

Abstract
Selective inhibition of neuronal nitric oxide synthase (nNOS) has been shown to prevent brain injury and is important for the treatment of various neurodegenerative disorders. This study shows that not only greater inhibitory potency and isozyme selectivity, but more drug-like properties can be achieved by fragment hopping. Based on the structure of lead molecule 6, fragment hopping effectively extracted the minimal pharmacophoric elements in the active site of nNOS for ligand hydrophobic and steric interactions and generated appropriate lipophilic fragments for lead optimization. More potent and selective inhibitors with better drug-like properties were obtained within the design of 20 derivatives (compounds 7-26). Our structure-based inhibitor design for nNOS and SAR analysis reveal the robustness and efficiency of fragment hopping in lead discovery and structural optimization, which implicates a broad application of this approach to many other therapeutic targets for which known drug-like small-molecule modulators are still limited.
Introduction
Fragment-based drug discovery has been recognized as a tangible alternative to more traditional methods of hit identification, such as high throughput screening (HTS).1 This approach was designed to screen a small collection of basic chemical building blocks (fragments) with molecular weights in the range of 120-250 Da and then to enlarge or to link them into high-affinity compounds.2 Recently we established a new fragment-based de novo design strategy,3 termed fragment hopping.4 The core of this approach is the determination of the minimal pharmacophoric elements, which are smaller than fragments, can be a cluster of atoms, virtual graphs and/or vectors. Based on each minimal pharmacophoric element, various fragments with different chemotypes can be generated with the aid of a basic fragment library and a bioisostere library. The fragments corresponding to each pharmacophoric element were then linked/hybridized into larger molecules. The advantages of our new approach are: (1) It can explore a much wider chemical space, and (2) it can effectively characterize and utilize the delicate differences in the active site that are responsible for isozyme selectivity. Consequently, it is able to generate more potent and selective inhibitors for isozymes.
Nitric oxide (NO),5 a highly reactive free radical, is an important and ubiquitous signaling molecule that plays a significant role in the regulation of a diverse set of mammalian physiological processes.6 Generally, NO acts as a potent activator of soluble guanylate cyclase (sGC) via its binding to the heme of the enzyme, which catalyzes the conversion of guanosine 5′-triphosphate (GTP) to an intramolecular second messenger, guanosine 3′,5′-cyclic monophosphate (cGMP).7 This NO/cGMP signaling pathway is essential in many physiological processes including vasodilation, neurotransmission, and platelet aggregation.8 It is also well established that NO has some cGMP-independent actions.9
Nitric oxide is synthesized by a family of enzymes called nitric oxide synthase (NOS, EC 1.14.13.39), which catalyzes an NADPH- and O2-dependent five-electron oxidation of L-arginine to L-citrulline and nitric oxide (NO) via the intermediate NG-hydroxy-L-arginine.10 Three distinct isozymes of NOS have been identified in mammals,11 products of different genes, with different subcellular localization, regulation, catalytic properties, and inhibitor sensitivity. Three mammalian NOSs show 50-60% sequence homology, and each is associated with a different physiological function: neuronal nitric oxide synthase (nNOS), which generates NO in the CNS, is involved in neurotransmission and long-term potentiation; endothelial nitric oxide synthase (eNOS) derived NO is involved in the regulation of smooth muscle relaxation and vascular tone; and a third, inducible isoform in macrophages (iNOS), is important in the immune system defense against pathogens and tumor cells.12
NOS is active as a homodimer with each subunit containing a C-terminal reductase domain (with binding sites for NADPH, FAD, and FMN) and a N-terminal oxygenase domain containing the heme prosthetic group. The substrate L-arginine and a redox cofactor, (6R)-5,6,7,8-tetrahydro-L-biopterin (H4B), both bind near the heme center in the oxygenase domain.13 nNOS and eNOS are constitutively expressed and intermittently produce small amounts of NO. In contrast, iNOS is inducible by cytokines and produces a large amount of NO for both a cytoprotective and cytotoxic effect. The constitutive enzymes bind calmodulin (CaM) reversibly in a Ca2+-dependent manner, and their activity is regulated by local Ca2+ concentration, while iNOS does not depend on the intracellular calcium level because iNOS carries CaM as a permanently bound subunit. Electrons are transferred from NADPH via FAD and FMN to the heme of the other subunit. This flow of electrons during catalysis occurs from the reductase domain of one monomer to the oxygenase domain of the other monomer, so an intact homodimeric form of the enzyme is essential for full enzyme activity.
Overproduction of NO from nNOS has been associated with harmful effects in the central nervous system, including acute (stroke)14 and chronic (Alzheimer’s disease, Parkinson’s disease, schizophrenia, and AIDS dementia) outcomes.15 Radical nitric oxide (NO·) can react rapidly with superoxide (O2·-) in aqueous media to generate the highly reactive peroxynitrite anion (ONOO-). Either nitric oxide or peroxynitrite causes oxidative damage and/or protein nitration and, therefore, leads to neurodegeneration.16 Overproduction of iNOS has been implicated in various pathological processes including septic shock, tissue damage following inflammation, and rheumatoid arthritis.17 In contrast, NO produced by eNOS in endothelial cells has mainly physiological roles, such as maintaining normal blood pressure and flow. 18 Accordingly, inhibition of nNOS or iNOS, but not of eNOS, could provide an effective therapeutic approach.19 On the other hand, selective inhibitors could also be useful tools for investigating other biological functions of NO. 20 The research goal of our laboratories has been to develop selective nNOS inhibitors.21
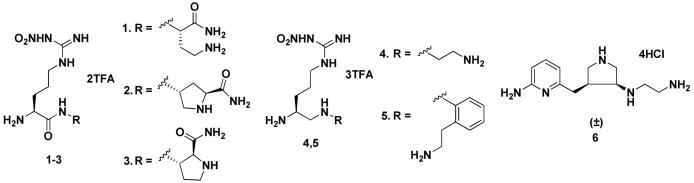
The crystal structures of the dimeric oxygenase domain for all three NOS isozymes have been solved,22 which provide the possibility for structure-based inhibitor design. However, this has proven to be a challenging problem because the active sites of NOS isozymes are highly conserved. Sixteen out of eighteen residues within 6 Å of the substrate binding site are identical, and the side chain of one of these two dissimilar amino acids points out of the substrate-binding site. 23 Previously, we synthesized and evaluated nitroarginine-containing dipeptide or peptidomimetic inhibitors and identified a family of compounds that had high potency and selectivity for inhibition of nNOS over eNOS and iNOS. The most potent nNOS inhibitors among these compounds were 1, 24 2, 25 3, 26 4, 27 and 5 28 (Table 1). The selectivity of the dipeptide/peptidomimetic inhibitors for nNOS over eNOS and/or iNOS was investigated by crystallographic analysis29 and by computer modeling (GRID/CPCA).30
Table 1.
The inhibitory activities of 1-6 toward three isozymes of nitric oxide synthase (NOS)
 | |||||
|---|---|---|---|---|---|
|
Ki (μM) |
Selectivitya |
||||
| No. | nNOS | eNOS | iNOS | n/e | n/i |
| 1 | 0.13 | 200 | 25 | 1538 | 192 |
| 2 | 0.10 | 128 | 29 | 1280 | 290 |
| 3 | 0.087 | 200 | 5.76 | 2299 | 66 |
| 4 | 0.10 | 314 | 39 | 2617 | 325 |
| 5 | 0.05 | 105 | 3.51 | 2121 | 70.2 |
| (±)-6 | 0.388 | 434.5 | 58.4 | 1114 | 150 |
n/e and n/i are the selectivity ratios of inhibition of nNOS/eNOS and nNOS/iNOS, respectively (inhibition of nNOS is greater than eNOS and iNOS in all examples).
Starting with nitroarginine-containing dipeptide inhibitors, fragment hopping derived the minimal pharmacophoric elements and utilized the minute structural differences of the active sites of the three NOS isozymes, leading to the design of a non-peptide small organic molecule 6, which exhibits nanomolar nNOS inhibitory potency and 1000-fold nNOS selectivity over eNOS (Table 1).4 The crystal structure of nNOS in complex with 6 showed a bioactive conformation of 6 as predicted and mimic the mode of action of the dipeptide nNOS inhibitors.4 As an initial attempt of the new methodology, test molecule 6 in Table 1 was successful, but rather polar and not optimized sufficiently to possess in vivo potency and efficacy. In this study, we continued to use fragment hopping to evolve more potent and selective nNOS inhibitors from 6 with increased lipophilicity. The aim of this research was to see whether or not fragment hopping was able to generate more potent and nNOS-selective inhibitors with improved physicochemical and pharmacokinetic profiles. The relationships between structure and activity and/or isozyme selectivity were addressed to interrogate the robustness and effectiveness of the methodology.
Results and Discussion
Inhibitor Design
In fragment hopping, two software programs GRID31 and multiple copy simultaneous search (MCSS)32 are the assistant tools used to explore potential pharmacophoric elements for the binding with the active site. In this study we continued to use these tools to investigate the active site of nNOS based on the scaffold of 6. As discussed earlier4,30, the active site of NOS can be divided into four pockets. The S pocket is above the heme ring, which is the catalytic site for the substrate, L-arginine. The M pocket is in between the substrate catalytic site and the substrate access channel. The C1 and C2 pockets are away from the substrate-binding cavity, and they constitute the substrate access channel. The schematic drawing shown in Figure 1 shows the NOS pockets targeted in this study. The residue numbering for rat nNOS, bovine eNOS, and murine iNOS are used in the following discussion because these are the sources of the NOS for the assay and NOS X-ray crystal structure data.
Figure 1.

Schematic drawing of the active site of nNOS. The binding mode of 6 and the amino acids and cofactors important for inhibitor binding are indicated. Numbering is for the sequence of rat nNOS. The H-bonds observed in the crystal structure are indicated by dotted lines (PDB id: 3B3N). Under physiological conditions, the nitrogen atoms of the pyridine ring, the pyrrolidine ring, and the terminal amino group of the ethylenediamine side chain of 6 in NOS are protonated, but the nitrogen atom attached to the pyrrolidine ring is unaffected.
One fundamental requirement for the development of potent inhibitors of nNOS is that they must cross the blood brain barrier (BBB), a natural bodyguard of the brain. As 6 is a very polar molecule, one of major requirements for the structural optimization of 6 is to increase its lipophilicity. In the GRID analysis, hydrophobic interactions are calculated with the DRY probe. Two hydrophobic areas were identified by the DRY probe (Figure 2A and Table 2). One is a hydrophobic cavity surrounded by M336, L337, Y706, and W306 (from the second monomer) in the C1 pocket. The other one is a small cavity lined with the side chains of P565, A566, V567, and F584 in the S pocket. The C3, C1=, and NM3 probes describe steric interactions. Two significant molecular interaction fields (MIFs) in the active site of nNOS for steric probes have been described.30 One is in the S pocket, and the other is located in the M pocket. However, compared to the position of the 2-aminopyridine group of 6 in the S pocket (Figure 2B), there is more space around position 4 of the 2-aminopyridine ring where the maximal molecular interaction energies are placed for the steric probes (-5.00 kcal/mol for the C3 probe, -5.30 kcal/mol for the C1= probe, and -12.50 kcal/mol for the NM3 probe). Furthermore, the cavity lined by M336, L337, Y706, and W306 in the C1 pocket is also favorable for van der Waals contact (the maximal interaction energies are -3.50 kcal/mol, -3.30 kcal/mol and -8.20 kcal/mol for the C3, C1= and NM3 probes, respectively), as shown in Figure 2B and Table 2. The N1+ and N2+ probes were utilized to explore the possibility of substitution on the amino groups of 6. It suggests that it is possible that the terminal amino group of the ethylenediamine side chain of 6 be modified into an sp3-hybridized NH cation (the maximal interaction energy is -10.00 kcal/mol in Table 2) or an sp3-hybridized NH2 cation (the maximal interaction energy is -10.50 kcal/mol in Table 2). Halogen atoms are often considered, in structural optimization, to enhance potency/selectivity and in vivo efficacy. Two organic halogen probes Cl and F were used to explore the potential MIFs in the active site of nNOS, as indicated in Table 2.
Figure 2.
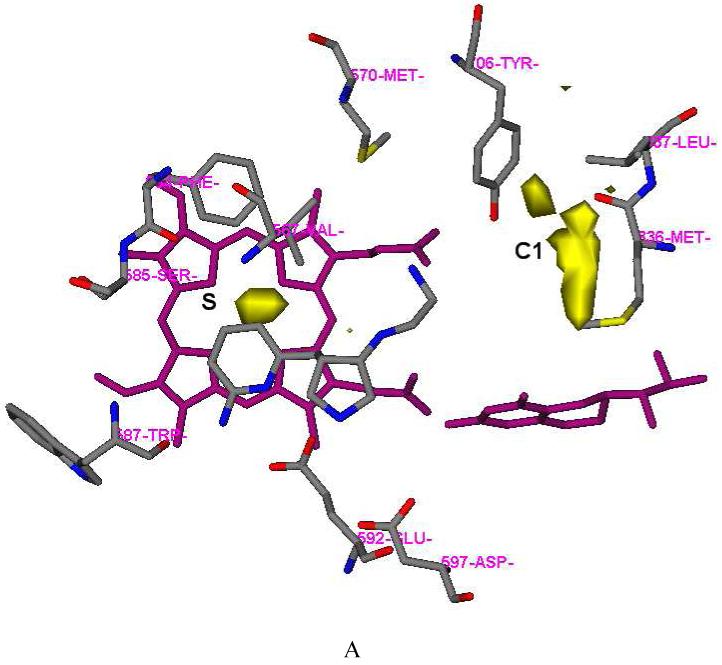
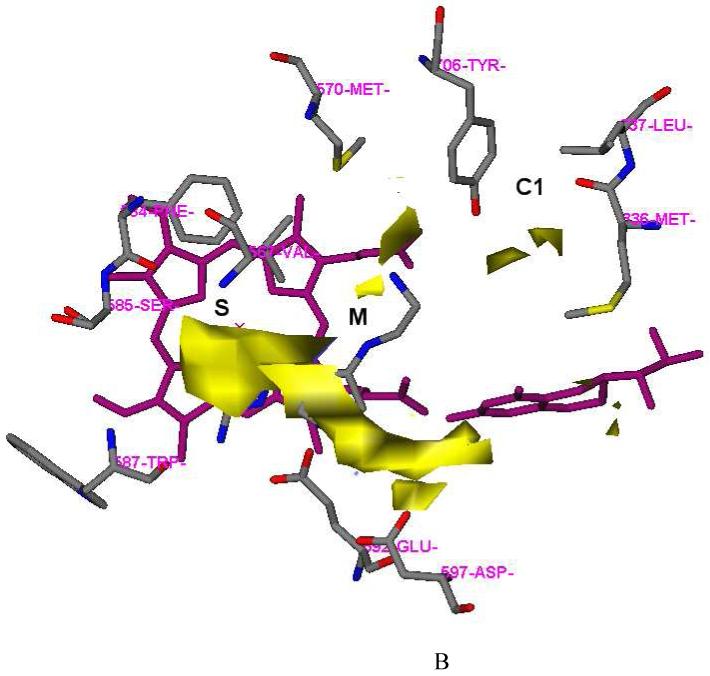
Results of GRID analysis of the substrate binding site of nNOS (PDB id: 1P6I). The residues and inhibitor 6 are represented in an atom-type style. Heme and H4B are colored magenta. The S, M, and C1 pockets are indicated. A: GRID contours of the DRY probe at an energy level of -0.75 kcal/mol. B: GRID contours of the C3+ probe at an energy level of -2.75 kcal/mol; “×” represents the position of the heme iron atom.
