

Abstract
A number of small organic molecules have been developed that bind to amyloid fibrils, a subset of which also inhibit fibrillization. Among these, the benzothiol dye, Thioflavin-T (ThT), has for decades been used in the diagnosis of protein-misfolding diseases and also in kinetic studies of self-assembly (fibrillization). Despite its importance, efforts to characterize the ThT binding mechanism at an atomic level have been hampered by the inherent insolubility and heterogeneity of peptide self-assemblies. To overcome these challenges, we have developed a minimalist approach to designing a ThT-binding site in a “peptide self-assembly mimic” (PSAM) scaffold. PSAMs are engineered water-soluble proteins that mimic a segment of β-rich peptide self-assembly, and they are amenable to standard biophysical techniques and systematic mutagenesis. The PSAM β-sheet contains rows of repetitive amino acid patterns running perpendicular to the strands (“cross-strand ladders”) that represent a ubiquitous structural feature of fibril-like surfaces. We successfully designed a ThT-binding site that recapitulates the hallmarks of ThT-fibril interactions by constructing a cross-strand ladder consisting of contiguous tyrosines. Their x-ray crystal structures suggest that ThT interacts with the β-sheet by docking on surfaces formed by a single tyrosine ladder, rather than in the space between adjacent ladders. Systematic mutagenesis further demonstrated that tyrosine surfaces across four or more β-strands formed the minimal binding site for ThT. Our work thus provides structural insights into how this widely used dye recognizes a prominent subset of peptide self-assemblies and proposes a strategy to elucidate the mechanisms of fibril-ligand interactions.
Keywords: amyloid fibrils, cross-β, β-sheet, tyrosine, protein engineering
The formation of β-rich peptide self-assemblies is associated with a diverse set of the so-called protein misfolding diseases.1; 2 Accordingly, there have been intense interest and research activities in the development of small organic molecules for the detection and inhibition of fibril formation.3–7 Although it is clear that understanding how these small molecules interact with fibrils would greatly aid the development of effective diagnostic and therapeutic reagents, there is a paucity of experimental data that provide such information at an atomic level. Fibrils and other peptide self-assemblies are insoluble and often heterogeneous in their composition, making it extremely difficult to characterize their interactions with ligands using high-resolution biophysical techniques. In this work, we report an alternative, protein-engineering approach to addressing this seemingly intractable problem.
Since its introduction in 1959, the benzothiol dye, Thioflavin-T (ThT), (Fig. 1a) has been a cornerstone in diagnosing amyloid fibrils.8 Its dramatic increase in fluorescence upon binding to β-sheet-rich deposits has made it an essential tool for staining amyloid in tissue samples, as well as in real-time monitoring of self-assembly kinetics in vitro.9–11 ThT represents a growing number of small organic compounds that bind to and sometimes inhibit amyloid fibrils.3; 6 Yet despite nearly 50 years of prominent use, the mechanism by which ThT recognizes and binds peptide self-assemblies remains poorly understood.
Figure 1.

PSAM model for ThT binding. (a) Chemical structure of ThT. (b) Adjacent cross-strand side-chain ladders of a self-assembled antiparallel β-sheet. The sheet backbone is depicted as lines between α-carbons. Side-chains indicated circles inscribed with the letter R. (c) Scheme illustrating the PSAM concept. A segment of peptide self-assembly is hypothetically excised, linked, and capped. The crystal structure of the 8-EK/KE PSAM is shown as a cartoon model on the right, with the N- and C-terminal caps colored black and the SLB colored grey. (d) Amino acid sequence of the wild-type PSAM SLB shown according to the β-sheet topology. The rectangles indicate β-strand residues. The side chains of the residues enclosed in the dashed lines point toward the reader, and those of the other β-strand residues point away from the reader. The designations of the four β-hairpin repeats (“0” – “3”) are shown. Hydrogen-bonding partners are depicted with arrows pointing from donor to acceptor. Note that 3-, 4-, 5-, and 8-YY/LL all contain a V201L mutation in a turn region of β-hairpin 0 (denoted with an asterisk), which occurred as an unintended consequence of gene construction. Leu is present in the homologous positions (L132, L155, and L178) of β-hairpins 1, 2 and 3, and therefore likely has a negligible effect on the structure, stability, and dye-binding properties of the PSAM.
Existing models of ThT binding are both incomplete and conflicting. Some research has suggested ThT binds promiscuously to a wide variety of substrates and that ThT micelles interact with fibrils.12 However, these models of ThT interaction cannot account for the chiral bias of bound ThT,13; 14 nor its preference to bind parallel to the long axis of fibrils.15 It is therefore increasingly accepted that ThT and other amyloid dyes bind to a common structural motif in fibrils.16; 17 Moreover, the fact that not all fibrils bind ThT further suggests that ThT recognizes certain structural and/or chemical features within fibrils.
