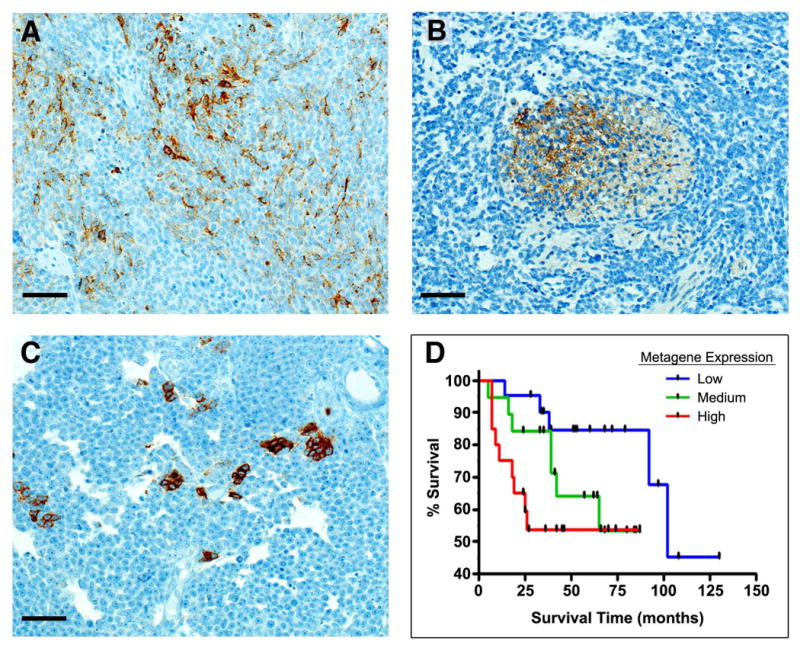
(A) Variable immunoreactivity for CD15 in a classic medulloblastoma. (B) CD15 immunoreactivity in the matrix of a nodule in a medulloblastoma with extensive nodularity. (C) Strong immunoreactivity for CD15 in clusters of tumor cells in a large cell medulloblastoma (Scale bars = 70 μm for all panels). (D) Increased probability of CD15+ signature correlates with poor survival in human medulloblastoma. Black tick marks represent surviving patients at time of last follow-up. Log rank for trend p = 0.02.
To gain insight into the clinical significance of CD15 expression, we took advantage of the gene expression analysis we had performed on murine tumors. Using the genes that were differentially expressed in CD15+ vs. CD15− cells from Ptc+/- tumors and published methods (Bild et al., 2005), we identified a set of genes (a CD15+ “signature”) whose expression robustly predicted the CD15 status of our samples (see Figure S6 and Table S4). The validity of this signature was confirmed in proper leave-one-out cross-validation of our original samples (see Supplementary Methods), and when applied to an independent set of tumors sorted for CD15 status. The CD15+ signature was then used to interrogate a database of gene expression profiles for human medulloblastoma samples (Pomeroy et al., 2002). Based on their gene expression profiles, tumors were scored as having “Low”, “Medium” or “High” expression of the CD15+ signature, and these groups were compared to determine whether expression of the signature was associated with tumor subtype or patient survival. Consistent with the immunohistochemical analysis above, we found no significant correlation between CD15+ signature expression and tumor subtype (data not shown). However, as shown in Figure 7D, expression of the CD15 signature was a strong predictor of patient survival: increased expression was associated with shorter survival time (p = 0.021 based on log-rank test for trend). When the same CD15 signature was applied to a second, albeit smaller, dataset (Thompson et al., 2006), elevated expression was again associated with shorter survival (Figure S7), although the results did not reach statistical significance (p = 0.16). Together these data indicate that a subset of human tumors contains CD15+ cells and suggest that the presence of a CD15+ gene expression signature may be an important predictor of prognosis for human medulloblastoma.
Discussion
The cancer stem cell hypothesis has gained a great deal of attention in recent years, with reports describing such cells in many different types of human cancer. But to date, there have been few studies of TPCs in animal models of the disease. In the current studies we identify TPCs in the most commonly used animal model of medulloblastoma, the Ptc mutant mouse. The cells we describe differ from previously identified brain tumor stem cells in that they do not express the stem cell marker CD133 and cannot form neurospheres. Instead, they express the neural progenitor marker Math1 and can be enriched based on their expression of the carbohydrate antigen CD15. Transplantation of Math1+CD15+ cells is sufficient to give rise to tumors in 100% of hosts, and these tumors resemble the original tumors in terms of histology and cellular heterogeneity. Based on these findings, we conclude that the Math1+CD15+ compartment contains TPCs.