Table 2.
Summary of the main probe interactions observed in the active site of rat nNOS. The relevant residues and cofactors and the maximal interaction energy involved in a particular interaction are given. Heme propionate A and D means the heme propionate of the pyrrole A ring and D ring, respectively
| Probe | Chemical group | Maximal energy (kcal/mol) | Main residues and cofactors | |
|---|---|---|---|---|
| DRY | hydrophobic probe | C1 | -1.85 | Y706, L337, M336 , W306 (the other NOS monomer ) |
| S | -0.80 | P565, A566, V567, F584, S585, G586, heme | ||
| C3 | methyl group | S | -5.00 | P565, A566, V567, F584, S585, G586, heme |
| M | -4.00 | D597, heme propionate A | ||
| C1 | -3.50 | Y706, L337, M336, W306 (the other NOS monomer) | ||
| C1= | sp2 CH aromatic or vinyl | S | -5.30 | P565, A566, V567, F584, S585, G586, heme |
| M | -4.0 | D597, heme propionate A | ||
| C1 | -3.30 | Y706, L337, M336, W306 (the other NOS monomer) | ||
| NM3 | trimethylammonium cation | S | -12.50 | P565, A566, V567, F584, S585, G586, heme |
| M | -10.00 | D597, heme propionate A | ||
| C1 | -8.20 | Y706, L337, M336, W306 (the other NOS monomer) | ||
| N1+ | sp3 amine NH cation | M | -11.00 | D597, Y588 |
| M | -10.00 | heme, Y706 | ||
| S | -9.50 | E592 | ||
| M | -8.50 | heme propionate A and D | ||
| N2+ | sp3 amine NH2 cation | M | -13.50 | D597, Y588 |
| S | -12.00 | E592, W587 | ||
| M | -10.50 | heme propionate A and D | ||
| Cl | organic chorine atom | S | -7.50 | P565, A566, V567, F584, S585, G586, heme |
| M | -6.00 | D597, heme propionate | ||
| C1 | -5.20 | Y706, L337, M336, W306 (the other NOS monomer) | ||
| F | organic fluorine atom | M | -4.80 | D597, heme propionate A |
| S | -4.30 | P565, A566, V567, F584, S585, G586, heme | ||
| C1 | -2.70 | Y706, L337, M336, W306 (the other NOS monomer) |
Five apolar and hydrophobic functional groups (benzene, cyclohexane, isobutane, n-butane, and propane) have been mapped into the active site of nNOS by MCSS calculations. As discussed previously,4 the minima with the most favorable interaction energies for benzene, cyclohexane, and isobutane are located in the S pocket. This study shows that the minima with the most favorable interaction energies for n-butane (minima no. 1-no. 11: -10.85 kcal/mol ∼ -8.58 kcal/mol) and propane (minima no. 1-no. 5: -9.99 kcal/mol ∼ -8.71 kcal/mol) are also located in the S pocket lined by P565, A566, V567, F584, S585, G586, and the heme (Figure 3 and Table 3). Comparing the relative positions of the 2-aminopyridine ring of 6, there is more space around position 4 of the 2-aminopyridine ring in the S pocket, as indicated by the heavy atom-only models (cyclohexane, isobutane, propane, and n-butane), which are appropriate to explore steric effects (Figure 3). Further analysis shows that there are minima displaying favorable interaction energies for the above five functional groups in the C1 pocket lined by M336, L337, Y706, and W306, as shown in Figure 3. Four of 27 minima of the benzene group are located in this pocket, and the lowest interaction energy is -13.47 kcal/mol (minimum no. 7). Others in this C1 pocket are 12 of 98 minima of the cyclohexane group (minimum no. 6: -7.08 kcal/mol), 4 of 22 minima of the isobutane group (minimum no. 13: -6.12 kcal/mol), 32 of 224 minima of the n-butane group (minimum no. 52: -5.70 kcal/mol), and 9 of 84 minima of the propane group (minimum no. 14: -5.71 kcal/mol). The trimethylamine cation and dimethylamine cation were also used to explore the possibility of substitution on the amino groups of 6 in the MCSS calculations. It is suggested in Table 3 that the terminal amino group of 6 can be modified to an sp3-hybridized NH cation (minima no. 2: -82.79 kcal/mol and no. 7: -73.98 kcal/mol for trimethylamine cation) or an sp3-hybridized NH2 cation (minima no. 1: -131.52 kcal/mol and no. 11: -102.92 kcal/mol for dimethylamine cation).
Figure 3.
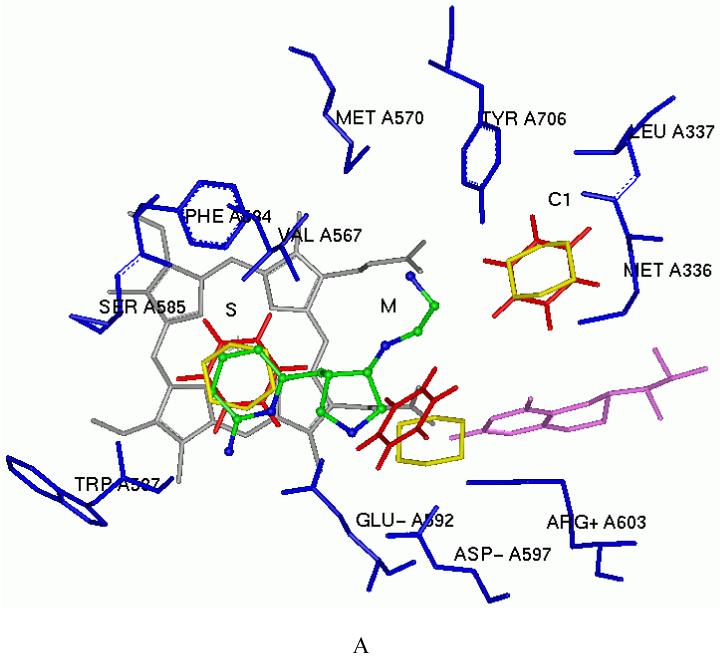
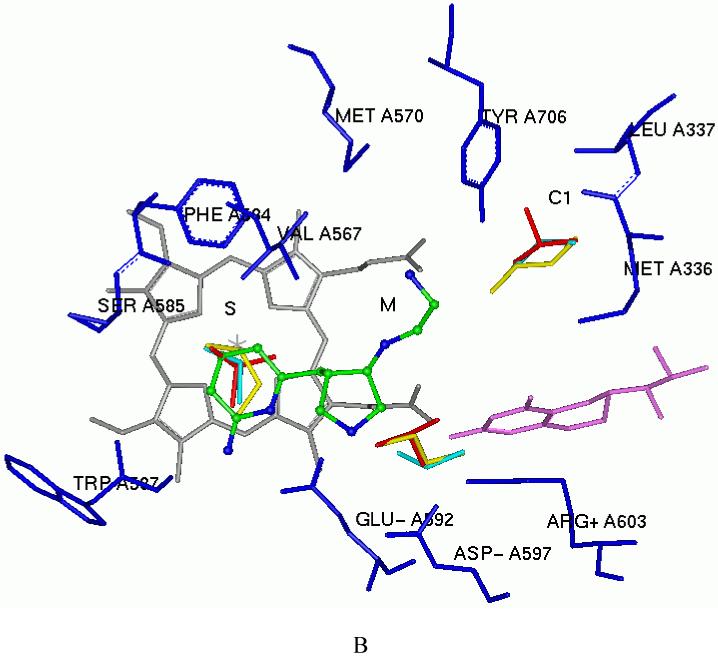
Representative MCSS-minimized positions of the functional groups in the active site of rat nNOS (PDB id: 1P6I). Cofactors heme and H4B are shown in gray and magenta, respectively. Inhibitor 6 is shown in the ball-and-stick model (an atom-type style). The S, M, and C1 pockets are indicated. A: Benzene (red, minimum No. 1 in the S pocket, minimum No. 7 in the C1 pocket, and minimum No. 8 in the M pocket); cyclohexane (yellow, minimum No. 1 in the S pocket, minimum No. 3 in the M pocket, and minimum No. 6 in the C1 pocket). B: isobutane (red, minimum No. 1 in the S pocket, minimum No. 3 in the M pocket, and No. 13 in the C1 pocket); n-butane (yellow, minimum No. 1 in the S pocket, minimum No. 12 in the M pocket, and minimum No. 52 in the Cl pocket); and propane (cyan, minimum No. 1 in the S pocket, minimum No. 6 in the M pocket and minimum No. 14 in the C1 pocket).
Table 3.
Minima found by MCSS in the active site of rat nNOS
| MCSS functional groups | Initial number of copies | Number of Minima | Range of Ubinda for minima (kcal/mol) | Selected minima | ||
|---|---|---|---|---|---|---|
| from | To | Ubind; Pocket | residues/cofactors involved | |||
| Benzene | 2500 | 27 | -1.75 | -24.16 | No.1: -24.16; S | V567, F584, heme |
| No.7: -13.47; C1 | M336, L337, Y706, W306 (the other NOS monomer) | |||||
| No.8: -12.99; M | D597, heme propionate A | |||||
| Cyclohexane | 5000 | 98 | -0.09 | -9.80 | No.1: -9.80; S | V567, F584, heme |
| No.3: -9.02; M | D597, heme propionate A | |||||
| No.6: -7.08; C1 | M336, L337, Y706, W306 (the other NOS monomer) | |||||
| isobutene | 5000 | 22 | -5.75 | -11.29 | No.1: -11.29; S | V567, F584, heme |
| No.3: -7.33; M | D597, heme propionate A | |||||
| No.13: -6.12; C1 | M336, L337, Y706, W306 (the other NOS monomer) | |||||
| n-butane | 5000 | 224 | -2.82 | -10.86 | No.1: -10.85; S | V567, F584, heme |
| No.12: -7.79; M | D597, heme propionate A | |||||
| No.52: -5.70; C1 | M336, L337, Y706, W306 (the other NOS monomer) | |||||
| Propane | 5000 | 84 | -1.55 | -9.99 | No.1: -9.99; S | V567, F584, heme |
| No.6: -6.50; M | D597, heme propionate A | |||||
| No.14: -5.71; C1 | M336, L337, Y706, W306 (the other NOS monomer) | |||||
| trimethylamine ion | 5000 | 28 | -2.18 | -87.43 | No.1: -87.43; M | D597, E592 |
| No.2: -82.79; M | heme propionate A and D | |||||
| No.4: -80.64; M | heme propionate A | |||||
| No.7: -73.98; M | heme propionate D | |||||
| No.22: -32.36; S | E592 | |||||
| dimethylamine ion | 5000 | 35 | -0.31 | -131.52 | No.1: -131.52; M | heme propionate A and D |
| No.6: -126.30; M | E592, D597 | |||||
| No.8: -110.59; M | heme propionate A | |||||
| No.11: -102.92; M | heme propionate D | |||||
Ubind, the binding energy for a given functional group in each minimized replica obtained from the MCSS calculations, is defined as Ubind = Uprotein-group+Ugroup0, where Uprotein-group represents the nonbonded interactions between nNOS and the given functional group. Ugroup represents the internal energy of the functional group within the complex, and Ugroup0 represents the internal energy of the isolated functional group in vacuum.
The above GRID and MCSS calculations show that there is a small steric and hydrophobic cavity around position 4 of the 2-aminopyridine ring of 6, which is lined with the side chains of P565, A566, V567, and F584 in the S pocket, and a hydrophobic cavity, lined with M336, L337, Y706, and W306 (from the second monomer) in the C1 pocket. These two hydrophobic/steric regions were used to generate the minimal pharmacophoric elements for better ligand potency, as well as better physicochemical and pharmacokinetic profiles. The structural modification targeting the S pocket is the introduction of a methyl group at position 4 of the 2-aminopyridine ring of 6. The structural modification for the C1 pocket is the introduction of a phenyl group or a substituted phenyl group at the ethylenediamine side chain of 6. Keeping the above minimal pharmacophoric elements in mind, several compounds were generated with the aid of the LUDI program33 using the focused fragment libraries generated in the previous study.4 To better understand the structure-activity relationship of this series of inhibitors, the design process was divided into three stages (Figure 4). Stage A is the continuing study of the structure-activity relationships based on the structure of 6; nine compounds were designed (Figure 4A). Stage B is the study of structure-activity relationships based on the structure of 15; thirteen compounds were designed (Figure 4B). Stage C is the structural modification of 16 and 26 to increase molecular lipophilicity and decrease molecular polar surface area (PSA); four compounds were designed (Figure 4C).
Figure 4.

Molecules designed and synthesized.
Chemistry
Scheme 1 shows the synthetic route for 7-14. The synthesis of 36 has been described in a previous paper.4 Direct reductive amination reactions were used to generate 37. When 3 Å molecular sieves were used as a water trap, anhydrous MeOH was used as the solvent, one equivalent of acetic acid was used as the proton source, and sodium cyanoborohydride (NaBH3CN) was used as the reducing agent, the yield of product was about 45-65%. The ratio of the cis to trans isomers was 45:55 for 7-9, 11, and 12, but 18:82 for 10, 13, and 14. When anhydrous MeOH was used as the solvent, one equivalent of acetic acid was used as the proton source, and catalytic hydrogenation (platinum IV oxide as catalyst) was used, the yield of product was still the same. However, the ratio of the cis and trans isomers was changed to 55:45 for 7-9. When 3 Å molecular sieves were used as the water trap, 1,2-dichloroethane was used as the solvent, and sodium triacetoxyborohydride [NaBH(OAc)3] was the reducing agent, the yield of product was increased to 80-95%. The ratio of the cis and trans isomers was changed to 75:25 for 7-9, 11, and 12, and 45:55 for 10, 13, and 14. The cis and trans isomers can be completely separated by silica gel column chromatography. As discussed earlier,4 the 1HNMR and 13CNMR spectra of the cis and trans isomers are quite different for each isomer. One prominent difference is that the 13C chemical shift of the methylene carbon atom attached to the pyridine ring is 29 ppm for the cis products, but 34 ppm for the trans isomers. The side chains of 41-43 were synthesized by Scheme 2B. The amino group of 38-40 was selectively protected with (Boc)2O in 1M NaOH aqueous solution. Then the hydroxyl group was converted into an azido group by a Mitsunobu reaction. After hydrolysis of the azido compounds with triphenylphosphine in a THF/H2O mixture or by catalytic hydrogenation using 10% Pd/C, the pure compounds (41-43) can be obtained by silica gel column chromatography using a mixture of CH2Cl2 and MeOH as eluent.