β-Rich peptide self-assemblies, such as fibrils, commonly adopt the “cross-β” structure, characterized by an extended β-conformation with the strands running perpendicular to the long axis of the assembly and lamination of multiple β-sheets.18 Both parallel and anti-parallel β-strand orientations give rise to a specific arrangement of side-chains in self-assemblies, termed “cross-strand ladders”.19 These cross-strand ladders consist of repeating side-chain interactions running across β-strands within a β-sheet layer (i.e. parallel to the long axis of the assembly; Fig. 1b) and arise from the inherently repetitive nature of self-assembly. Recent structures of short fibril-forming peptides demonstrate that these neighboring rows of cross-strand side-chains occur regardless of peptide sequence, and form extended channel-like motifs along solvent-exposed surfaces of the assembly into which linear dyes could bind (Fig. 1b).20–23 Our hypothesis is that ThT binds to the β-sheet surface along motifs formed by cross-strand ladders of certain amino acid composition, hence rationalizing the ThT-binding properties of many peptide self-assemblies.
Unfortunately, characterizing the role the side-chains of cross-strand ladders play in ThT binding is hampered by the experimental intractability of fibrils. Previous models of ThT binding are therefore based on macroscopic observations and are ambiguous at the molecular level. Though recent work has yielded atomic-resolution insight into microcrystalline self-assemblies, which could in principle be used as the starting structure for systematic mutagenesis experiments for ThT binding, the surface properties of the short peptide subunits cannot be precisely manipulated without disrupting, destabilizing or altering the mode of assembly.20; 21 Recently, the x-ray crystal structure of ThT bound to acetylcholine esterase was determined 24. Although this structure reveals interactions responsible for extremely high affinity (Kd in the low nM range), the binding site is constructed with residues in mostly α-helical regions of the enzyme, providing little insights into the mechanism of ThT-fibril interactions. Thus it remains a major challenge to elucidate the minimal chemical and structural requirements for ThT binding to fibrils.
In an attempt to overcome many of these experimental limitations of short peptide studies, we employed a complementary approach by examining ThT binding using a “peptide self-assembly mimic” (PSAM) derived from Borrelia outer surface protein A (OspA) (Figs. 1c and 1d).25 PSAMs covalently link a fixed number of β-hairpin units into a single-layer β-sheet (SLB) scaffold, thereby eliminating the heterogeneity in composition of self-assembly. This sheet is capped by N- and C-terminal globular domains that prevent lateral assembly and lamination. Unlike fibrils, PSAMs are water-soluble, crystallization-efficient and highly amenable to site-directed mutagenesis, allowing us to precisely control the surface properties of the β-sheet.26; 27 A peptide corresponding to the β-hairpin unit readily forms β-rich self-assemblies, suggesting that the underlying subunit is primed for self-assembly and that the combination of the PSAM and the peptide can serve as a model for studying mechanisms of peptide self-assembly.28; 29 Although our current PSAM system mimics only anti-parallel self-assemblies due to the strand topology of OspA, it can still provide fundamental information that would be otherwise unattainable. Using PSAMs as “host” scaffolds, we can introduce a variety of “guest” mutations and study their effects on ThT binding in solution using well-established biophysical methods, thus providing a unique opportunity to systematically define chemical and structural requirements for the interaction of ThT with self-assembly-like materials.
Results
Design of cross-strand ladders
We selected a PSAM scaffold containing four homologous β-hairpins comprising a total of eight β-strands (Figs. 1c and 1d),30 which we will refer to it as the “wild type” hereafter. This scaffold’s large SLB minimizes the context effects from the N- and C-terminal globular domains, and at the same time its sequence can be manipulated efficiently. Furthermore, we have successfully crystallized this PSAM,25; 27 suggesting a good likelihood of obtaining atomic structures of mutants.
The PSAM β-sheet contains several endogenous cross-strand ladders and can be considered a structural mimic of the surface of fibrils (Fig. 1d). However, ThT did not appreciably bind to the wild-type PSAM (Figs. 2b and 2c). This scaffold contains mostly polar and charged amino acids in the SLB (Fig. 1d), and its lack of ThT binding agrees with ThT’s low affinity to highly charged fibrils.17; 31 Thus, the PSAM provides an inert platform on which we can attempt to design a ThT-binding site.
Figure 2.