The nature of the cells that initiate and propagate medulloblastoma has been the subject of debate for many years (Eberhart, 2007; Katsetos et al., 2003; Read et al., 2006). The fact that these tumors are frequently found on the surface of the cerebellum and express markers associated with the granule lineage has led many investigators to speculate that these tumors arise from GNPs (Kadin et al., 1970; Miyata et al., 1998). On the other hand, the demonstration that many medulloblastomas express stem cell markers, form neurospheres and are propagated by CD133+ cells has raised the possibility that these tumors may arise from, and be propagated by, neural stem cells (Hemmati et al., 2003; Singh et al., 2003; Singh et al., 2004). Recent studies by our lab and others (Schuller et al., 2008; Yang et al., 2008) have shed light on the cell of origin for medulloblastoma, demonstrating that Hh pathway activation in either stem cells or GNPs can lead to medulloblastoma. But while these studies provided insight into the normal cells that can give rise to tumors (the cells of origin), they did not identify the cells are critical for propagation of tumors once they are established (the TPCs). In the current study we show that tumors from Ptc+/- mice are propagated by GNP-like cells, and in particular, by a subset of these cells that expresses CD15.
CD15 is expressed on both progenitors and stem cells in the embryonic and adult central nervous system (Capela and Temple, 2002; Capela and Temple, 2006). In light of this, one possible interpretation of our findings is that CD15+ cells in Ptc+/- tumors might represent neural stem cell-like cells. However, several observations argue against this notion. First, the fact that CD15 is expressed on a subset of GNPs in the neonatal cerebellum (Figure S2 and (Marani and Tetteroo, 1983)) indicates that CD15 expression is not exclusive to stem cells. The fact that the CD15+ cells in our tumors co-express Math1, are unable to form neurospheres and show no evidence of multi-lineage differentiation also suggests that these are not stem-like cells. Thus, we believe that the CD15+ cells we have isolated are progenitor-like cells with a unique capacity for tumor propagation.
CD15+ cells propagate tumors following transplantation, and when they represent a small subset of the cells in a tumor, isolating them significantly enriches for tumor propagating capacity. However, it is important to note that CD15+ cells sometimes represent a fairly large fraction of the tumor. The high percentage of cells expressing a marker of TPCs is not unprecedented: in human brain tumors, CD133 has been reported to be found on up to 40% of cells (Singh et al., 2003; Singh et al., 2004). Whether this means that a large percentage of cells in the tumor are capable of tumor propagation (Kelly et al., 2007), or whether the true TPCs represent a subset of the CD133+ or CD15+ cells, will need to be examined by sub-fractionating each population with additional markers.
To investigate the molecular basis for the tumor-propagating capacity of CD15+ cells, we performed microarray analysis on CD15+ and CD15− cells from Ptc+/- tumors. This analysis revealed that CD15+ cells have a distinct gene expression profile characterized by increased expression of genes associated with proliferation and self-renewal and decreased expression of genes involved in apoptosis and differentiation. Consistent with this expression profile, we have also observed that CD15+ cells are more proliferative than CD15− cells, both in vitro and in vivo. This property distinguishes CD15+ cells from the tumor-propagating cells found in human leukemia, which are more quiescent than the bulk of the tumor (Bonnet and Dick, 1997; Holyoake et al., 1999). On the other hand, CD133+ cells from human brain tumors have been reported to be more proliferative than their CD133− counterparts (Singh et al., 2004), perhaps indicating a distinction between TPCs in the nervous and hematopoietic systems. Interestingly, we have found that CD15+ cells in the neonatal cerebellum also have higher proliferative capacity or higher intrinsic Hh responsiveness than CD15− cells. It is possible that the tumors in Ptc mutant mice arise from these cells and retain this proliferative capacity. Alternatively, it is possible that tumor cells acquire these properties, and concomitantly acquire expression of CD15. Further studies will be necessary to determine the relationship between normal and neoplastic CD15+ cells and the mechanisms that regulate CD15 expression.