Scheme 1.

Scheme 2.

The synthesis of 15-26 and 29 is shown in Scheme 2. The synthesis of 46 was described in a previous paper.23 The amine compounds (47) were prepared by reductive amination using NaBH(OAc)3 as the reducing agent as mentioned above. The cis and trans isomers can be completely separated by silica gel column chromatography. The ratio of the cis and trans isomers was still 75:25. As discussed above, the cis and trans isomers were characterized and assigned by 1HNMR and 13CNMR spectra. Deprotection of the Boc groups of 47 afforded final products 15-26 and 29. The synthesis of the amine side chains 53 are shown in Scheme 3B. The substituted phenylacetaldehydes were condensed with ethanolamine and then reduced with sodium borohydride (NaBH4) to generate 48 in high yields (≥ 94%), whereas direct reductive amination affords much lower yields (about 67%) if NaBH3CN is used as the reducing agent, 3 Å molecular sieves as a water trap, one equivalent of acetic acid as the proton source, and anhydrous MeOH as the solvent. In the synthesis of 50, direct reductive amination affords quantitative yields of the desired product. Benzyl protection of ethanolamine is necessary; otherwise, the dibenzylethyl substituted product will be obtained after reductive amination.
Scheme 3.

The syntheses of 30 and 31 were achieved by two different synthetic routes shown in Scheme 3A. By one way, 47b or 47m was subjected to reductive amination to generate methylated derivative 54 using formaldehyde as the carbon source, acetic acid as the proton source, and NaBH3CN as the reducing reagent. The second approach was a direct reductive amination using NaBH(OAc)3 as the reducing reagent. 46 was allowed to react with 57 or 60 to generate cis isomer 55 exclusively. The synthesis of 32 was only carried out by the latter synthetic route. The synthesis of amine side chain 57 is shown in Scheme 3B. Alcohol 53 was oxidized to 56 with Dess-Martin periodinane. Then the aldehyde was converted to the secondary amine by an indirect reductive amination in almost a quantitative yield. In Scheme 3C, N1,N2-dimethyl-ethane-1,2-diamine was first monoprotected with Boc anhydride. Then it was treated with aldehyde 49 to generate 59; the Boc group of 59 was deprotected with TFA, and then the compound was basified to afford 60.
In the early stage of this study, we discovered that trans alcohol 35 or 45a were not able to be converted into the cis amine (37 or 47) by any SN2 reaction, for example a Mitsunobu reaction or attempts to activate the hydroxyl group of 35 or 45a as a tosylate, methylate, or triflate followed by displacement with a nitrogen-containing nucleophile. The only product generated from these attempted reactions is a new cis five-membered ring (a quarternary ammonium) that occurs by intramolecular attack of the nitrogen atom of the 2-aminopyridine on the carbon atom attached to the hydroxyl group. Consequently, alcohol 35 or 45a were oxidized to ketones, and then reductive amination was carried out to generate 37, 47, and 55 as shown in Schemes 1-3. The disadvantage of these synthetic routes is that both the cis and trans isomers are generated, although they can be effectively separated by silica gel column chromatography. Later, we discovered that the Mitsunobu reaction can proceed smoothly when the Boc protected 2-amino group of the 2-aminopyridine moiety of 35 is further protected with a benzyl group.34 In this study, we continued to use a benzyl group to protect 45a to generate 61. 61 was successfully converted to azide 62 by a Mitsunobu reaction (Scheme 4). Azide was chosen as the nitrogen source in the Mitsunobu reaction because the benzyl group can be removed concurrently with reduction of the azide to amine 63 when Pd(OH)2 is used as the catalyst in a hydrogenation reaction. 63 was allowed to react with 56 to afford 47 by reductive amination. Compounds 16 and 26 were re-synthesized by this synthetic route.
Scheme 4.

Compound 45b can be directly converted to 64 by a Mistunobu reaction because the potential cyclization reaction discussed above is unfavorable for a seven-membered ring (Scheme 5). Azide 64 can be reduced to 65 with triphenylphosphine in THF and water. The reductive amination of 65 with 56 afforded 66. The final products (27 and 28) were generated after standard deprotection of 66 using 4M HCl in 1,4-dioxane.
Scheme 5.

All of the above chemical structures were confirmed by 1HNMR, 13CNMR, and high-resolution mass spectra as well as some by elemental analysis. All of the products are racemic mixtures.
NOS Inhibition and Structure-Activity Relationships
Table 4 shows the results of the NOS enzyme inhibition assays. None of compounds 7-14 is more potent than lead compound 6. However, the nNOS/iNOS selectivities of 7, 9, and 10-12 are comparable to that of 6, and the nNOS/eNOS selectivities of 7-9 are similar to that of 6. The nNOS potency and selectivity of 7 is higher than those of 8 and 9. Based on molecular docking, the dimethyl-substituted derivative (10) can form a H-bond interaction with the heme propionate through its amine attached to the pyrrolidine ring, and charge-charge interactions with heme propionates through the terminal tertiary amine when protonated in the active site of nNOS, just as 6 does. However, tertiary terminal amine 10 is not able to undergo H-bond interactions with heme propionate D and a conserved structural water molecule observed in the crystal structure of 6 in complex with nNOS (PDB id: 3B3N).35 Based on the enzyme assay results in Table 4 these interactions appear to be important for inhibitor binding. On the other hand, conformationally-constrained molecules were designed to bind to nNOS because they can minimize the entropic penalty typically associated with binding of a flexible inhibitor and can enhance the free binding energy of the inhibitor. Compared to the dipeptide or peptidomimetic inhibitors, such as 1-5 in Table 1, some parts of the structure of 6 have been conformationally constrained.4 A further modification for conformational constraint was the introduction of a ring at the terminal amino group of 6, generating 11-14. The enzyme assay results in Table 4 show that this structural modification does not work well.
Table 4.
Inhibition of NOS isozymes by synthetic inhibitors
|
Ki (μM)* |
Selectivity |
||||
|---|---|---|---|---|---|
| Compounds | nNOS | eNOS | iNOS | n/e | n/i |
| (±)-7 | 1.18 | 1450 | 161 | 1230 | 136 |
| (±)-8 | 2.42 | 1450 | 238 | 600 | 98 |
| (±)-9 | 2.3 | 1450 | 297 | 632 | 129 |
| (±)-10 | 3.33 | 140 | 195 | 42 | 59 |
| (±)-11 | 1.36 | 424 | 200 | 312 | 147 |
| (±)-12 | 1.26 | 243 | 174 | 193 | 138 |
| (±)-13 | 2.74 | 181 | 425 | 66 | 155 |
| (±)-14 | 3.34 | 108 | 222 | 32 | 66 |
| (±)-15 | 0.098 | 283 | 5.84 | 2890 | 60 |
| (±)-16 | 0.085 | 85 | 8.95 | 1000 | 106 |
| (±)-17 | 0.104 | 83 | 9.48 | 800 | 91 |
| (±)-18 | 0.197 | 84 | 14.1 | 428 | 72 |
| (±)-19 | 0.144 | 130 | 7.97 | 905 | 55 |
| (±)-20 | 0.206 | 116 | 13.6 | 564 | 66 |
| (±)-21 | 0.159 | 72 | 9.01 | 451 | 57 |
| (±)-22 | 0.157 | 41 | 3.56 | 260 | 23 |
| (±)-23 | 0.110 | 30 | 7.7 | 272 | 70 |
| (±)-24 | 0.024 | 78 | 5.4 | 3230 | 222 |
| (±)-25 | 0.027 | 18 | 4.6 | 656 | 170 |
| (±)-26 | 0.014 | 28 | 4.06 | 2000 | 290 |
| (±)-27 | 18.8 | 141 | 232 | 7.5 | 12 |
| (±)-28 | 0.551 | 164 | 10.6 | 297 | 19 |
| (±)-29 | 0.136 | 40.0 | 12.2 | 294 | 90 |
| (±)-30 | 0.393 | 125 | 22.8 | 319 | 58 |
| (±)-31 | 0.326 | 92.4 | 38.2 | 284 | 117 |
| (±)-32 | 1.21 | 48.9 | 56.6 | 40 | 47 |
The apperent Ki values are represented as the mean from two or more independent experiments preformed in duplicate with five or six data points each and correlation coefficients of 0.89-0.99. The experimental standard deviations were less than 10%.
The structural modification made to target the steric and hydrophobic cavity defined by F584, P565, V567, and A566 in the S pocket was the addition of a methyl group at position 4 of the 2-aminopyridine ring of 6 to generate 15. The nNOS potency and nNOS selectivity over eNOS of 15 quadrupled and doubled, respectively, by introduction of only one carbon atom.
The active site of nNOS contains many polar residues, cofactors, and clusters of acidic residues.23 It is reasonable that the environment in the active site of NOS is both polar and acidic, because the natural substrate L-arginine, a polar and basic residue, needs the polar and acidic environment to stabilize it in the active site. However, the requirement of an inhibitor to bind to a polar and acidic environment raises a large problem for the design of an exogenous inhibitor that also can pass the BBB by passive absorption. The polar residues in the NOS active site require the inhibitor to contain polar functional groups, and the acidic environment requires the inhibitor to contain positively charged functional groups, which are generally inappropriate for BBB penetration. Further structural modification of 6 was directed at this problem. When 7 is docked into the active site of nNOS, Figure 5 shows that the terminal phenyl group is close to the hydrophobic cavity defined by Y706, M336, L337, and W306. A secondary amino group in 7 is more lipophilic and has less polar surface area, compared to a primary amino group.36 This functional group has much better physicochemical properties for passing biomembranes than 6, as shown in Table 5. Further structural modifications are based on the chemical structure of 7. A molecular docking study of 7 showed that the benzyl group can adopt several different orientations in the C1 pocket because it is not long enough to form perfect interactions with the hydrophobic cavity in the C1 pocket. Two strategies were employed in the modification of the ethylenediamine side chain: (1) substituted benzyl groups were introduced at the terminal amino group of 15 to generate 16-23 in Figure 4; (2) one more carbon atom was inserted between the terminal amino group of 15 and the benzyl group to form a phenylethyl group (24-26 in Figure 4).
Figure 5.
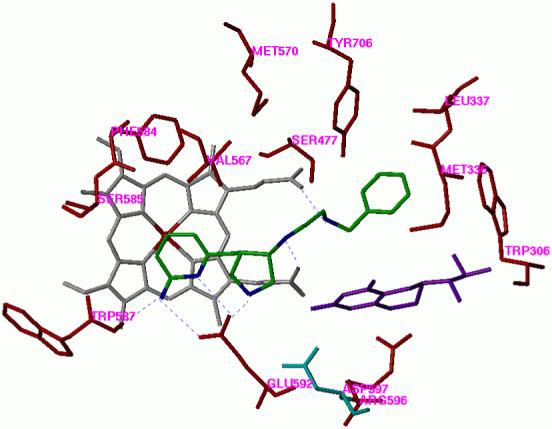
The predicted binding conformation of 7 in the active site of nNOS. Cofactors heme and H4B are shown in gray and purple, respectively. Inhibitor 7 is shown in an atom-type style. The potential H-bonds are indicated by dotted lines.
Table 5.
The physicochemical data related to the absorption and biomembrane permeability of inhibitors
| Compounds | ClogP | TPSA | ClogD (pH=7.4) | Hb-A | Hb-D | MW | RB |
|---|---|---|---|---|---|---|---|
| 1 | -7.000 | 217.968 | -5.42 | 10 | 10 | 318.338 | 9 |
| 2 | -6.267 | 203.972 | -6.57 | 10 | 9 | 330.349 | 7 |
| 3 | -6.172 | 203.972 | -6.4 | 10 | 9 | 330.349 | 7 |
| 4 | -5.787 | 157.803 | -5.88 | 7 | 8 | 247.303 | 8 |
| 5 | -3.270 | 157.803 | -4.07 | 6 | 8 | 323.401 | 8 |
| 6 | -1.329 | 88.992 | -5.16 | 4 | 6 | 235.331 | 5 |
| 7 | 1.364 | 74.996 | -2.82 | 4 | 5 | 325.456 | 8 |
| 16 | 2.576 | 74.996 | -1.81 | 4 | 5 | 373.928 | 8 |
| 17 | 2.576 | 74.996 | -1.72 | 4 | 5 | 373.928 | 8 |
| 24 | 2.052 | 74.996 | -2.44 | 4 | 5 | 353.509 | 9 |
| 25 | 2.195 | 74.996 | -2.38 | 4 | 5 | 371.5 | 9 |
| 26 | 2.195 | 74.996 | -2.36 | 4 | 5 | 371.5 | 9 |
| 29 | 2.885 | 74.996 | -1.49 | 4 | 5 | 387.955 | 8 |
| 30 | 3.220 | 66.207 | -0.91 | 4 | 4 | 387.955 | 8 |
| 31 | 2.838 | 66.207 | -1.81 | 4 | 4 | 385.527 | 9 |
| 32 | 3.432 | 57.418 | -1 | 4 | 3 | 399.554 | 9 |
Hb-A: sum of H-bond acceptors; Hb-D: sum of H-bond donors; MW: Molecular Weights; RB: number of freely rotating bonds. ClogP values for the inhibitors were calculated by ClogP program version 4.2 in the SYBYL 6.8 environment with a special license from BioByte Corporation.37 Log D values for the inhibitors were calculated by ACD/logD at pH 7.4.38 The topological molecular polar surface areas (TPSA) were calculated by the atom-based method of Ertl, et al.36
Among the derivatives 16-21, the 4-chloro substituted derivative (16) and the 3-chloro substituted derivative (17) show good inhibitory potency for nNOS. Compared to 15, compound 16 shows twice the selectivity for nNOS over iNOS. The greater iNOS selectivity is probably triggered by the polarity difference between the corresponding residues at that site of the isozymes, rat nNOS L337 and murine iNOS N115 (human iNOS T121). The nNOS inhibition and selectivity of the dichloro-substituted analogues 22 and 23 are not as good as those of 16, possibly due to the steric crowding. The crystal structures of 16 in complex with nNOS and eNOS have been solved. The binding conformation of 16 observed in the crystal structure of nNOS is very similar to that predicted (PDB id: 3B3O),23,35 and is very similar to the binding mode of dipeptide inhibitors 1, 2, and 4 in nNOS.29
The phenylethyl-substituted derivative 24 shows higher inhibitory potency for nNOS compared to 16. The Ki value for the racemic mixture is 24 nM and its nNOS selectivity over eNOS is over 3000 fold. The nNOS/iNOS selectivity of 24 is increased four times compared with that of 15. That is also caused by the selectivity of residue nNOS L337 versus iNOS N115. The hydrophilicity of asparagine is incompatible with the phenyl group of 24. The nNOS/eNOS selectivity of 24 is also increased in comparison with those of 15 and 16. The docking mode of 24 in the active site of nNOS is shown in Figure 6A.