Design of single cross-strand ladders in the PSAM and their ThT binding properties. (a) Schemes of single Tyr-ladders of different lengths used in this work. Note that the locations of the Tyr ladder mutations were dictated by our limited ability to specifically introduce mutations at certain positions due to the redundancy of the PSAM gene. (b) Fluorescence emission spectra of 10 μM ThT in the absence and presence of 100 μM 8-YY/KE or 100 μM 8-EK/KE. (c) ThT fluorescence emission as a function of the number of contiguous cross-strand Tyr residues. Fluorescence intensity at 485 nm is shown after subtracting a blank (no protein) spectra of 10 μM ThT, and are normalized relative to the 8-YY/KE signal.
We selected 16 residues, two adjacent ladders each containing 8 residues, in the center of the SLB as “host” positions into which amino acid substitutions were introduced (Fig. 1d). These positions exhibit particularly regular β-sheet geometry because they are distant from the turn regions of the scaffold. These ladders are referred to as Ladder1 and Ladder2 (Fig. 1d). We emphasize that these ladder represent motifs across β-strands, not continuous segments along the peptide chain. Because PSAMs are designed to mimic peptide self-assemblies, each ladder always contains four repeats of a two-residue basic unit. In the wild-type PSAM, Ladder1 consists of four repeats of Glu and Lys (EK), while Ladder2 consists of four repeats of Lys and Glu (KE) (Fig. 1d). We hereafter will refer to these mutants in the form of [ladder length]−[basic unit for Ladder1]/[basic unit for Ladder2]. Hence, the wild-type pattern is denoted 8-EK/KE (Figs. 1d and 2a).
Single Tyr ladders define ThT-binding threshold
As aromatic amino acids are often present in self-assembling peptides32 and have been implicated in computational models of ThT binding,16 we initially investigated whether a cross-strand ladders consisting solely of Tyr could promote interactions between the PSAM and ThT. We had previously designed the 8-YY/KE mutant by replacing all residues in Ladder1 with Tyr in order to study the structural and stability effects of long aromatic ladders (Fig. 2a).27 Because this PSAM was already well characterized and it is soluble and monomeric,27 we used it in our initial screen for ThT binding.
Surprisingly, ThT bound to 8-YY/KE with many of the hallmarks of ThT binding to true peptide self-assemblies, including a dramatic increase in fluorescence emission at 485 nm (Fig. 2b). To define the minimal length required for ThT binding to the Tyr ladders, we constructed smaller ladders in the 8-EK/KE scaffold, termed 2-, 3-, 4-, and 6-YY/KE (Fig. 2a). Due to restrictions in mutagenesis imposed by repetitive DNA sequences for the β-hairpin repeats, the positions of the Tyr ladders in these mutants varied for different ladder lengths. ThT bound at comparably low levels to PSAMs containing Tyr ladders shorter than four residues. ThT binding became significant when the Tyr ladder was lengthened between four and six residues (Fig. 2c), suggesting the minimal ladder length for ThT binding lies between these values (see below for further investigation). While we observed a substantial range of ThT binding to these PSAMs, fluorescence emission measured as a function of PSAM concentration did not show a sign of saturation even at a PSAM concentration of 100 μM (data not shown). This indicates the affinity of ThT for these PSAMs is much weaker than the 1–5 μM Kd of the dye to many amyloid fibrils.3; 17 Therefore, additional factors must contribute to the higher-affinity interactions between amyloid fibrils and ThT.
Side-by-side ladders create high-affinity ThT-binding sites
The discovery that the Tyr-rich cross-strand ladders confer ThT binding on otherwise inert PSAMs suggest that linear stretches of solvent-exposed residues in peptide self-assemblies are important determinants for ThT binding. This view is consistent with the channel model in which ThT binds between two adjacent ladders on the surface of the self-assembly. To assess the role of neighboring ladders in the ThT binding mechanism, we introduced eight Leu residues into Ladder2 to create the 8-YY/LL mutant (Fig. 3a). These side-by-side ladders of Tyr and Leu are homologous to the alternating Val-Phe ladders that have been suggested as a possible ThT-binding site on amyloid-β.16 We additionally designed smaller versions of the YY/LL ladders termed 3-, 4-, and 5-YY/LL, which contain cross-strand ladders stretching only three, four, and five residues (Fig. 3a). These mutants were constructed by disrupting the continuous Tyr and Leu ladders of 8-YY/LL with the wild-type, E/K (or K/E) pattern within a β-strand (Fig. 3a). The lack of ThT binding to the 8-EK/KE ladders (Figs. 2b and 2c) suggested that the intra-strand E/K (or K/E) pattern would effectively bisect the YY/LL ladders into shorter ones. The 3-, 4-, and 5-YY/LL PSAMs were expressed as monomeric and soluble proteins as judged by size-exclusion chromatography (data not shown). In contrast, 8-YY/LL did not exhibit a monodispersed elution profile, and thus we eliminated it from further analysis.
Figure 3.