Our observation that CD15+ cells recapitulate the heterogeneity within the original tumor, and in particular, generate CD15+ as well as CD15− cells, suggests that these cells may sit at the top of a hierarchy of differentiation within the tumor. Although our data indicate that many CD15− cells represent tumor cells (based on expression of Math1 and loss of Ptc), these cells appear to be less proliferative than their CD15+ counterparts. However, as shown in Figure 6 and Figure S5, these cells are not completely quiescent. Indeed, since CD15− cells are frequently much more abundant than CD15+ cells, they may contribute substantially to the growth of tumors in vivo. In the context of a rapidly growing tumor, it may be critical to eliminate both the tumor-propagating cells and their more differentiated progeny in order to effectively eradicate the tumor.
The identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma raises the question of whether it is relevant for human medulloblastoma as well. Our observation that CD15 is expressed in a subset of human medulloblastomas is consistent with this possibility. Since we identified CD15 as a marker of TPCs in tumors from Ptc+/- mice, which are thought to be genetically similar to human nodular/desmoplastic medulloblastomas (Goodrich et al., 1997; Pietsch et al., 1997; Pomeroy et al., 2002), we expected that CD15 expression might be more common in this subtype of tumor. However, our immunohistochemical and flow cytometric analysis suggest that CD15 expression is not restricted to such tumors, and can occur in a variety of medulloblastoma subtypes. These findings indicate that tumor morphology and the presence of Hh pathway mutations may not be critical determinants of CD15 expression, and suggest that the role of CD15 should be investigated in all medulloblastoma subtypes.
Support for the notion that CD15 may have significance in human medulloblastoma comes from our demonstration that a CD15+ gene expression signature can predict survival in medulloblastoma patients. One interpretation of this finding is that tumors with strong expression of the CD15+ signature contain a large percentage of CD15+ cells, and that having many such cells makes them particularly aggressive. Alternatively, it is possible that expression of the CD15+ signature does not reflect expression of CD15 itself, but rather the presence of properties (increased proliferative capacity, decreased tendency to differentiate or die) that are associated with CD15+ cells. In either case, the signature we have derived from our studies of TPCs in mice may be a valuable tool for predicting outcome in human medulloblastoma patients. Such tools are critical, since most medulloblastoma patients are treated with an aggressive regimen of radiation and chemotherapy; while this results in high cure rates, it is associated with severe side effects including cognitive deficits, endocrine disorders and an increased risk of secondary tumors. Stratification of patients based on predictors of outcome such as the CD15+ signature may allow clinicians to limit use of the most aggressive therapies to patients with poor prognosis, and to use less toxic but equally effective treatments for patients whose prognosis is more favorable.
In summary, our studies identify a population of TPCs in a mouse model of medulloblastoma. The fact that these cells express markers of progenitors and cannot form neurospheres indicates that TPCs need not resemble normal stem cells. Rather, some tumors may be propagated by cells that resemble neural progenitors. Identification of TPCs in a mouse model of human medulloblastoma will allow us to study the mechanisms by which they propagate tumors and test approaches to targeting them in vivo. Ultimately, such studies will be necessary to determine whether eradication of TPCs cells is critical for effective therapy of brain tumors.
Experimental Procedures
Animals
Ptc+/- mice (Goodrich et al., 1997) were maintained by breeding with 129X1/SvJ mice from Jackson Laboratory. Math1-GFP/Ptc+/− mice were generated by crossing Ptc+/- mice with Math1-GFP mice (Lumpkin et al., 2003) from Jane Johnson at UT Southwestern Medical Center. SCID/Beige mice were obtained from Taconic Farms. Mice were maintained in the Cancer Center Isolation Facility at Duke University Medical Center, and all experiments were performed using procedures approved by the Duke University IACUC.