Figure 6.

A. The docking conformation of 24 in the active site of rat nNOS. Inhibitor 24 is shown in an atom-type style. B. The superimposition of the docking conformation of 25 and 26 in the active site of rat nNOS. Inhibitor 25 is colored yellow and 26 colored blue. Cofactors heme and H4B are shown in gray and purple, respectively. The potential H-bonds are indicated by dotted lines.
One potential metabolism problem of 24 is its electron-rich phenyl ring, which can be metabolized in vivo by cytochrome P-450 enzymes to generate the 4-hydroxyl substituted derivative, as predicted by Metasite 2.5.3.39 The introduction of a halogen atom at position 4 of the ring will help the metabolic stability. Computer modeling shows that the enzyme at position 4 of the bound compound is not large enough to accommodate even a fluorine atom. The superimposition of the docking conformations of 25 and 26 clearly shows that the ethylenediamine side chain of 25 has to adopt a curled conformation to accommodate its 4-fluorophenyl group into the hydrophobic cavity defined by Y706, L337, M336, and W306 (Figure 6B). However, position 3 of the phenyl group contains an extra small pocket defined by L337 and Y706, which is appropriate for a fluorine atom. The result of the NOS inhibitory activity assay (Table 4) confirms that 26 is a better nNOS inhibitor than 25. The nNOS/eNOS selectivity of 26 is also much better than that of 25. Compared to 25, 26 might be a little more vulnerable to metabolism, but it is much more stable than 24.
Compounds 27 and 28 are the 2-amino-6-methylpyridino analogues of 16 and 26, respectively. The NOS enzyme assay shows that the nNOS inhibitory activities and selectivities are decreased dramatically in 27 and 28. Compared to 16 and 26, the 6-methyl groups of 27 and 28 can form van der Waals contacts with the hydrophobic cavity defined by F584, P565, V567, and A566 in the S pocket, and the 2-amino group can also form H-bond interactions with the backbone amide of W587. A more than 200-fold drop in inhibitory potency in comparing 27 with 16 suggests that the charge-charge and H-bond interactions between the protonated pyridine nitrogen atom and E592 are important for the binding of this series of inhibitors with nNOS.
To provide inhibitors with better biomembrane permeability, methylation of one of two amino groups of the ethylenediamine side chain of 16 and 26 to generate compounds 30-32 was carried out. Compound 30 is the methylated derivative of 16. The nNOS potency is decreased four-fold, while the nNOS/eNOS and nNOS/iNOS selectivities are decreased three-fold and two-fold, respectively. The nNOS inhibitory activities and nNOS selectivities of 31 and 32 are decreased dramatically compared to those of 26, although they have improved physiochemical properties for biomembrane penetration. Compound 29 was designed to increase lipophilicity (see Table 5) by introduction of an alkyl group on the carbon atom adjacent to the amino group. The nNOS inhibition of 29 is comparable to that of 16. However, the selectivity between nNOS and eNOS is decreased by three fold, while the selectivity between nNOS and iNOS is maintained.
Conclusion
This study of structure-activity relationships of 2-aminopyridine analogues demonstrates that a significant improvement in nNOS inhibitory activity, nNOS/eNOS selectivity, and physicochemical/pharmacokinetic properties can be achieved by our structure-based de novo design strategy, fragment hopping. Using the extracted minimal pharmacophoric elements for hydrophobic and steric interactions, fragments were generated from focused fragment libraries, and lead compound 6 was evolved into high potent and selective inhibitors with better drug-like properties. Two hydrophobic/steric pockets that were identified by fragment hopping were verified by inhibitor design.
Halogen-substituted phenyl fragments were introduced at the terminal amino group of the ethylenediamine fragment for three reasons: (1) The phenyl group can be stabilized in a hydrophobic pocket defined by M336, L337, Y706, and W306. The residues that correspond to rat nNOS L337 are N115 in murine iNOS and T121 in human iNOS, respectively. Therefore, the hydrophobic aryl group could be used to differentiate the hydrophobic L337 in nNOS from the polar N115 and T121 residues in murine and human iNOS, respectively. (2) The introduction of the phenylalkyl group renders the amino group of the ethylenediamine fragment a secondary amine and decreases its polar surface area, which is favorable for biomembrane permeability, compared to the primary amino group in 6.36 (3) The introduction of a halogen atom at the para or meta positions of the phenyl group blocks/decreases the potential for metabolic degradation of the phenyl group as well as increases its lipophilicity. Compounds 16 and 26, which are the best of the substituted benzyl and substituted phenethyl derivatives, respectively, were tested in a rabbit model for cerebral palsy and found to prevent hypoxia-ischemia induced deaths and to reduce the number of newborn kits exhibiting symptoms of cerebral palsy.23 Following maternal administration in a rabbit model, these two compounds were found to distribute to fetal brain, to be non-toxic, without cardiovascular effects, inhibit fetal brain NOS activity in vivo and dramatically ameliorate deaths and the number of newborn kits exhibiting signs of cerebral palsy without affecting the eNOS regulated blood pressure.23
In a previous study4 we demonstrated that fragment hopping can be used to generate a series of new inhibitors with different chemical structures. This study demonstrates that fragment hopping also can facilitate a structural optimization process. Greater inhibitory potency, isozyme selectivity, and improved drug-like properties can be achieved by a couple rounds of substitution of multiple functionality or cores onto a single high-affinity lead. The structure-activity relationship analysis matched the extracted pharmacophoric elements. The robustness and efficiency of fragment hopping in these studies show that it can be widely applicable to expedite the lead discovery and structural optimization efforts for many other targets for which known small molecule modulators are limited.
Recently, a crystallography-based approach for the design of selective iNOS inhibitors called anchored plasticity was reported.40 The general idea is that the “head” of the inhibitor binds directly over the heme, where it is anchored in place by the isozyme-invariant Glu residue. The “tail” of the inhibitor then extends out of the active site, where subtle differences in the NOS isoforms lead to isoform selectivity. This is basically the same approach we took in our earlier studies using nitroarginine as the anchor for the design of nNOS-selective inhibitors, which became the basis for fragment hopping.41 However, unlike fragment hopping, in the anchored plasticity model, conformational changes, which involve residues that both directly contact the bulky tail of the inhibitor as well as second and third tier amino acids, are required to control isoform selectivity. Some key third tier amino acids that differ between eNOS and iNOS were shown to be critical in controlling greater potency in iNOS inhibition compared to eNOS. Although the two approaches share common features, there are distinct differences. Unlike anchored plasticity, our fragment hopping approach does not require prior identification of isozyme-selective inhibitors. The minimal pharmacophoric elements for ligand isozyme selectivity can be derived directly by comparison of the isozyme structures with the use of the GRID/CPCA approach. Another difference is that anchored plasticity requires the crystallographic determination of isoform-selective conformational changes while this is not the case with fragment hopping.
Experimental Section
Computer Modeling
Protein Structures
The crystallographic coordinates for NOS from the Research Collaboratory for Structural Bioinformatics (RCSB) protein database are as follows: rat nNOS1: 1P6I (1.90 Å resolution, Rcryst = 0.228); 3B3N (1.98 Å resolution, Rcryst = 0.230). The amino acid sequences of NOS were retrieved from the PIR protein sequence database. The sequences are human nNOS (entry P29475), rat nNOS (entry P29476), human eNOS (entry P29474), bovine eNOS (entry P29473), human iNOS (entry P35228), and murine iNOS (entry P29477). All of the computational work was performed on Silicon Graphics Octane 2 Workstations with an IRIX 6.5 operating system. The molecular modeling was achieved with commercially available InsightII 200042 and SYBYL 6.843 software packages. The NOS model was prepared by first adding H-atoms using the Accelrys/InsightII 2000 software, and the protonation states of the residues were set to pH 7.0. The cvff force field of the Discover 98.0 program within Insight II was used to optimize the orientation of hydrogen atoms of the protein and of structural waters. The ligands and solvent molecules of the protein structures were removed, but the heme and H4B were retained near the active site.
GRID Calculations
The calculations were performed with version 22 of the GRID software.44 Hydrogens were added with the program GRIN. The GRID box dimensions were chosen to encompass all of the active site residues. This resulted in a box size of 31 × 28 × 31 Å. The grid spacings were set to 1 Å (directive NPLA = 1) and 0.5 Å (directive NPLA = 2), respectively. The amino acids in the active site were considered rigid (directive move = 0). The directives NETA and ALMD were set to 120 and 1, respectively, to include the atoms of heme and H4B in calculations and to interpret which atom(s) in the active site contribute(s) to the interaction with a specific probe atom. The following single atom probes were used in the calculation: DRY, C3, C1=, NM3, N1+, N2+, Cl and F.
MCSS Calculations
The functional groups chosen for the MCSS calculations were benzene (all hydrogen model), cyclohexane, isobutene, n-butane, propane, trimethylamine cation, and dimethylamine cation. The binding site area in the MCSS simulation was defined as an approximately 27 Å × 26 Å × 31 Å box. Replicas of a given functional group were randomly distributed inside the binding site and then simultaneously and independently energy minimized. Pairs of molecules were considered to be identical if the root-mean-square deviation (RMSD) between them was less than 0.2 Å, and in such cases one of the pairs was eliminated. The above protocol was repeated 10 times for each of the functional groups to allow complete searching of the active site. The minima with the binding energy lower than 0 kcal/mol were retrieved in the analysis. All of the above calculations were performed using the CHARMM 22 force field and the MCSS 2.1 program.45
LUDI Ligand Design
LUDI33 was first applied to generate the interaction sites for each pharmacophore. The residues inside an 8.0 Å radius sphere, which centered on the centroid of the minimal pharmacophoric elements, were used to generate the interaction sites. The link library switch was turned off, and the target mode switch was turned on. The LUDI scoring function was set to Energy-estimate-2.46 Other settings were kept with the standard default parameters. To convert the above determined fragments into a molecule, the LUDI link library was used in the connection operation. The hydrogen atoms in the above fragment structure were replaced by a link fragment to create a new compound. The switch for the target mode was turned off, but the switch for the link library was turned on. The linkage parameter can be set to 1 (the link fragment fit at least one link site), 2 (the link fragment simultaneously fit at least two link sites), or be specified (the link site was specifically assigned) according to the actual requirements. The other settings are the standard default parameters.
AutoDock Analysis
AutoDock 3.0.5 was employed to perform the docking calculations.47 For the protein structure (PDB id: 1P6I), polar hydrogen atoms were added, and Kollman united atom charges were assigned.48 Hydrogens were also added to the heme and H4B, and charges were calculated by the Gasteiger-Marsili method.49 The charge of the Fe atom bound to heme was assigned +3. The nonpolar hydrogen atoms of heme and H4B were removed manually, and their charges were united with the bonded carbon atoms. Atomic solvation parameters and fragmental volumes were assigned using the AddSol utility. The 3D structures of the ligands were built and partial atomic charges were also calculated using the Gasteiger-Marsili method. The rotatable bonds in the ligands were defined using another AutoDock 3.0 auxiliary program, AutoTors, which also unites the nonpolar hydrogens and partial atomic charges to the bonded carbon atoms. The grid maps were calculated using AutoGrid. The dimensions of the grid box was 27 × 26 × 31 Å, and the grid spacing was set to 0.375 Å. Docking was performed using the Lamarckian genetic algorithm (LGA), and the pseudo-Solis and Wets method were applied for the local search. The procedure in detail used was that previously described.4,30
Chemical Synthesis
General Methods, Reagents and Materials
All reagents were purchased from Aldrich, Acros Organics, or Fisher Scientific and were used without further purification unless stated otherwise. NADPH, calmodulin, and human ferrous hemoglobin were obtained from Sigma Chemical Co. Tetrahydrobiopterin (H4B) was purchased from Alexis Biochemicals. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury 400 MHz or a Varian Inova 500 MHz spectrometer (100.6, or 125.7 MHz, for 13C NMR spectra, respectively) in CDCl3, DMSO-d6, CD3OD or D2O. Chemical shifts are reported as values in parts per million and the reference resonance peaks set at 0 ppm [TMS(CDCl3)], 2.50 ppm [(CD2H)2SO], 3.31 ppm (CD2HOD), and 4.80 ppm (HOD) respectively for 1H NMR and 77.23 ppm (CDCl3), 39.52 ppm (DMSO-d6) and 49.00 ppm (CD3OD) for 13C NMR spectra. An Orion research model 701H pH meter with a general combination electrode was used for pH measurements. Mass spectra were performed on a Micromass Quattro II triple quadrupole HPLC/MS mass spectrometer with an electrospray ionization (ESI) source or atmospheric pressure chemical ionization (APCI) source. High-resolution mass spectra were carried out using a VG70-250SE mass spectrometer. Chemical ionization (CI) or electron impact (EI) was used as the ion source. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA. Thin-layer chromatography was carried out on E. Merck pre-coated silica gel 60 F254 plates with visualization accomplished with phosphomolybdic acid or ninhydrin spray reagent or with a UV-visible lamp. Column chromatography was performed with E. Merck silica gel 60 (230-400 mesh) or Sorbent Technologies 250 mesh silica gel. Tetrahydrofuran (THF) and ethyl ether were redistilled under nitrogen using sodium and benzophenone as the indicator. Dichloromethane was redistilled from CaH2 under nitrogen. Other dry solvents were directly purchased from the companies. N1, N1-dimethylethane-1,2-diamine, (S)-(-)-1-Boc-3-aminopyrrolidine, (R)-(+)-1-Boc-3-aminopyrrolidine, (S)-(+)-1-benzyl-3-aminopyrrolidine, (R)-(-)-1-benzyl-3-aminopyrrolidine, and N-Boc-1,2-diaminoethane were purchased from Aldrich.