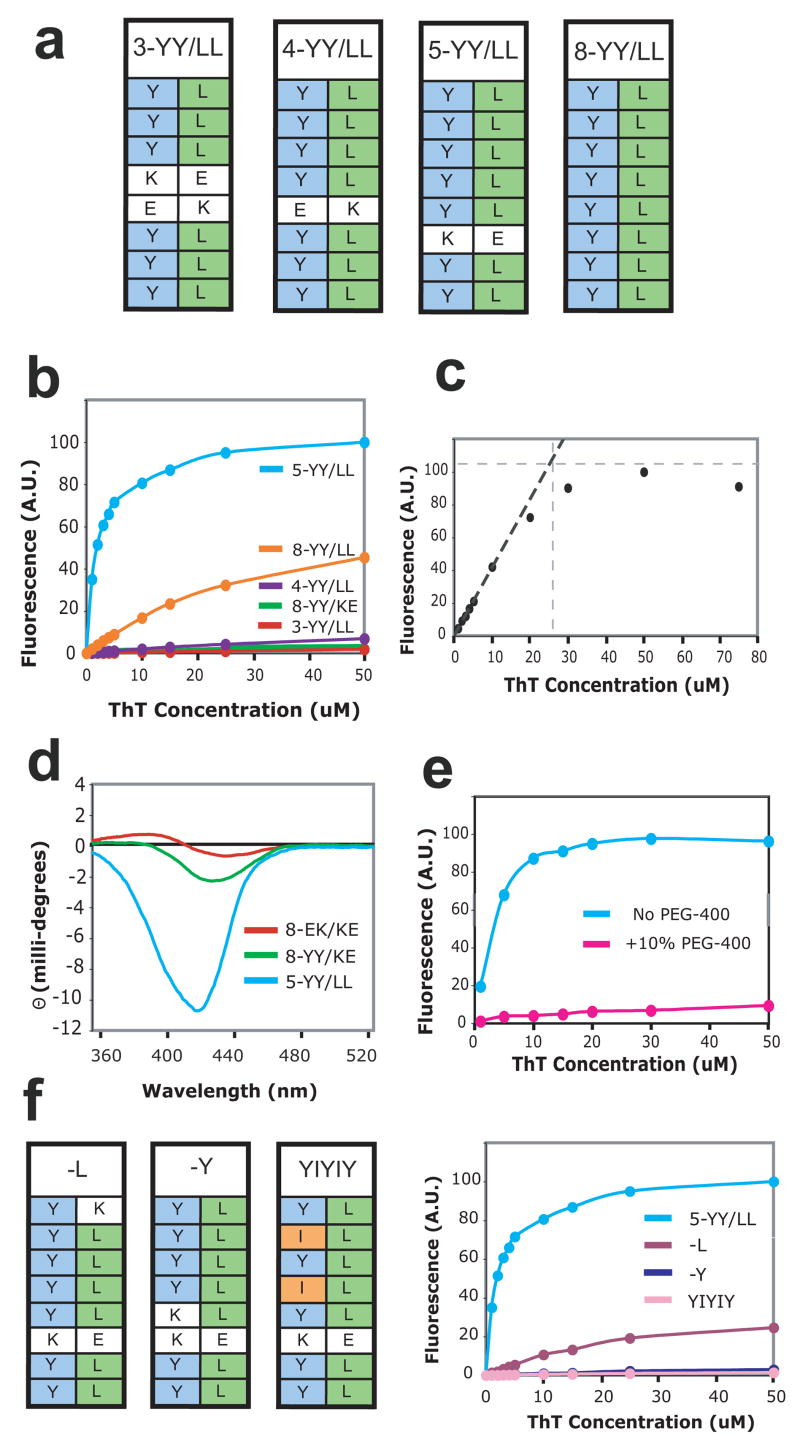
Design of side-by-side ladders and their ThT binding. (a) Schemes of the side-by-side ladders used in this work. (b) ThT binding titrations measuring ThT fluorescence emission at 485 nm. Protein concentrations were 10 μM. (c) Determination of the stoichiometry of ThT binding to the PSAM by binding site titration. ThT fluorescence emission intensity at 485 nm is plotted as a function of ThT concentration. The binding titration was performed with 25 μM 5-YY/LL PSAM, i.e. a protein concentration much higher than the Kd of the binding reaction. Extrapolation of the linear portion of the curve near the origin is shown as the black dashed line. The gray horizontal dashed line is a saturation value of ThT fluorescence estimated at ~110 A.U. The intercept of the two lines indicates that the saturating ThT concentration is ~25 μM, i.e. a 1:1 binding stoichiometry between ThT and the PSAM. (d) CD spectra of 0.5 mM ThT in the presence of 0.5 mM of the PSAM variants. (e) Crystallization conditions impede observation of bound ThT. ThT binding titrations to 10 μM 5-YY/LL in the presence (pink) and absence (blue) of 10% v/v PEG-400. Note that a 40% v/v PEG-400 solution was used in crystallization. (f) Schemes of point mutants of 5-YY/LL, termed –Y, –L, and YIYIY. On the right are shown ThT binding titrations of these mutants, measured as described in (b).