Stereotaxic implantation of tumor cells
SCID/beige mice were anesthetized using 100 mg/kg ketamine (Fort Dodge Animal Health) + 9 mg/kg xylazine (Ben Venue Laboratories), and positioned in a stereotaxic frame with a mouse adapter (Kopf Instruments). An incision was made in the midline of the scalp over the cerebellum, and small hole was made in the skull (1 mm lateral to midline) using a beveled (sharp point) 18-gauge needle. A 24-gauge Hamilton syringe loaded with cells was mounted on a micromanipulator and introduced through the hole at a 30-degree angle to the surface of the cerebellum, at a depth of 1 mm. Freshly-sorted (uncultured) cells were injected over the course of 2 minutes, and the needle was left in place for two more minutes to avoid reflux. After removing the mouse from the frame, 1-2 drops of 0.25% (2.5mg/mL) Bupivicaine (Hospira) were applied along the incision for postoperative analgesia and the skin was closed with 6-0 fast absorbing plain gut suture using a 3/8 PC-1 cutting needle (Ethicon).
Antibodies
Antibodies used for FACS and immunostaining include CD15 (clone MMA), CD57/HNK-1, CD24 (clone 30F1), CD117/c-kit (clone 2B8) and anti-BrdU-APC from BD Biosciences; CD44 (clone IM7) and Sca-1 (clone D7) from eBioscience; CD133 (clone 13A4) PSA-NCAM (clone 2-2B) and A2B5 (clone A2B5-105) from Millipore; GD3 (Clone R24) from EMD Biosciences; anti-BrdU (rat IgG) from Abcam; and anti-Ki67 (Clone TEC-3) from Dako. Secondary antibodies included Fluorescein (DTAF)- or phycoerythrin-conjugated F(ab′)2 fragments of goat-anti-mouse IgM or goat-anti-mouse IgG (Jackson ImmunoResearch), and anti-rat IgG Alexa Fluor-568 and anti-mouse IgM-Alexa Fluor-488 (Molecular Probes).
Cell and tissue staining
Dissociated tumor cells were plated in poly-D-lysine coated chamber slides for 6 hours prior to fixation in 4% paraformaldehyde (PFA) and blocking with PBS+10% normal goat serum. Anti-CD15 antibody was applied for 2h at RT and Alexa Fluor 568-conjugated anti-mouse IgM was applied for 1h at RT.
For staining of frozen tissue, brains were fixed in 4% PFA, cryoprotected in 25% sucrose, embedded in Tissue Tek-OCT (Sakura Finetek) and sectioned saggitally at a thickness of 10 μm. Sections were permeabilized with PBST (PBS+0.1% Triton-X 100), blocked with PBST+ 10% normal goat serum, and incubated with primary antibodies for 24h at 4°C and secondary antibodies for 1h at 37°C. Sections were counterstained with DAPI (Molecular Probes) and mounted in Fluoromount G (Southern Biotechnology Associates). For detection of BrdU+ cells, animals were pulsed with BrdU before staining, and sections were treated with DNase (Sigma) prior to antibody staining.
For staining of human and mouse paraffin sections, tissue was fixed overnight in 10% formalin, transferred to 70% ethanol, paraffin-embedded and sectioned at a thickness of 5 μm. Sections were stained with Hematoxylin and Eosin (Sigma) or with Hematoxylin and rat anti-mouse Ki-67 (Dako) or anti-CD15 (BD Biosciences). Ki-67 was detected using biotinylated rabbit-anti-rat IgG followed by Streptavidin-conjugated horseradish peroxidase and DAB+ as a chromogen (Dako). CD15 staining was visualized using the Ventana IVIEW Detection kit (biotinylated Ig, Ventana Medical Systems). Sections were mounted in permanent mounting medium and imaged using a Nikon TE200 inverted microscope and OpenLab software (Improvision).
Expression of CD15 in human medulloblastoma
Human medulloblastoma samples were obtained from tissue repositories at Duke University Medical Center and St. Jude Children's Research Hospital. Informed consent was obtained from all patients prior to acquisition of tissue, and patient identifying information was protected in accordance with the Duke University and St. Jude Institutional Review Boards. Immunohistochemical analysis was performed on paraffin sections as described above.
Microarray analysis
Ptc+/- tumor cells were sorted into CD15+ and CD15- populations, and RNA was isolated, labeled and hybridized to Affymetrix Mouse Genome 430 2.0 arrays. Microarray data were preprocessed using robust multi-chip analysis (RMA). Principal components analysis (PCA) and hierarchical clustering were performed with Partek Genomics Suite 6.3. PCA was performed on samples based on normalized expression of all genes. Unsupervised hierarchical clustering of differentially expressed genes was carried out using Average Linkage with Euclidean Distance as the similarity measurement. Genes differentially expressed between CD15+ and CD15- samples were selected based upon both relative and statistical criteria (1.8-fold change cutoff; pairwise t-test FDR of <0.05).