(±)-cis-tert-Butyl 3-{2′-[benzyl(tert-butoxycarbonyl)amino]ethylamino}-4-{[6′-(tert-butoxycarbonylamino)pyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (37a)
A mixture of 36 (0.196 g, 0.5 mmol),4 a substituted amine such as 41 (0.138 g, 0.55 mmol), sodium triacetoxyborohydride [NaBH(OAc)3 (0.149 g, 0.7 mmol)], and 3Å molecular sieves (0.5 g) in dry 1,2-dichloroethane (10 mL) was stirred at room temperature under a nitrogen atmosphere for 14 h (TLC used to monitor the completion of the reaction). To the reaction mixture was then added 1 M NaOH (15 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (15 mL × 2). The combined organic layers were washed with brine (20 mL) and dried over MgSO4. The solvent was concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 9 : 1 : 0.5) to afford a pale-yellow oil (0.199 g, 85%, diastereomer ratio: cis : trans = 75 : 25). 1H NMR (CDCl3, 500 MHz): δ 7.783-7.748 (m, 1H), 7.556-7.512 (m, 1H), 7.337-7.234 (m, 6H), 6.798-6.775 (m, 1H), 4.569-4.438 (m, 2H), 3.340-3.109 (m, 7H), 2.774-2.573 (m, 5H), 1.506-1.454 (m, 27H). 13C NMR (CDCl3, 125.7 MHz): δ (158.450+158.318) (1C), (154.856+154.775) (1C), (152.844+152.763) (1C), (151.974+151.834) (1C), (138.761+138.684) (1C), (138.808+138.781) (1C), 128.525 (2C), 127.947 (1C), 127.247 (2C), (117.685+117.484) (1C), (109.870+109.789) (1C), 80.716 (1C), (80.108+79.986) (1C), 79.184 (1C), (59.569+58.594) (1C), (50.869+50.362) (1C), (50.686+50.182) (1C), (49.731+49.398) (1C), (48.191+48.157) (1C), (46.267+46.220) (1C), (42.514+41.462) (1C), (35.045+34.983) (1C), 28.658 (3C), 28.426 (3C), 28.264 (3C). MS (ESI, CH3OH): [C34H51N5O6] m/z 626.5 ([M+H]+).
N1-{(±)-4′-[(6”-aminopyridin-2”-yl)methyl]pyrrolidin-3′-yl}-N2-benzylethane-1,2-diamine tetrachloride (7)
A solution of 4M HCl in 1,4-dioxane (4 mL) was added to 37a (0.125 g, 0.2 mmol) at 0 °C under an argon atmosphere. The ice-water bath was removed after 3 h, and the reaction mixture was stirred at room temperature for 36 h. After the completion of the reaction, liquids were evaporated under reduced pressure, and the residue was partitioned between water (10 mL) and ethyl acetate (10 mL). The aqueous layer was then washed with ethyl acetate (5 mL × 2). After evaporation of water by high-vacuum rotory evaporation, the residue was dried with a lyophilizer to afford a hygroscopic white solid (0.0943 g, quantitative yield). 1H NMR (D2O, 500 MHz): δ 7.843 (t, 1H, J=9Hz), 7.544-7.516 (m, 5H), 7.063 (d, 1H, J=9Hz), 6.8235 (d, 1H, J=6.5Hz), 4.344 (s, 2H), 4.287-4.089 (m, 1H), 3.895-3.785 (m, 1H), 3.750-3.154 (m, 9H), 2.914-2.849 (m, 1H). 13C NMR (D2O, 125.7 MHz): δ 154.843 (1C), 144.819 (2C), 130.238 (1C), 130.114 (1C), 130.041 (2C), 129.542 (2C), 112.535 (2C), 58.524 (1C), 51.831 (1C), 47.475 (1C), 46.005 (1C), 43.324 (2C), 38.964 (1C), 29.369 (1C). MS (ESI, CH3OH): [C19H27N5] m/z 326.3 ([M+H]+). HRMS (CI+, CH3OH) Calc.: 326.2339, Found: 326.2334. Comb. Anal. (C19H27N5·4HCl·1.75H2O), Calcld: C, 45.39; H, 6.92; N, 13.93, Found: C, 45.38; H, 6.78; N, 13.66.
tert-Butyl benzyl(2-hydroxyethyl)carbamate (38)
A solution of di-tert-butyl dicarbonate (1.094 g, 0.005 mol) in CH2Cl2 (20 mL) was added dropwise to a solution of 2, benzylaminoethanol (0.756 g, 0.005 mol) in CH2Cl2 (20 mL) and 1M NaOH (16 mL). The reaction mixture was then stirred at room temperature for 24 h, and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with water (10 mL × 2) and dried over Na2SO4. The solvent was evaporated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 7 : 3) to afford a colorless oil (1.256 g, quantitative yield). 1H NMR (CDCl3, 500 MHz): δ 7.339-7.234 (m, 5H), 4.474 (m, 2H), 3.693 (m, 2H), 3.399 (m, 2H), 1.459 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 154.500 (1C), 138.366 (1C), 128.733 (1C), 127.453 (2C), 127.329 (1C), 80.743 (1C), (62.227+61.334) (1C), (52.184+51.155) (1C), (49.956+49.113) (1C), 28.535 (3C). MS (ESI, CH3OH): [C14H21NO3] m/z 252.3 ([M+H]+); m/z 274.4 ([M+Na]+); m/z 525.0 ([2M+Na]+).
tert-Butyl 2-aminoethyl(benzyl)carbamate (41)
To a solution of triphenylphosphine (Ph3P, 1.64 g, 0.00625 mol) in dry THF (10 mL) was added 38 (1.256 g, 0.005 mol) in dry THF (10 mL) at 0 °C under a N2 atmosphere via a cannula. Diisopropyl azodicarboxylate (DIAD, 1.315 g, 1.30 mL, 0.0065 mol) was added dropwise, and the solution was stirred for 20 min at 0 °C. Diphenylphosphoryl azide (DPPA, 1.72 g, 1.347 mL, 0.00625 mol) was added at 0 °C, and the solution was stirred overnight at room temperature. The reaction mixture was then concentrated in vacuo. The crude residue was purified by column chromatography (silica gel, hexanes : EtOAc = 9.5 : 0.5) to yield a colorless oil.
The above azide product was dissolved in THF (5 mL) and was put into an ice-cold bath, then Ph3P (1.574 g, 0.006 mol) and H2O (0.324 g, 0.018 mol) were added at 0 °C. The reaction mixture was stirred for 2 h at 0 °C and for 21 h at room temperature. The solvent was evaporated in vacuo, and the residue was partitioned between water (10 mL) and CH2Cl2 (10 mL). The aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, and the solvent was evaporated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2 : MeOH = 9 : 1) to afford a colorless oil (0.751 g, 60%). 1H NMR (CDCl3, 500 MHz): δ 7.334-7.238 (m, 5H), 4.464 (m, 2H), 3.289-3.204 (m, 2H), 2.782 (m, 2H), 1.456 (s, 9H), 1.049 (brs, 2H). 13C NMR (CDCl3, 125.7 MHz): δ 156.268 (1C), 138.607 (1C), 128.688 (2C), 127.860 (1C), 127.368 (2C), 80.027 (1C), (51.294+50.737) (1C), 50.118 (1C), 40.628 (1C), 28.596 (3C). MS (ESI, CH2Cl2): [C14H22N2O2] m/z 251.2([M+H]+), 501.0([2M+H]+).
The amine product can also be prepared by hydrogenation of the azide. The azide product was dissolved in MeOH (15 mL), and to the reaction solution was added a catalytic amount of 10% Pd/C. The solution was stirred under a H2 atmosphere at room temperature for 24h. The solution was then filtered through Celite, and the Celite pad was washed with MeOH (10 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2 : MeOH = 9 : 1) to afford a colorless oil (0.688 g, 55%).
(±)-tert-Butyl 3-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}-4-[2′-(tert-butoxycarbonylamino)ethylamino]pyrrolidine-1-carboxylate (47a)
A mixture of 46 (0.203 g, 0.5 mmol),23 N-Boc-1,2-diaminoethane (0.089 g, 0.55 mol), NaBH(OAc)3 (0.149 g, 0.7 mmol), and 3 Å molecular sieves (0.5 g) in dry 1,2-dichloroethane (10 mL) was stirred at room temperature under a N2 atmosphere for 16 h. The reaction mixture was then filtered through Celite, and the Celite pad was washed with CH2Cl2 (5 mL × 2). To the filtrate was added 1M NaOH (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with brine (10 mL) and dried over MgSO4. The solvent was evaporated, and the residue was purified by column chromatography (silica gel, hexane: EtOAc : Et3N = 6: 4: 0.25) to afford a colorless oil (0.196 g, 95%, diastereomer ratio: cis : trans = 75 : 25). 1H NMR (CDCl3, 500 MHz): δ 7.635-7.622 (s, 1H), 7.373 (m, 1H), 6.657 (s, 1H), 5.451 (brs, 1H), 3.444-3.346 (m, 3H), 3.224-3.076 (m, 4H), 2.863-2.803 (m, 2H), 2.588-2.562 (m, 3H), 2.321-2.299 (m, 3H), 1.523 (s, 9H), 1.455 (s, 18H). 13C NMR (CDCl3, 125.7 MHz): δ 158.521 (1C), 156.374 (1C), (155.035+154.973) (1C), 152.706 (1C), 151.670 (1C), 150.126 (1C), 119.280 (1C), 110.498 (1C), 81.006 (1C), 79.408 (2C), (59.023+57.913) (1C), (51.007+50.640) (1C), (49.526+49.228) (1C), 47.436 (1C), (42.941+42.125) (1C), 40.623 (1C), (35.099+35.006) (1C), 28.680 (3C), 28.595 (3C), 28.468 (3C), 21.465 (1C). MS (ESI, CH3OH): [C28H47N5O6] m/z 550.4 ([M+H]+).
2-(3′-chlorobenzylamino)ethanol (48a)
3-Chlorobenzaldehyde (1.4 g, 0.01 mol) was dissolved in dry ethanol (30 mL), and ethanolamine (0.61 g, 0.01 mol) was added. The reaction mixture was stirred overnight at 60 °C. The ethanol was evaporated under reduced pressure to give a colorless oil. The oil was then dissolved in dry methanol (30 mL) and cooled in ice. Sodium borohydride (NaBH4, 0.57g, 0.015 mol) was added slowly in portions, and the resulting solution was left overnight at room temperature. The solvent was evaporated in vacuo, and the residue was dissolved in water and extracted with CH2Cl2 (3 × 20 mL). The organic layers were combined and dried with Na2SO4, filtered and evaporated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2 : MeOH = 9.5 : 0.5) to generate a pale-green oil (1.795 g, 97%). 1H NMR (CDCl3, 500 MHz): δ 7.322 (m, 1H), 7.271-7.221 (m, 2H), 7.199-7.186 (m, 1H), 3.785 (s, 2H), 3.662 (t, 2H, J=5.5Hz), 2.788 (t, 2H, J=5.5Hz), 2.079 (brs, 2H). 13C NMR (CDCl3, 125.7 MHz): δ 142.374 (1C), 134.513 (1C), 129.919 (1C), 128.378 (1C), 127.439 (1C), 126.388 (1C), 61.200 (1C), 53.155 (1C), 50.731 (1C). MS (ESI, CH3OH): [C9H12ClNO] m/z 186.5 ([M+H]+); m/z 371.2 ([2M+H]+).
2-(4′-fluorophenyl)acetaldehyde (49a)
To a suspension of Dess-Martin periodinane (2.04 g, 0.0048 mol) in anhydrous CH2Cl2 (50 mL) was added dropwise 4-fluorophenethyl alcohol (0.56 g, 0.5 mL, 0.004 mol). The reaction mixture was stirred at room temperature for 16 h under a N2 atmosphere. Na2S2O3 (1M, 40 mL) was then added to the reaction mixture, and after being stirred for 15 min, the mixture was separated. The aqueous layer was extracted with CH2Cl2 (20 mL × 2). The combined organic layers were washed with 5% NaHCO3 (20 mL × 2) and brine (10 mL) and dried over Na2SO4. The solvent was concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 8 : 2) to afford a pale-yellow volatile oil (0.437, 79%). 1H NMR (CDCl3, 500 MHz): δ 9.747-9.743 (m, 1H), 7.195-7.167 (m, 2H), 7.076-7.041 (m, 2H), 3.683 (s, 2H). 13C NMR (CDCl3, 125.7 MHz): δ 199.225 (1C), (163.354+161.393) (1C), (131.394+131.327) (2C), (127.721+127.696) (1C), (116.154+115.984) (2C), 49.823 (1C).
2-[benzyl(phenethyl)amino]ethanol (50a)
A mixture of commercially-available phenylacetaldehyde (90%, 0.40 g, 0.003 mol), N-benzylethanolamine (0.5 g, 0.0033 mol), and NaBH(OAc)3 (0.90 g, 0.0042 mol) in dry 1,2-dichloroethane (60 mL) was stirred at room temperature under a N2 atmosphere for 14 h. To the reaction mixture was then added 1M NaOH (40 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (30 mL × 2). The combined organic layers were washed with brine (20 mL) and dried over MgSO4. The solvent was concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 5 : 5) to afford a pale-yellow oil (0.766 g, quantitative yield). 1H NMR (CDCl3, 500 MHz): δ 7.289-7.105 (m, 10H), 3.675 (s, 2H), 3.509 (m, 2H), 2.766 (m, 4H)-2.673 (t, 2H, J=5Hz), 2.559 (brs, 1H). 13C NMR (CDCl3, 125.7 MHz): δ 140.240(1C), 138.898 (1C), 129.020(2C), 128.850 (2C), 128.552 (1C), 128.528 (1C), 127.302 (1C), 126.239 (1C), 58.627 (1C), 58.390 (1C), 55.306 (1C), 55.251 (1C), 33.624 (1C). MS (ESI, CH3OH): [C17H21NO] m/z 256.4 ([M+H]+); m/z 272.2 ([M+Na]+).
2-(phenethylamino)ethanol (51a)
A suspension of 50a (0.766 g, 0.003 mol) and 10% Pd/C (450 mg) in MeOH (30 mL) was stirred at room temperature under a hydrogen atmosphere overnight. The catalyst was then filtered through Celite. The Celite pad was washed with MeOH (10 mL × 3), and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2 : MeOH = 8.5 : 1.5) to yield a pale-green oil (0.382 g, 77%). 1H NMR (CDCl3, 500 MHz): δ 7.309-7.199 (m, 5H), 3.663 (t, 2H, J=5Hz), 2.931 (t, 2H, J=7Hz), 2.849 (t, 2H, J=7Hz), 2.818 (t, 2H, J=4.5Hz), 3.194 (brs, 2H). 13C NMR (CDCl3, 125.7 MHz): δ 139.493 (1C), 128.868 (2C), 128.735 (2C), 126.850 (1C), 60.467 (1C), 51.086 (1C), 50.685 (1C), 35.956 (1C). MS (ESI, CH3CN): [C10H15NO] m/z 166.4 ([M+H]+).