Of all PSAMs constructed to date, 5-YY/LL displayed the highest affinity for ThT, with a Kd of ~2.5 μM that is comparable to that observed for amyloid fibrils (Fig. 3b). ThT binding titrations performed at a high concentrations of 5-YY/LL with respect to the Kd determined the ThT:PSAM binding stoichiometry to be ~1:1 (Fig. 3c). Moreover, ThT titration data for 5-YY/LL (Fig. 3b) show no significant deviation from a hyperbolic binding curve near the critical micelle concentration of ThT, reported previously as between 4–8 μM in a solution of comparable composition 12; 17. Along with stoichiometry data, these results indicate that a single ThT molecule binds to each PSAM. Circular dichroism (CD) measurements of ThT in complex with 5-YY/LL revealed the Cotton Effect peak near 450 nm, similar to ThT bound to fibrils (Fig. 3d). The Cotton Effect indicates that ThT is bound in a chiral environment and is twisted in terms of the rotation about the bond between the two rings.13; 14; 17 This immobilization has been viewed as the cause of the increase in quantum yield responsible for ThT’s characteristic fluorescence enhancement upon fibril binding.33 Together, these results demonstrate that 5-YY/LL fully recapitulates a fibril-like ThT-binding site.
In corroboration of our findings on the ladder length requirement from the single Tyr ladder (Fig. 2c), 3-YY/LL lost nearly all ability to bind ThT, while 4-YY/LL showed a diminished, but still observable interaction with the dye (Fig. 3b). The YY/LL mutants therefore refine our knowledge of the ladder-length dependence for ThT binding, demonstrating that while low-affinity interactions can be mediated by ladders stretching four cross-strand residues, ladders across five or more β-strands are required for fibril-like affinity.
As side-by-side ladders may comprise generic binding sites for small molecules on peptide self-assemblies, we further investigated the ability of 5-YY/LL to bind a variety of common amyloid dyes (BTA-1, Congo Red and bis-ANS; Fig. 4a). The naphthalene sulfonate dye, bis-ANS, showed a substantial increase in fluorescence upon binding to 5-YY/LL (Fig. 4b) and competed with ThT (Fig. 4c), suggesting these dyes share a common binding site on the PSAM. In contrast, we did not observe binding of the uncharged ThT derivative BTA-1 (data not shown), which differs from the parent molecule by a several exo-cyclic methyls.3 Our results are consistent with previous work showing that, despite their structural similarity, ThT and BTA bind to distinct sites on fibrils.3 We also found no evidence for binding of the amyloid dye Congo Red to 5-YY/LL (data not shown). This may be because the designed ThT-binding site is too small for Congo Red (the tip-to-tail length of Congo Red is ~26 Å, compared to ~15 Å for ThT; Fig. 4a), or it requires a distinct binding motif.17