Derivation of CD15+ Signature and Survival Analysis
The CD15+ signature was developed from gene expression differences between CD15+ and CD15− tumor cells (n = 6 for each), using established methods (Bild et al., 2005). Briefly, t-tests and FDR estimates from RMA post-processed data (Bolstad et al., 2003) identified genes with the strongest levels of differential expression. The expression profile of the gene set, termed a ‘metagene’, is summarized by the top principle component of a singular value decomposition (SVD), and then applied to a Bayesian probit regression model to generate the signature. The internal accuracy of the predictive signature is assessed through proper leave-one-out cross-validation. For all analysis, SVD was performed using only training data. The CD15+ signature was applied to three independent microarray datasets: A) 10 samples of CD15+ and CD15− tumor cells (n = 5 for each); B) data from (Pomeroy et al., 2002) and C) data from (Thompson et al., 2006). During this process, the CD15+ signature was applied to each sample and a probability of CD15+ status was assigned (range 0 (CD15-) to 1 (CD15+)). For datasets B and C, where the species differed between the training samples (murine) and the test samples (human), Chip Comparer (http://tenero.duhs.duke.edu/genearray/perl/chip/chipcomparer.pl) was used to find common probes across species and microarray platforms and File Merger (http://tenero.duhs.duke.edu/genearray/perl/filemerger.pl) was used to bring together the training and test microarray data; all duplicate gene features were removed prior to analysis.
Supplementary Material
Further information on cell isolation, flow cytometry, neurosphere assays, proliferation assays, RNA isolation, real-time RT-PCR, and microarray analysis is available in Supplementary Methods.
Acknowledgments
The authors thank Jack Dutton for assistance with animal colony maintenance, Beth Harvat for help with flow cytometry, Tannishtha Reya for helpful discussions and Jeremy Rich for critical review of the manuscript. We are also grateful to Tania Contente-Cuomo and Elizabeth Salomon the NIH Neuroscience Microarray Consortium at TGEN for microarray processing and analysis. This work was supported by a fellowship from the University of Bergen (TAR) and by funds from the Duke Stem Cell Research Program, the Pediatric Brain Tumor Foundation, Golfers Against Cancer and NINDS grant number NS052323-01 (RWR).
Abbreviations
- SSEA-1
stage-specific embryonic antigen-1
- TPC
tumor-propagating cell
- GNP
granule neuron precursor
- EGF
epidermal growth factor
- bFGF
basic fibroblast growth factor
- Hh
Hedgehog
- FACS
fluorescence-activated cell sorting
Footnotes
Accession Number: Microarray data are available in NCBI-GEO, under accession number GSE12430.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendoerfer KL, Magnani JL, Patterson PH. FORSE-1, an antibody that labels regionally restricted subpopulations of progenitor cells in the embryonic central nervous system, recognizes the Le(x) carbohydrate on a proteoglycan and two glycolipid antigens. Mol Cell Neurosci. 1995;6:381–395. doi: 10.1006/mcne.1995.1029. [DOI] [PubMed] [Google Scholar]
- Ashwell KW, Mai JK. A transient CD15 immunoreactive sling in the developing mouse cerebellum. Int J Dev Neurosci. 1997;15:883–889. doi: 10.1016/s0736-5748(97)00036-1. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science (New York, NY. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2005 doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics (Oxford, England) 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Capela A, Temple S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron. 2002;35:865–875. doi: 10.1016/s0896-6273(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Capela A, Temple S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Developmental biology. 2006;291:300–313. doi: 10.1016/j.ydbio.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Eberhart CG. In search of the medulloblast: neural stem cells and embryonal brain tumors. Neurosurg Clin N Am. 2007;18:59–69. viii–ix. doi: 10.1016/j.nec.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Fox N, Damjanov I, Knowles BB, Solter D. Immunohistochemical localization of the mouse stage-specific embryonic antigen 1 in human tissues and tumors. Cancer research. 1983;43:669–678. [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York, NY. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- Hall PA, D'Ardenne AJ. Value of CD15 immunostaining in diagnosing Hodgkin's disease: a review of published literature. J Clin Pathol. 1987;40:1298–1304. doi: 10.1136/jcp.40.11.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. 1999;94:2056–2064. [PubMed] [Google Scholar]