(±)-tert-Butyl 3-{{2′-[tert-butoxycarbonyl(4”-chlorobenzyl)amino]ethyl}(methyl)amino}-4-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (54a or 55a)
To a solution of 47b 0.337 g, 0.5 mmol) dissolved in MeOH (10 mL) at 0 °C was added a 37% formaldehyde solution (0.405 g, 0.375 mL, 0.005 mol), sodium cyanoborohydride [NaBH3CN (0.165 g, 0.0025 mol)], and acetic acid (0.06 g, 0.06 mL, 0.001 mol). The reaction mixture was then stirred at room temperature overnight. The solvent was removed under reduced pressure. The residue was dissolved in water, extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous MgSO4, and evaporated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 6 : 4) to generate a colorless oil (54a, 0.325 g, 94%). 1H NMR (CDCl3, 500 MHz): δ 7.594-7.579 (m, 1H), 7.288-7.266 (m, 2H), 7.178-7.152 (m, 3H), 6.664-6.613 (s, 1H), 4.522-4.401 (m, 2H), 3.614-3.522 (m, 1H), 3.418-3.078 (m, 5H), 2.953-2.838 (m, 2H), 2.695-2.516 (m, 2H), 2.474-2.170 (m, 8H), 1.518-1.389 (m, 27H). 13C NMR (CDCl3, 125.7 MHz): δ (159.189+159.068+158.928) (1C), (155.929+155.528) (1C), (155.006+154.903) (1C), 152.547 (1C), 151.521 (1C), (149.882+149.791) (1C), (137.320+137.071) (1C), 133.112 (1C), (129.226+128.807+128.540) (4C), (119.919+119.822) (1C), 110.095 (1C), (80.921+80.830) (1C), 80.114 (1C), 79.391 (1C), (66.429+65.797) (1C), (54.104+53.752) (1C), (50.965+50.643+50.260+49.872) (1C), (49.775+49.338) (1C), (48.743+48.396) (1C), (44.201+44.007) (1C), (40.631+40.285+39.902) (2C), 33.898 (1C), (28.707+28.652+28.561+28.415) (9C), 21.415 (1C). MS (ESI, CH3OH): [C36H54ClN5O6] m/z 688.5 ([M+H]+).
The title compound can also be made by the following synthetic route
To a solution of 46 (0.203 g, 0.5 mmol), 57a (0.149 g, 0.5 mmol), acetic acid (0.045 g, 0.043 mL, 0.75 mmol), and 3 Å molecular sieves (0.5 g) in dry MeOH (5 mL) was added NaBH3CN (0.063 g, 0.001 mol). The reaction mixture was stirred at room temperature under a N2 atmosphere for 36 h (TLC monitor the completion of reaciton). The reaction mixture was then filtered. The filtrate was concentrated in vacuo, and the residue was diluted with 1M NaOH (15 mL), extracted with CH2Cl2 (15 mL × 2). The organic layers were washed with brine, dried over anhydrous MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 6 : 4) to produce a colorless oil (55a, 0.327 g, 95%).
tert-Butyl 4-chlorobenzyl(2′-oxoethyl)carbamate (56a)
To a suspension of Dess-Martin periodinane (0.509 g, 0.0012 mol) in anhydrous CH2Cl2 (10 mL) was added a solution of 52b (0.285 g, 0.001 mol) in anhydrous CH2Cl2 (5 mL). Then the reaction mixture was stirred at room temperature for 21 h under a N2 atmosphere. Na2S2O3 (1 M, 10 mL) was added to the reaction mixture, and after stirring for 10 min, the reaction mixture was extracted with CH2Cl2 (10 mL × 3). The combined organic layers were washed with 5% aqueous NaHCO3 (20 mL) and brine (20 mL). The organic layer was dried over Na2SO4 and evaporated in vacuo. The residue was then purified by column chromatography (silica gel, hexanes : EtoAc = 8 : 2) to afford a colorless oil (0.20 g, 72%). 1H NMR (CDCl3, 500 MHz): δ (9.492+9.430) (s, 1H), 7.294 (m, 2H), 7.196-7.154 (m, 2H), 4.489-4.451 (m, 2H), 3.938 (m, 1H), 3.798 (m, 1H), 1.465 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ (198.988+198.897+198.502+198.405+197.998) (1C), (155.783+155.510) (1C), (136.063+135.905) (1C), (133.671+133.567) (1C), (129.585+129.038+128.971) (4C), 81.371 (1C), 56.684 (1C,), (51.560+51.068) (1C), (28.421+28.330) (3C).
tert-Butyl 4-chlorobenzyl[2′-(methylamino)ethyl]carbamate (57a)
To a solution of 56a (0.566 g, 0.002 mol) in MeOH (30 mL) was added MeNH2 (2M in MeOH, 2 mL, 0.004 mol) at room temperature under a N2 atmosphere. The reaction mixture was heated at reflux for 6 h and then cooled to -20 °C. Then NaBH4 (0.15 g, 0.004 mol) was added in portions. After being stirred for 1 h at -20 °C, the reaction mixture was stirred for 3 h at room temperature. The solvent was then evaporated under reduced pressure. The residue was dissolved in water (20 mL) and extracted with CH2Cl2 (20 mL × 3). The combined organic layers were washed with brine and dried over MgSO4. The solvent was evaporated in vacuo, and the residue was purified by column chromatography (silica gel, CH2Cl2 : MeOH = 9:1) to afford a colorless oil (0.59 g, 99%). 1H NMR (CDCl3, 500 MHz): δ 7.287 (d, 2H), 7.174 (m, 2H), 4.428 (s, 2H), 3.348-3.260 (m, 2H), 2.710 (m, 2H), 2.412 (s, 3H), 1.568-1.444 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 155.993 (1C), 137.216 (1C), 133.076 (1C), (129.220+128.795+128.577) (4C), 80.241 (1C), (50.825+50.115) (2C), 46.551 (1C), 36.423 (1C), 28.567 (3C). MS (ESI, CH2Cl2): [C15H23ClN2O2] m/z 299.1 ([M+H]+).
tert-Butyl methyl[2-(methylamino)ethyl]carbamate (58)
A solution of di-tert-butyl dicarbonate (2.183 g, 0.01 mol) in CH2Cl2 (120 mL) was added dropwise to a solution of N,N’-dimethylethane-1,2-diamine (1.763 g, 0.02 mol) in CH2Cl2 (40 mL) over 6 h with vigorous stirring. The reaction mixture was stirred for an additional 18 h at room temperature. Then the solvent was concentrated in vacuo to give an oily residue, which was dissolved in 60 mL of 2 M aqueous Na2CO3 and extracted with CH2Cl2 (30 mL × 2). The combined organic layers were washed with 2 M Na2CO3 (30 mL × 2) and dried over anhydrous MgSO4. The solvent was evaporated in vacuo to yield the product, which was purified by column chromatography (silica gel, CH2Cl2 : MeOH, 9:1) to afford a colorless oil (1.15 g, 61%). 1H NMR (CDCl3, 500 MHz): δ 3.347 (m, 2H), 2.893-2.882 (m, 3H), 2.744-2.734 (m, 2H), 2.466-2.452 (m, 3H), 1.625 (brs, 1H), 1.474-1.459 (m, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 156.014 (1C), 79.452 (1C), 49.720 (1C), (48.475+48.202) (1C), 36.308 (1C), 34.632 (1C), 28.452 (1C). MS (ESI, CH3OH): [C9H20N2O2] m/z 189.2 ([M+H]+); m/z 377.3 ([2M+H]+).
tert-Butyl 2-[(3′-fluorophenethyl)(methyl)amino]ethyl(methyl)carbamate (59a)
A mixture of 58 (0.941 g, 0.005 mol), 3-fluoro-phenyl-acetaldehyde (0.76 g, 0.0055 mol), and NaBH(OAc)3 (1.48 g, 0.007 mol) in dry 1,2-dichloroethane (50 mL) was stirred overnight at room temperature under a N2 atmosphere. To the reaction mixture was added 1 M NaOH (30 mL), and then the mixture was extracted with CH2Cl2 (30 mL × 3). The combined organic layers were washed with brine and dried over MgSO4. The solvent was then evaporated in vacuo to give the product, which was purified by column chromatography (silica gel, hexanes : EtOAc = 5:5) to afford a colorless oil (1.0 g, 65%). 1H NMR (CDCl3, 500 MHz): δ 7.235-7.222 (m, 1H), 6.975-6.960 (m, 1H), 6.915-6.862 (m, 2H), 3.322-3.277 (m, 2H), 2.856-2.831 (m, 3H), 2.771-2.740 (t, 2H, J=7 Hz), 2.652-2.621 (t, 2H, J=7.5 Hz), 2.546 (m, 2H), 2.330 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ (163.937+161.982) (1C), 155.765 (1C), 143.063 (1C), 129.840 (1C), 124.491 (1C), (115.717+115.547) (1C), (113.064+112.906) (1C), 79.440 (1C), (59.604+59.441) (1C), (55.306+55.033) (1C), (47.134+46.587) (1C), 42.495 (1C), (35.003+34.778) (1C), 33.825 (1C), 28.579 (3C). MS (ESI, CH3OH): [C17H27FN2O2] m/z 311.2 ([M+H]+).
N1-(3-fluorophenethyl)-N1,N2-dimethylethane-1,2-diamine (60a)
To a mixture of 59a (0.93 g, 0.003 mol) in anhydrous CH2Cl2 (4.5 mL) was added trifluoroacetic acid (TFA, 4.5 mL) at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and 1 h at room temperature. After the reaction was finished, 2 M aqueous NaOH (30 mL) was added to adjust the pH value to basic. The mixture was stirred for an additional 10 min. The organic layer was separated. The aqueous layer was extracted with CH2Cl2 (30 mL × 2). The combined organic layers were washed with 2 M aqueous NaOH (30 mL × 2). The solvent was evaporated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2: MeOH: Et3N = 9.75: 0.25: 0.25) to afford a colorless oil (0.59 g, 93%). 1H NMR (CDCl3, 500 MHz): δ 7.255-7.212 (m, 1H), 6.971-6.956 (m, 1H), 6.907-6.866 (m, 2H), 2.757 (t, 2H, J=8Hz), 2.622-2.596 (m, 4H), 2.538 (t, 2H, J=5.5 Hz), 2.379 (s, 3H), 2.339 (brs, 1H), 2.330 (s, 3H). 13C NMR (CDCl3, 125.7 MHz): δ (163.943+161.994) (1C), (143.391+143.337) (1C), (129.858+129.791) (1C), (124.545+124.521) (1C), (115.735+115.572) (1C), (113.021+112.851) (1C), 59.277 (1C), 56.805 (1C), 49.344 (1C), 42.307 (1C), 36.423 (1C), 33.722 (1C). MS (ESI, CH3OH): [C12H19FN2] m/z 211.2 ([M+H]+).
(±)-tert-Butyl 3-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}-4-hydroxypyrrolidine-1-carboxylate (61)
To an ice-cooled solution of sodium hydride (0.08 g, 0.002 mol, 60% dispersion in mineral oil) in anhydrous DMF (2 mL) was added dropwise 45a (0.81 g, 0.002 mol) in anhydrous DMF (8 mL). The suspension was stirred vigorously for 30 min at 0 °C. The color of the reaction mixture changed from colorless to pale red. Benzyl bromide (0.34 g, 0.24 mL, 0.002 mol) was then added dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 30 min and was brought to room temperature over 1 h; stirring continued at room temperature for an additional hour. H2O (5 mL) was added dropwise to quench the reaction. The reaction mixture was concentrated in vacuo, and the residue was diluted with H2O (25 mL) and EtOAc (30 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (30 mL × 2). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 6 : 4) to afford a colorless oil (0.84g, 84%). 1H NMR (CDCl3, 500 MHz): δ 7.338-7.178 (m, 6H), 6.713-6.696 (m, 1H), 5.168-5.079 (m, 2H), (4.164+3.869) (brs, 1H), 4.061-4.042 (m, 1H), 3.726-3.528 (m, 2H), 3.211-3.144 (m, 1H), 3.094-3.044 (m, 1H), 2.812-2.765 (m, 2H), 2.488-2.364 (m, 1H), 2.296-2.289 (m, 3H), 1.452-1.416 (m, 18H). 13C NMR (CDCl3, 125.7 MHz): δ 157.398 (1C), (154.727+154.666) (1C), 154.405 (1C), 154.016 (1C), (149.463+149.341) (1C), (139.530+139.445) (1C), 128.340 (2C), 127.180 (1C), 126.871 (1C), (120.914+120.696) (1C), (118.577+118.358) (1C), 81.516 (1C), 79.416 (1C), (75.287+74.577) (1C), (52.798+52.367) (1C), (50.619+50.461) (1C), (49.817+49.404) (1C), (45.403+44.638) (1C), 39.368 (1C), 28.634 (3C), 28.294 (3C), 21.257 (1C). MS (ESI, CH3OH): [C28H39N3O5] m/z 498.4 ([M+H]+); m/z 995.2 ([2M+H]+).
(±)-tert-Butyl 3-azido-4-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (62)
To Ph3P (0.33 g, 0.00125 mol) in a dry THF (5 mL) solution was added 61 (0.5 g, 0.001 mol) in dry THF (10 mL) at 0 °C under a N2 atmosphere via cannula. DIAD (0.26 g, 0.26 mL, 0.0013 mol) was added dropwise, and the solution was stirred at 0 °C for 20 min. DPPA (0.36g, 0.28 mL, 0.0013 mol) was added dropwise at 0 °C, and the reaction mixture was stirred for 22 h at room temperature. The solvent was concentrated in vacuo. The crude residue was directly purified by column chromatography (silica gel, hexanes : EtOAc = 9 : 1) to afford a colorless oil (0.5 g, 95%). 1H NMR (CDCl3, 500MHz): δ 7.494-7.466 (m, 1H), 7.262-7.173 (m, 5H), 6.689 (m, 1H), 5.174 (m, 2H), 3.779-3.735 (m, 1H), 3.579-3.468 (m, 1H), 3.443-3.367 (m, 1H), 3.322-3.280 (m, 1H), 3.030-2.966 (m, 1H), 2.879-2.834 (m, 1H)-2.745-2.692 (m, 1H), 2.669-2.590 (m, 1H), 2.302-2.289 (m, 3H), 1.462-1.443 (m, 9H), 1.417 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ (156.603+156.585) (1C), (154.338+154.302+154.089) (2C), 153.943 (1C), 148.710 (1C), (139.827+139.754) (1C), (128.115+128.085) (2C), (126.743+126.634) (2C), (126.555+126.458) (1C), 119.931 (1C), (117.089+117.041) (1C), (81.249+81.201) (1C), 79.489 (1C), (63.229+62.470) (1C), (51.487+51.201) (1C), 49.908 (1C), (48.846+48.445) (1C), (42.513+41.942) (1C), (34.972+34.912) (1C), 28.452 (3C), 28.130 (3C), 21.117(1C). MS (ESI, CH3OH): [C28H38N6O4] m/z 523.2([M+H]+); m/z 1066.9 ([2M+Na]+).