Figure 4.
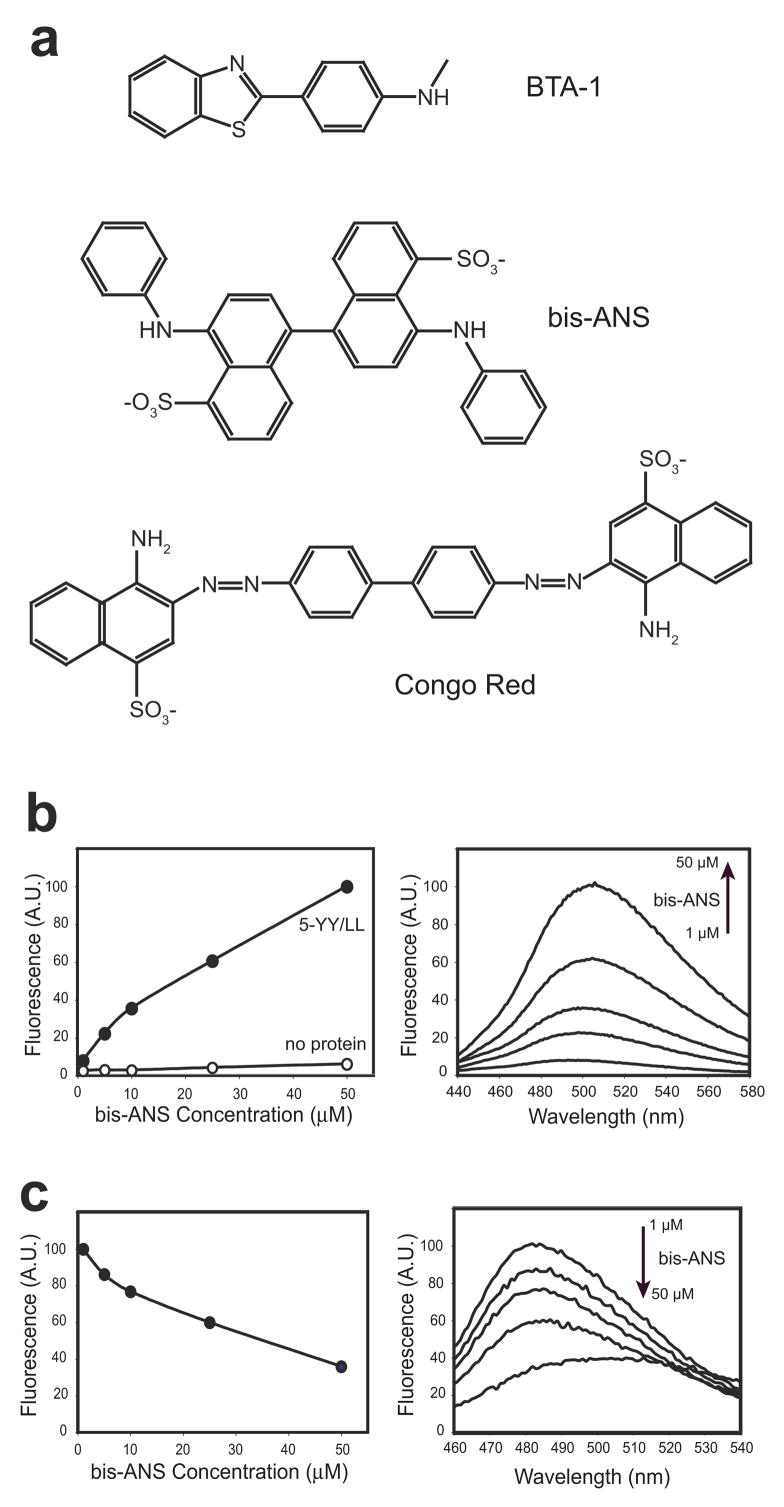
Binding of other dyes to side-by-side ladders. (a) Structures of dyes used. (b) Bis-ANS titration in the presence (solid circles) and absence (open circles) of 10 μM 5-YY/LL. Fluorescence emission intensity at 505 nm is plotted as a function of bis-ANS concentration. Spectra of 1, 5, 10, 25 and 50 μM bis-ANS in the presence of 5-YY/LL are shown on the right. (c) Competition of ThT binding by bis-ANS, conducted in the presence of 1 μM 5-YY/LL and 5 μM ThT. Fluorescence emission at 485 nm is plotted as a function of bis-ANS concentration. Spectra from this competition experiment are shown on the right. The same concentrations of bis-ANS were used as in (b). Note the gradual decrease in the ThT peak (maximum near 485 nm) and corresponding increase in the bis-ANS peak (maximum near 505 nm).
High-resolution x-ray crystal structures of ThT-binding PSAMs
To further understand the structural basis for ThT binding, we determined the x-ray crystal structures of the strongest ThT binders from both the single- and side-by-side ladder mutants. We introduced a set of surface mutations into the N- and C-terminal globular domains of the PSAM, which facilitates crystallization without perturbing the structure of the SLB region (see Methods).25; 30 The structure of 5-YY/LL was determined at a 1.75 Å resolution (Table 1), while the structure of 8-YY/KE at 1.50 Å has been reported (previously referred to as (YY)4).27
Table 1.
Statistics for crystal structure of 5-YY/LL (PDB 3EC5).
| Data collection statistics | |
| Space group | P 1 21 1 |
| Cell parameters | a=53.073 b=35.876 c=92.606 β=105.85 |
| Beamline | APS 23-ID |
| Wavelength | 0.9793 |
| Resolution (Å) a) | 20-1.75 (1.81–1.75) |
| Completeness(%) | 99.8 (97.6) |
| I/σ(I) | 14.81 (1.74) |
| Rmerge b) | 0.087 (0.663) |
| Average redundancy | 3.6 (3.5) |
| Refinement statistics | |
| Resolution range (Å) | 20.00-1.75 |
| Reflections used (free) | 30820 (3435) |
| R factor c) | 0.22311 |
| Rfree d) | 0.27388 |
| RMS deviations | |
| Bonds (Å) | 0.014 |
| Angles (°) | 1.504 |
| No. protein residues | 315 |
| No. waters | 249 |
| Average B factor (Å2) | 9.558 |
| Ramachandran plot statistics | |
| Most favored (%) | 91.4 |
| Additionally allowed (%)8.6 | |
| Generally allowed (%) | 0.0 |
Highest resolution shell is shown in parenthesis.