- Huntly BJ, Gilliland DG. Cancer biology: summing up cancer stem cells. Nature. 2005;435:1169–1170. doi: 10.1038/4351169a. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH, Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science (New York, NY. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- Kadin ME, Rubinstein LJ, Nelson JS. Neonatal cerebellar medulloblastoma originating from the fetal external granular layer. J Neuropathol Exp Neurol. 1970;29:583–600. doi: 10.1097/00005072-197010000-00005. [DOI] [PubMed] [Google Scholar]
- Katsetos CD, Del Valle L, Legido A, de Chadarevian JP, Perentes E, Mork SJ. On the neuronal/neuroblastic nature of medulloblastomas: a tribute to Pio del Rio Hortega and Moises Polak. Acta Neuropathol (Berl) 2003;105:1–13. doi: 10.1007/s00401-002-0618-5. [DOI] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science (New York, NY. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Lee A, Kessler JD, Read TA, Kaiser C, Corbeil D, Huttner WB, Johnson JE, Wechsler-Reya RJ. Isolation of neural stem cells from the postnatal cerebellum. Nat Neurosci. 2005;8:723–729. doi: 10.1038/nn1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C, Zindy F, Roussel MF, Curran T, Gilbertson RJ, McKinnon PJ. A molecular fingerprint for medulloblastoma. Cancer research. 2003;63:5428–5437. [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, Johnson JE. Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns. 2003;3:389–395. doi: 10.1016/s1567-133x(03)00089-9. [DOI] [PubMed] [Google Scholar]
- Marani E, Tetteroo PA. A longitudinal band-pattern for the monoclonal human granulocyte antibody B4,3 in the cerebellar external granular layer of the immature rabbit. Histochemistry. 1983;78:157–161. doi: 10.1007/BF00489494. [DOI] [PubMed] [Google Scholar]
- McCarthy NC, Simpson JR, Coghill G, Kerr MA. Expression in normal adult, fetal, and neoplastic tissues of a carbohydrate differentiation antigen recognised by antigranulocyte mouse monoclonal antibodies. J Clin Pathol. 1985;38:521–529. doi: 10.1136/jcp.38.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H, Ikawa E, Ohama E. Medulloblastoma in an adult suggestive of external granule cells as its origin: a histological and immunohistochemical study. Brain Tumor Pathol. 1998;15:31–35. doi: 10.1007/BF02482098. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4048–4053. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, Henk B, Schmandt N, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer research. 1997;57:2085–2088. [PubMed] [Google Scholar]
- Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development. 2006;133:3929–3937. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Read TA, Hegedus B, Wechsler-Reya R, Gutmann DH. The neurobiology of neurooncology. Ann Neurol. 2006;60:3–11. doi: 10.1002/ana.20912. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Scott MP. Patching the gaps in Hedgehog signalling. Nat Cell Biol. 2007;9:1005–1009. doi: 10.1038/ncb435. [DOI] [PubMed] [Google Scholar]
- Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, Curran T. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer research. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Skubitz KM, Snook RW., 2nd Monoclonal antibodies that recognize lacto-N-fucopentaose III (CD15) react with the adhesion-promoting glycoprotein family (LFA-1/HMac-1/gp 150,95) and CR1 on human neutrophils. J Immunol. 1987;139:1631–1639. [PubMed] [Google Scholar]
- Solter D, Knowles BB. Monoclonal antibody defining a stage-specific mouse embryonic antigen (SSEA-1) Proceedings of the National Academy of Sciences of the United States of America. 1978;75:5565–5569. doi: 10.1073/pnas.75.11.5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes & development. 2005;19:2656–2667. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer research. 2001;61:513–516. [PubMed] [Google Scholar]
- Xin L, Lawson DA, Witte ON. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6942–6947. doi: 10.1073/pnas.0502320102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Further information on cell isolation, flow cytometry, neurosphere assays, proliferation assays, RNA isolation, real-time RT-PCR, and microarray analysis is available in Supplementary Methods.