(±)-tert-Butyl 3-amino-4-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (63)
A solution of 62 (0.52 g, 0.001 mol) in EtOH (20 mL) was treated with 20% wt Pd(OH)2 on carbon (300 mg). The reaction mixture was stirred at 60 °C under a hydrogen atmosphere for 36 h. The catalyst was filtered through Celite. The Celite pad was washed with EtOH (10 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2 : CH3OH = 9.5: 0.5) to afford a pale-green oil (0.28 g, 69%). 1H NMR (CDCl3, 500MHz): δ 7.690 (brs, 1H), 7.635-7.626 (m, 1H), 6.645 (m, 1H), 3.563-3.173 (m, 4H), 2.832-2.772 (m, 1H), 2.720-2.659 (m, 2H), 2.478 (m, 1H), 2.307-2.296(m, 3H), 1.518(s, 9H), 1.454-1.446 (m, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 157.860 (1C), (154.745+154.678) (1C), 152.602 (1C), 151.600 (1C), 150.270 (1C), 118.868 (1C), 110.447 (1C), (80.836+80.782) (1C), 79.294 (1C), (54.274+53.891) (1C), (52.513+51.365) (1C), (48.815+48.396) (1C), (43.904+43.199) (1C), (35.264+35.082) (1C), 28.561 (3C), 28.336 (3C), 21.378 (1C). MS (ESI, CH3OH): [C21H34N4O4] m/z 407.4([M+H]+); m/z 429.2([M+Na]).
(±)-tert-Butyl 3-{2′-[tert-butoxycarbonyl(4”-chlorobenzyl)amino]ethylamino}-4-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (47b) or (±)-tert-Butyl 3-{2′-[tert-butoxycarbonyl(3”-fluorophenethyl)amino]ethylamino}-4-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (47m)
To a mixture of 63 (0.203 g, 0.5 mmol), NaBH(OAc)3 (0.127 g, 0.6 mmol), and 3 Å molecular sieves (0.5 g) in dry 1,2-dichloroethane (10 mL) were added 56a (0.142 g, 0.5 mmol) or 56b (0.141 g, 0.5 mmol) in dry 1,2-dichloroethane (5 mL) via cannula under a N2 atmosphere. The reaction mixture was stirred at room temperature under a N2 atmosphere for 16 h and then was filtered through Celite, and the Celite pad was washed with CH2Cl2 (5 mL × 2). To the filtrate was then added 1 M aqueous NaOH (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with brine (10 mL) and dried over MgSO4. The solvent was evaporated, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 8: 2: 0.25) to afford a colorless oil (47b, 0.306 g, 91%). 1H NMR (CDCl3, 500 MHz): δ 7.625-7.608 (m, 1H), 7.478 (brs, 1H), 7.281 (s, 2H), 7.187 (s, 2H), 6.642 (s, 1H), 4.569-4.339 (m, 2H), 3.339-3.110 (m, 7H), 2.764-2.567 (m, 5H), 2.300-2.286 (m, 3H), 1.508-1.452 (m, 27H). 13C NMR (CDCl3, 125.7 MHz): δ 158.210 (1C), 155.818 (1C), (154.792+154.695) (1C), (152.539+152.503) (1C), 151.453 (1C), 149.819 (1C), 137.191 (1C), 132.892 (1C), (129.073+128.655+128.618+128.412) (4C), 119.159 (1C), 110.264 (1C), 80.691 (1C), (80.108+79.986) (1C), 79.106 (1C), (59.295+58.245) (1C), (50.686+50.182) (2C), (49.326+49.022) (1C), (47.310+46.873) (1C), (46.236+46.114) (1C), (42.538+41.646) (1C), (35.034+34.882) (1C), 28.519 (3C), 28.422 (3C), 28.264 (3C), 21.288 (1C). MS (ESI, CH3OH): [C35H52ClN5O6] m/z 674.3 ([M+H]+). Or a colorless oil (47m, 0.295 g, 88%). 1H NMR (CDCl3, 500 MHz): δ 7.611-7.596 (m, 1H), 7.376 (brs, 1H), 7.266-7.223 (m, 1H), 6.945-6.902 (m, 3H), 6.636 (s, 1H), 3.424-3.115 (m, 9H), 2.819-2.785 (m, 4H), 2.619-2.523 (m, 3H), 2.295-2.280 (m, 3H), 1.511-1.441 (m, 27H). 13C NMR (CDCl3, 125.7 MHz): δ), (163.907+161.952) (1C), 158.272 (1C), 155.656 (1C), (154.866+154.769) (1C), (152.589+152.547) (1C), 151.466 (1C), (149.900+149.869) (1C), (141.825+141.770) (1C), 129.961 (1C), (124.636+124.618) (1C), (119.239+119.202) (1C), (115.857+115.693) (1C), (113.337+113.167) (1C), (110.295+110.235) (1C), (80.788+80.727) (1C), 79.744 (1C), 79.161 (1C), (59.465+58.384) (1C), (50.801+50.303) (1C), (49.372+49.064) (2C), 47.486 (1C), (46.879+46.551+46.253) (1C), (42.483+41.621) (1C), (35.088+34.918+34.329) (2C), 28.567 (3C), 28.464 (3C), 28.318 (3C), 21.342 (1C). 19F NMR (CDCl3, 376.5 MHZ): δ -113.945 (Ar-F). MS (ESI, CH3OH): [C36H54FN5O6] m/z 672.4 ([M+H]+).
(±)-tert-Butyl 3-azido-4-{[2′-(tert-butoxycarbonylamino)-6′-methylpyridin-4′-yl]methyl}pyrrolidine-1-carboxylate (64)
To Ph3P (0.984 g, 0.00375 mol) in dry THF (5 mL) solution was added 45b (1.222 g, 0.003 mol) in THF (10 mL) at 0 °C under a nitrogen atmosphere via a cannula. DIAD (0.789 g, 0.778 mL, 0.0039 mol) was then added dropwise, and the solution was stirred for 20 min at 0 °C. After addition of DIAD, DPPA (1.032 g, 0.808 mL, 0.00375 mol) was added at 0 °C, and the solution was stirred overnight at room temperature. The solution was concentrated, and the crude residue was purified by column chromatography (silica gel, hexanes: EtOAc = 8 : 2) to afford a colorless oil (0.674 g, 52%). 1H NMR (CDCl3, 500MHz): δ (8.325+8.275) (s, 1H), (7.666+7.654) (s, 2H), (6.688+6.670) (s, 1H), 3.977-3.945 (m, 1H), (3.724+3.699+3.633+3.609) (m, 1H), 3.528-3.438 (m, 2H), 3.103-3.050 (m, 1H), 2.839-2.791 (m, 1H), 2.712-2.653 (m, 1H), 2.579-2.522 (m, 1H), (2.430+2.411) (s, 3H), 1.493 (s, 9H), 1.460 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ (157.059+157.029) (1C), (154.275+154.092) (1C), (152.738+152.709) (1C), (152.047+151.983) (1C), 151.066 (1C), 118.485 (1C), 109.140 (1C), (80.676+80.634) (1C), 79.785 (1C), (63.102+62.206) (1C), (51.452+51.125) (1C), (48.762+48.405) (1C), (44.373+43.737) (1C), (33.390+33.288) (1C), 28.450 (3C), 28.259 (3C), 23.824 (1C).
(±)-tert-Butyl 3-amino-4-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (65)
To an ice-cooled solution of 64 (0.649 g, 0.0015 mol) in THF (5 mL) was added Ph3P (0.472 g, 0.0018 mol) and water (0.081 g, 0.081 mL, 0.0045 mol). The reaction mixture was stirred for 2 h at 0 °C and for 21 h at room temperature. The solvent was removed in vacuo, and the residue was purified by column chromatography (silica gel, CH2Cl2 : MeOH = 9 : 1) to afford a pale-green oil (0.537 g, 88%). 1H NMR (CDCl3, 500MHz): δ (8.494+8.430) (s, 1H), (7.548+7.537) (s, 2H), (6.563+6.549) (s, 1H), 3.379-3.243 (m, 3H), 3.178-3.105 (m, 1H), 3.071-3.014 (m, 1H), 2.717-2.679 (m, 1H), 2.522-2.466 (m, 1H), 2.400-2.305 (m, 1H), (2.305+2.285) (s, 3H), 1.371 (s, 9H), 1.329 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 156.706 (1C), (154.593+154.551) (1C), 152.696 (1C), (151.991+151.924) (1C), (151.817+151.745) (1C), 118.239 (1C), (109.217+109.166) (1C), (80.426+80.400) (1C), (79.136+79.114) (1C), (54.932+54.427) (1C), (52.666+51.652) (1C), (48.329+47.930) (1C), (44.047+43.435) (1C), (33.114+32.919) (1C), 28.395 (3C), 28.170 (3C), 23.727 (1C). MS (ESI, CH3OH): [C21H34N4O4] m/z 407.3([M+H]+); m/z 813.1([2M+H]+); m/z 835.2([2M+Na]+).
(±)-tert-Butyl 3-{2-[tert-butoxycarbonyl(4-chlorobenzyl)amino]ethylamino}-4-{[2-(tert-butoxycarbonylamino)-6-methylpyridin-4-yl]methyl}pyrrolidine-1-carboxylate (66a)
To a mixture of 65 (0.203 g, 0.5 mmol), NaBH(OAc)3 (0.127 g, 0.6 mmol), and 3 Å molecular sieves (0.5 g) in dry 1,2-dichloroethane (10 mL) was added 56a (0.142 g, 0.5 mmol) in dry 1,2-dichloroethane (5 mL) via cannula under a N2 atmosphere. The reaction mixture was stirred at room temperature under a N2 atmosphere for 16 h and then was filtered through Celite, and the Celite pad was washed with CH2Cl2 (5 mL × 2). To the filtrate was then added 1 M aqueous NaOH (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with brine (10 mL) and dried over MgSO4. The solvent was evaporated, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 9 : 1 : 0.25) to afford a colorless oil (0.283 g, 84%). 1H NMR (CDCl3, 500 MHz): δ (8.209+8.152) (s, 1H), (7.641+7.636) (s, 1H), 7.301-7.285 (m, 2H), 7.199 (m, 2H), 6.642 (s, 1H), 4.454 (s, 2H), 3.370-3.092 (m, 7H), 2.796 (m, 2H), 2.660 (m, 1H), 2.446 (m, 2H), (2.413+2.391) (s, 3H), 1.523-1.417 (m, 27H). 13C NMR (CDCl3, 100.7 MHz): δ 156.659 (1C), 155.901 (1C), (154.780+154.727) (1C), 152.629 (1C), 152.235 (1C), (151.857+151.781) (1C), (137.209+137.057) (1C), 132.914 (1C), (129.074+128.658+128.430) (4C), 118.645 (1C), (109.374+109.298) (1C), 80.517 (1C), 80.070 (1C), 79.298 (1C), (59.264+58.401) (1C), 50.819 (1C), 50.183 (1C), (48.858+48.646) (1C), (47.222+47.063) (1C), 46.358 (1C), (43.139+42.473) (1C), (32.854+32.664) (1C), 28.491 (6C), 28.249 (3C), 23.826 (1C). MS (ESI, CH3OH): [C35H52ClN5O6] m/z 674.5 ([M+H]+).
N1-{(±)-4′-[(2”-amino-6”-methylpyridin-4”-yl)methyl]pyrrolidin-3′-yl}-N2-(4′-chlorobenzyl)ethane-1,2-diamine tetrahydrochloride (27)
The procedure to prepare 27 is the same as that to prepare 7 except using 66a (0.135 g, 0.2 mmol) instead of 37a, affording a hygroscopic white solid (0.104 g, quantitative yield). 1H NMR (D2O, 500 MHz): δ 7.4455 (d, 2H, J = 8.5 Hz), 7.3765 (d, 2H, J = 8.5 Hz), 6.700 (s, 1H), 6.632 (s, 1H), 4.374-4.334 (m, 1H), 4.317 (s, 2H), 3.990-3.948 (m, 1H), 3.781-3.741 (m, 1H), 3.678-3.604 (m, 4H), 3.443-3.405 (m, 1H), 3.305-3.266 (m, 1H), 3.161-3.140 (m, 2H), 2.720-2.665 (m, 1H), 2.355 (s, 3H). 13C NMR (D2O, 125.7 MHz): δ 155.550 (1C), 153.724 (1C), 147.332 (1C), 135.357 (1C), 131.639 (2C), 129.389 (2C), 128.768 (1C), 113.279 (1C), 109.818 (1C), 58.746 (1C), 51.089 (1C), 47.454 (1C), 45.219 (1C), 43.523 (1C), 42.977 (1C), 39.622 (1C), 31.624 (1C), 18.453 (1C). MS (ESI, CH3OH): [C20H28ClN5] m/z 374.81 ([M+H]+). HRMS (ESI, CH3OH) Calc.: 374.2111, Found: 374.2106.
N1-{(±)-4′-[(2”-amino-6”-methylpyridin-4”-yl)methyl]pyrrolidin-3′-yl}-N2-(3′-fluorophenethyl)ethane-1,2-diamine tetrahydrochloride (28)
The procedure to prepare 28 is the same as that to prepare 7 except using 66b (0.134 g, 0.2 mmol) instead of 37a, affording a hygroscopic white solid (0.103 g, quantitative yield). 1H NMR (D2O, 500 MHz): δ 7.316-7.306 (m, 1H), 7.086-7.076 (m, 1H), 7.029-6.971 (m, 2H), 6.708 (s, 1H), 6.632 (s, 1H), 4.351 (m, 1H), 3.972-3.960 (m, 1H), 3.776-3.751 (m, 1H), 3.610 (m, 4H), 3.407 (m, 3H), 3.292-3.272 (m, 1H), 3.161-3.142 (m, 2H), 3.030 (m, 2H), 2.721-2.670 (m, 1H), 2.357 (s, 3H). 13C NMR (D2O, 125.7 MHz): δ (163.889+161.473) (1C), 155.557 (1C), 153.717 (1C), 147.309 (1C), (138.485+138.410) (1C), (130.889+130.805) (1C), (124.966+124.943) (1C), (115.832+115.620) (1C), (114.377+114.173) (1C), 113.279 (1C), 109.795 (1C), 58.746 (1C), 49.226 (1C), 47.446 (1C), 45.234 (1C), 43.523 (2C), 39.622 (1C), 31.601 (2C), 18.445 (1C). 19F NMR (D2O, 376.5 MHZ): δ -113.609 (Ar-F). MS (ESI, CH3OH): [C21H30FN5] m/z 372.6 ([M+H]+). HRMS (ESI, CH3OH) Calc.: 372.2563, Found: 372.2556.