R-merge = ShklSi| I(hkl)i − <I(hkl)> |/ShklSi<I(hkl)i> over i observations of a reflection hkl.
R-factor = S ||F(obs)| − |F(calc)||/S|F(obs)|.
Rfree is R with 10% of reflections sequestered before refinement.
Crystals were obtained using the hanging-drop vapor diffusion method. A protein-ThT was first prepared in 10 mM Tris-Cl pH 8.0 to a final concentration of 29 mg/mL protein and 2 mM ThT. One μL of the protein-ThT complex was mixed with 1 μL of a well solution containing 40% PEG-400, 2% glycerol, 0.85 M Tris-Cl pH 9.0, 0.25 M (NH4)2SO4, and inverted over the well. Crystals grew in 3–4 days, and the mother liquor was used as a cryo-protectant.
Although crystallization of 5-YY/LL was performed in the presence of excess ThT, no electron density interpretable as ThT was observed. This is probably due to inhibitory effects of the 40% (v/v) PEG-400 precipitant required for crystallizing 5-YY/LL. We found that even 10% PEG-400 ablated ThT binding (Fig. 3e). Two well-resolved portions of PEG molecules were observed near the presumed ThT-binding site (Fig. 5b), suggesting direct competition between ThT and PEG-400. Small molecules such as glycerol are known to compete with ThT for binding to fibrils.34; 35 Although no bound ThT was observed, the putative ThT-binding site of 5-YY/LL is exposed to a large solvent-channel in the crystal, and is not involved in crystal contacts (Fig. 5a). Therefore, the 5-YY/LL crystal structure likely represents a dominant structure in solution of the ThT-binding cross-strand ladders.
Figure 5.

Crystal structures of ThT-binding PSAMs. (a) Packing of 5-YY/LL monomers in the crystal creates large solvent-channels. The location of the designed ThT-binding site is shown as the purple curves along the central β-sheet of two PSAMs. The N- and C-terminal caps are denoted. (b) Structures of 5-YY/LL and 8-YY/KE. Only the central β-sheet of each PSAM is shown, drawn to scale with ThT. The side chains of Ladder1 and Ladder2 are shown as sticks. The ladder mutations and their solvent-exposed surfaces are colored blue for Tyr and green for Leu, while wild-type side chains are in dark grey. The two PEG-400 molecules found near the putative ThT-binding site on 5-YY/LL are shown as red sticks. (c) The side chain conformations of the 5-YY/LL ladders. The ladders are shown looking down the long axis of the PSAM from C- to N-terminus (top) and looking straight down on the SLB (bottom). The backbone is depicted using ribbons, with turns omitted for clarity. At right, a model of ThT bound to the Tyr surface in an orientation consistent with our results is shown using the same depiction. The ThT ring conformation is derived from that observed in acetylcholinesterase. (d) The ThT-binding pocket of acetylcholinesterase shown in orthogonal views (PDB 2J3Q).24 All residues within 4 Å of ThT are shown as sticks, except for Tyr 121, which is omitted for clarity. The Tyr and Trp residues forming the orthogonal binding motif similar to that observed in 5-YY/LL are labeled (top). (e) The backbone conformations of PSAMs. The eight-stranded β-sheet used as the host in this work is shown looking from the C- to N-terminus, with turns omitted for clarity. The two ladders would be located above the β-sheet as shown. (f) Values of the three parameters Twist, Bend, Bend’ describing the 3-dimensional rotations of each β-hairpin in the SLB.25 The types of rotations are schematically shown above the graphs. Zero values define a perfectly flat, rectangular β-sheet.
As expected, 8-YY/KE and 5-YY/LL retained the overall structure of the PSAM scaffold. Both structures have nearly identical N- and C-terminal domains (Cα RMSD = 0.7 Å and 0.5 Å, respectively). While the topology of the SLB region was identical in these proteins, a larger Cα RMSD (2.1 Å) indicated conformational differences in their SLBs. We found that the 8-YY/KE β-sheet has a mild degree of twist, while the 5-YY/LL β-sheet is almost perfectly flat (Fig. 5e). A quantitative analysis of these differences further supports this notion (Fig. 5f).
Clear electron density was observed for all of the introduced ladder residues, though the Tyr182 side chain in 8-YY/KE had two equally-occupied rotamers. In analyzing 5-YY/LL below, we will focus solely on the five consecutive Tyr and Leu residues in the ladder and ignore the smaller two-residue Tyr and Leu ladder, an artifact of bisecting 8-YY/LL with Glu and Lys (Fig. 3a). This simplification is justified because we have shown that ladders shorter than three residues are incapable of binding ThT (Fig. 3b).