Enzyme and assay
All of the NOS isozymes used were recombinant enzymes overexpressed in E.coli. The murine macrophage iNOS was expressed and isolated according to the procedure of Hevel et al.50 The rat nNOS was expressed51 and purified as described.52 The bovine eNOS was isolated as reported.53 Nitric oxide formation from NOS was monitored by the hemoglobin capture assay as described at 30 °C.54 A typical assay mixture for nNOS and eNOS contained 10 μM L-arginine, 1.0 mM CaCl2, 600 unit/mL calmodulin (Sigma, P-2277), 100 μM NADPH, 0.125 mg/mL hemoglobin-A0 (ferrous form, Sigma, H0267), 10 μM H4B, in 100 mM HEPES (pH 7.5). A typical assay mixture for iNOS contained 10 μM L-arginine, 100 μM NADPH, 0.125 mg/mL hemoglobin-A0 (ferrous form), 10 μM H4B, in 100 mM HEPES (pH 7.5). All assays were in a final volume of 600 μL and were initiated by addition of enzyme. NOS assays were monitored at 401 nm on a Perkin-Elmer Lamda 10 UV-visible spectrophotometer.
Determination of Ki values
The apparent Ki values were obtained by measuring the percent enzyme inhibition in the presence of 10 μM L-arginine with at least five concentrations of inhibitor. The parameters of the following inhibition equation55 were fitted to the initial velocity data: % inhibition = 100[I]/{[I] + Ki(1+[S]/Km)}. Km values for L-arginine were 1.3 μM (nNOS), 8.2 μM (iNOS), and 1.7 μM (eNOS). The selectivity of an inhibitor was defined as the ratio of the respective Ki values.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health, GM 49725 to RBS, GM57353 to TLP, and GM52419 to Dr. Bettie Sue Masters, with whose lab PM and LJR are affiliated, and Grant No. AQ1192 from The Robert A. Welch Foundation to BSM. P.M. is also supported by Grant KAN200200651 from Academy of Science, Czech Republic.
Abbreviations
- NO
nitric oxide
- NOS
nitric oxide synthase
- nNOS
neuronal NOS
- iNOS
inducible NOS
- eNOS
endothelial NOS
- SAR
structure-activity relationship
- HTS
high throughput screening
- sGC
soluble guanylate cyclase
- GTP
guanosine 5′-triphosphate
- cGMP
guanosine 3′,5′-cyclic monophosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- H4B
(6R)-5,6,7,8-tetrahydro-L-biopterin
- CaM
calmodulin
- CPCA
consensus principle component analysis
- MCSS
multiple copy simultaneous search
- BBB
blood brain barrier
- MIFs
molecular interaction fields
- PSA
polar surface area
- RMSD
root-mean-square deviation
- DIAD
diisopropyl azodicarboxylate
- DPPA
diphenylphosphoryl azide
References
- 1.(a) Erlanson DA, McDowell RS, O’Brien T. Fragment-based drug discovery. J. Med. Chem. 2004;47(14):3463–3482. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]; (b) Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004;3(8):660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]; (c) Erlanson DA. Fragment-based lead discovery: a chemical update. Curr. Opin. Biotechnol. 2006;17(6):643–652. doi: 10.1016/j.copbio.2006.10.007. [DOI] [PubMed] [Google Scholar]; (d) Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J. Med. Chem. 2008;51(13):3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 2.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007;6(3):211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 3.(a) Mauser H, Guba W. Recent developments in de novo design and scaffold hopping. Curr. Opin. Drug Discov. Devel. 2008;11(3):365–374. [PubMed] [Google Scholar]; (b) Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005;4(8):649–663. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- 4.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J. Am. Chem. Soc. 2008;130(12):3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Bredt DS, Snyder SH. Nitric oxide: A Physiologic Messenger Molecule. Annu. Rev. Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]; (b) Kerwin JF, Jr., Lancaster JR, Jr., Feldman PL. Nitric oxide: A New Paradigm for Second Messengers. J. Med. Chem. 1995;38(22):4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 6.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. The Chemical Biology of Nitric Oxide: Implications in Cellular Signaling. Free Radic. Biol. Med. 2008;45(1):18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cary SPL, Winger JA, Derbyshire ER, Marletta MA. Nitric Oxide Signaling: No Longer Simply On or Off. Trends Biochem. Sci. 2006;31(4):231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Moncada S, Palmer RM, Higgs EA. Nitric Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 9.Domenico R. Pharmacology of Nitric Oxide: Molecular Mechanisms and Therapeutic Strategies. Curr. Pharm. Des. 2004;10(14):1667–1676. doi: 10.2174/1381612043384709. [DOI] [PubMed] [Google Scholar]
- 10.Rosen GM, Tsai P, Pou S. Mechanism of Free-Radica Generation by Nitric Oxide Synthase. Chem. Rev. 2002;102(4):1191–1199. doi: 10.1021/cr010187s. [DOI] [PubMed] [Google Scholar]
- 11.Knowles RG, Moncada S. Nitric Oxide Synthases in Mammals. Biochem. J. 1994;298(2):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alderton WK, Cooper CE, Knowles RG. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem. J. 2001;357(3):593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roman LJ, Martásek P, Masters BSS. Intrinsic and Extrinsic Modulation of Nitric Oxide Synthase Activity. Chem. Rev. 2002;102(4):1179–1189. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 14.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of Cerebral Ischemia in Mice Deficient in Neuronal Nitric Oxide Synthase. Science. 1994;265(5180):1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 15.(a) Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of Neuronal Nitric Oxide in 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Dopaminergic Neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 1996;93(10):4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of Neuronal Nitric Oxide Synthase prevents MPTP-induced Parkinsonism in Baboons. Nat. Med. 1996;2(9):1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 16.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441(7092):513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 17.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310(5756):1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 18.(a) Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in Mice Lacking the Gene for Endothelial Nitric Oxide Synthase. Nature. 1995;377(6546):239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]; (b) Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for Stroke Protection. Trends Neurosci. 2004;27(5):283–289. doi: 10.1016/j.tins.2004.03.009. [DOI] [PubMed] [Google Scholar]; (c) Braam B, Verhaar MC. Understanding eNOS for Pharmacological Modulation of Endothelial Function: A Translational View. Curr. Pharm. Des. 2007;13(17):1727–1740. doi: 10.2174/138161207780831275. [DOI] [PubMed] [Google Scholar]
- 19.(a) Salerno L, Sorrenti V, Di Giacomo C, Romeo G, Siracusa MA. Progress in the development of selective nitric oxide synthase (NOS) inhibitors. Curr. Pharm. Des. 2002;8(3):177–200. doi: 10.2174/1381612023396375. [DOI] [PubMed] [Google Scholar]; (b) Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Selective neuronal nitric oxide synthase inhibitors. Curr. Top. Med. Chem. 2005;5(7):603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]; (c) Tafi A, Angeli L, Venturini G, Travagli M, Corelli F, Botta M. Computational studies of competitive inhibitors of nitric oxide synthase (NOS) enzymes: towards the development of powerful and isoform-selective inhibitors. Curr. Med. Chem. 2006;13(16):1929–1946. doi: 10.2174/092986706777585031. [DOI] [PubMed] [Google Scholar]
- 20.Vallance P, Leiper J. Blocking NO Synthesis: How, Where and Why? Nat. Rev. Drug Discov. 2002;1(12):939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 21.(a) Silverman RB, Huang H, Marletta MA, Martásek P. Selective Inhibition of Neuronal Nitric Oxide Synthase by Nω-Nitroarginine-and Phenylalanine-Containing Dipeptides and Dipeptide esters. J. Med. Chem. 1997;40(18):2813–2817. doi: 10.1021/jm970200u. [DOI] [PubMed] [Google Scholar]; (b) Zhang HQ, Fast W, Marletta MA, Martásek P, Silverman RB. Potent and Selective Inhibition of Neuronal Nitric Oxide Synthase by Nω-propyl-L-arginine. J. Med. Chem. 1997;40(24):3869–3870. doi: 10.1021/jm970550g. [DOI] [PubMed] [Google Scholar]
- 22.(a) Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279(5359):2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]; (b) Raman CS, Li H, Martásek P, Kral V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95(7):939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]; (c) Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat. Struct. Biol. 1999;6(3):233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]; (d) Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-alkyl-N’-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41(47):13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 23.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martásek P, Roman LJ, Vásquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors and the prevention of cerebral palsy. Ann. Neurol. 2008 doi: 10.1002/ana.21555. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Martásek P, Roman LJ, Masters BS, Silverman RB. Nω-Nitroarginine-containing dipeptide amides. Potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 1999;42(16):3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 25.Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Potent and selective conformationally restricted neuronal nitric oxide synthase inhibitors. J. Med. Chem. 2004;47(3):703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 26.Ji H, Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Confomationally restricted dipeptide amide as potent and selective neuronal nitric oxide synthase inhibitors. J. Med. Chem. 2006;49(21):6254–6263. doi: 10.1021/jm0604124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hah J-M, Roman LJ, Martásek P, Silverman RB. Reduced amide bond peptidomimetics. (4S)-N-(4-amino-5-[aminoakyl]aminopentyl)-N’-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2001;44(16):2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 28.Hah J-M, Martásek P, Roman LJ, Silverman RB. Aromatic reduced amide bondpeptidomimetics as selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2003;46(9):1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]
- 29.(a) Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah J-M, Gómez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat. Struct. Mol. Biol. 2004;11(1):54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]; (b) Flinspach M, Li H, Jamal J, Yang W, Huang H, Silverman RB, Poulos TL. Structures of the Neuronal and Endothelial Nitric Oxide Synthase Heme Domain with D-Nitroarginine-Containing Dipeptide Inhibitors Bound. Biochemistry. 2004;43(18):5181–5187. doi: 10.1021/bi0361867. [DOI] [PubMed] [Google Scholar]; Li H, Flinspach ML, Igarashi J, Jamal J, Yang W, Gómez-Vidal JA, Litzinger EA, Huang H, Erdal EP, Silverman RB, Poulos TL. Exploring the binding conformations of bulkier dipeptide amide inhibitors in constitutive nitric oxide synthases. Biochemistry. 2005;44(46):15222–15229. doi: 10.1021/bi0513610. [DOI] [PubMed] [Google Scholar]
- 30.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. Computer modeling of selective regions in the active site of nitric oxide synthases: implication for the design of isoform-selective inhibitors. J. Med. Chem. 2003;46(26):5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 31.(a) Wade RC, Goodford PJ. Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 1. Ligand and probe groups with the ability to form two hydrogen bonds. J. Med. Chem. 1993;36(1):140–147. doi: 10.1021/jm00053a018. [DOI] [PubMed] [Google Scholar]; (b) Wade RC, Goodford PJ. Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 2. Ligand and probe groups with the ability to form more than two hydrogen bonds. J. Med. Chem. 1993;36(1):148–156. doi: 10.1021/jm00053a019. [DOI] [PubMed] [Google Scholar]
- 32.Miranker A, Karplus M. Functionality maps of binding sites: a multiple copy simultaneous search method. Proteins. 1991;11(1):29–34. doi: 10.1002/prot.340110104. [DOI] [PubMed] [Google Scholar]
- 33.(a) Böhm H-J. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J. Comput.-Aided Mol. Des. 1992;6(1):61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]; (b) Böhm H-J. LUDI: Rule-Based Automatic Design of New Substitutents for Enzyme Inhibitor Leads. J. Comput.-Aided Mol. Des. 1992;6(6):593–606. doi: 10.1007/BF00126217. [DOI] [PubMed] [Google Scholar]
- 34.Lawton GR, Ji H, Silverman RB. Remote protection prevents unwanted cyclizations with 2-aminopyridines. Tetrahedron Lett. 2006;47(34):6113–6115. [Google Scholar]
- 35.Igarashi J, Li H, Jamal J, Ji H, Fang J, Silverman RB, Poulos TL. Crystal structures of constitutive nitric oxide synthases in complex with de novo designed inhibitors. J. Biol. Chem. 2008 doi: 10.1021/jm900007a. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000;43(20):3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 37.BioByte Corp. 201 W. Fourth Street, Suite 204, Claremont, CA 91711.( http://www.biobyte.com/)
- 38.ACD/log D suite, Advanced Chemistry Development Inc. 110 Yonge Street, 14th floor, Toronto-Ontario, Canada M5C 1T4. ( http://www.acdlabs.com)
- 39.Cruciani G, Carosati E, De Boeck B, Ethirajulu K, Mackie C, Howe T, Vianello R. MetaSite: Understanding Metabolism in Human Cytochromes from the Perspective of the Chemist. J. Med. Chem. 2005;48(22):6970–6979. doi: 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]
- 40.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nature Chem. Biol. 2008;4(11):700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silverman RB. Design of Selective Neuronal Nitric Oxide Synthase Inhibitors for the Prevention and Treatment of Neurodegenerative Diseases. Acc. Chem. Res. doi: 10.1021/ar800201v. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Insight II 2000: Accelrys Inc. 10188 Telesis Court, Suite 100, San Diego, CA 92121, Phone: (858) 799-5000 Fax: (858)799-5100. http://www.accelrys.com/
- 43.SYBYL 6.8: Tripos, Inc. 1699 South Hanley Road St. Louis, MO 63144-2319 Phone: (800) 323-2960. Fax: (314) 647-9241. http://www.tripos.com/
- 44. GRID 22: Molecular Discovery, Via Stoppani, 38, 06087 - Ponte San Giovanni - PG, ITALY. Telephone and fax: +39-075-397411. ( http://www.moldiscovery.com/)
- 45.Evensen ER, Joseph-McCarthy D, Karplus M. MCSS, version 2.1. Harvard University; Carmbridge, MA: 1997. [Google Scholar]
- 46.Böhm H-J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J. Comput.-Aided Mol. Des. 1994;8(3):243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- 47.Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 1998;19(14):1639–1662. [Google Scholar]
- 48.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. A new force field for molecular mechanical simulation of nucleic acid and proteins. J. Am. Chem. Soc. 1984;106(3):765–784. [Google Scholar]
- 49.Gasteiger J, Marsili M. Iterative paratial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron. 1980;36(22):3219–3228. [Google Scholar]
- 50.Hevel JM, White KA, Marletta M. A purification of the inducible murine macrophage nitric oxide synthase. J. Biol. Chem. 1991;266(34):22789–22791. [PubMed] [Google Scholar]
- 51.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. High-level expression of functional rat neuronal nitric oxide synthase in escherichia coli. Proc.Natl. Acad. Sci. U.S.A. 1995;92(18):8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerber NC, Ortiz de Montellano PR. Neuronal nitric oxide synthase: expression in Escherichia coli, irreversible inhibition by phenyldiazene, and active site topology. J. Biol. Chem. 1995;270(30):17791–17796. doi: 10.1074/jbc.270.30.17791. [DOI] [PubMed] [Google Scholar]
- 53.Martasek P, Liu Q, Roman LJ, Gross SS, Sessa WC, Masters BSS. Characterization of bovine endothelial nitric oxide synthase expressed in Escherichia coli. Biochem. Biophys. Res. Commun. 1996;219(2):359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- 54.Hevel JM, Marletta MA. Nitric oxide synthase assays. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 55.Segel IH. Enzyme Kinetics. John Wiley and Sons; NewYork: 1975. p. 105. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.