In both 8-YY/KE and 5-YY/LL, Tyr residues grafted into Ladder1 predominantly exhibit the trans side-chain rotamer, which due to the anti-parallel arrangements of adjacent β-strands resulted in alternating side-chain orientations along the Tyr ladders (Figs. 5b and 5c). Because of this alternation, the planes of neighboring Tyr rings are essentially orthogonal and give rise to extended channels across the β-strands of the SLB (Fig. 5b and 5c). These Tyr channels in 8-YY/KE and 5-YY/LL present 380 and 240 Å2 of hydrophobic surface to the solvent, respectively. In contrast, unlike the Tyr side chains, the Ladder2 residues do not present an obvious solvent-accessible surface channel. In particular, the row of Leu of 5-YY/LL is well-buried by adjacent residues within the ladder as well as by neighboring Tyr (Figs. 5b and 5c). The PSAM structures thus give no indication of a channel formed between adjacent ladders, as has been proposed.15
We attempted to confirm the putative ThT-binding site suggested by the PSAM crystal structures using solution NMR spectroscopy. Though HSQC spectra of 8-YY/KE and 5-YY/LL displayed several peak shifts upon addition ThT (data not shown), extensive overlap of resonances and the presence of four copies of the nearly identical β-hairpin repeats prevented us from unambiguously identifying peaks for the ThT-binding site. Nevertheless, these spectra confirm a modest-affinity interaction between ThT and the PSAMs that is consistent with the results of fluorescence dye-binding assays.
The dominant role of the Tyr ladder in ThT binding
To further test the mode of ThT-binding, we designed a set of additional point-mutations to 5-YY/LL. Replacing a single Tyr at one end of the 5-YY ladder (“-Y” mutant) with Lys dramatically reduced ThT binding, but an equivalent mutation in the 5-LL ladder (“-L” mutant) had a much smaller effect (Fig. 3f). These results demonstrate that the Tyr ladder is particularly important for ThT interactions and the Leu ladder may play an accessory role in ThT-binding.
We additionally constructed the YIYIY mutant, which replaced two Tyr residues of 5-YY/LL with Ile, to determine whether hydrophobic residues could substitute for Tyr in this context. These Ile mutations abolished ThT binding (Fig. 3f). Thus, data from the -Y, -L and YIYIY variants of 5-YY/LL collectively support that the Tyr ladder forms the dominant binding motif in the PSAMs.
Discussion
The PSAM system allowed us to systematically manipulate the surface properties of a self-assembly-like extended β-sheet and demonstrate that cross-strand ladders of certain amino acid compositions are sufficient for recapitulating the characteristic high-affinity interactions between ThT and amyloid fibrils (Fig. 3b). In particular, we demonstrated that cross-strand ladders consisting of Tyr may be important in mediating a ThT interactions with a subset of peptide self-assemblies. While our results do not exclude the possibility of alternative ThT binding modes to fibrils, such as binding to β-sheet edges and to the “dry” face between β-sheet layers, a large degree of structural variation among different amyloid models20; 21; 36–38 suggest that alternative binding modes do not explain the broad binding spectrum of ThT. The cross-strand ladders represent common surface motifs of cross-β fibrils, and therefore provide an attractive model for the general ability of ThT to bind to peptide self-assemblies.
The transition from a non-binding to a binding-competent surface produced by extending the Tyr/Leu ladders (e.g. 3-, 4-, and 5-YY/LL; Fig. 3a) down the long axis of the β-sheet argues that ThT binds preferentially parallel to the long axis of peptide self-assemblies, which is consistent with previous spectroscopic studies.15 ThT binding data suggest that a five-residue ladder is necessary for achieving fibril-like affinity for ThT (Fig. 3), though four cross-strand Tyr residues form the minimal, weak binding site. These requirements are consistent with the tip-to-tail length of ThT (~15 Å) and the Cα-Cα distance of a four-residue ladder (~14 Å). In the context of the single-ladder Tyr mutants, lengthening the ladder increased the fluorescence intensity with no enhancement of binding affinity, suggesting that the fluorescence increase is due to an increase of the number of ThT-binding sites but not due to affinity increase. This is reasonable because a longer ladder encodes multiple copies of the minimal ThT binding ladder.
In light of the common (though not identical) capacity of 8-YY/KE and 5-YY/LL to bind ThT, the alternating Tyr ladder motif shared by them, and the absence of an apparent channel between the Tyr and Leu ladders in 5-YY/LL (Fig. 5), we propose that the side-chain surfaces formed by the cross-strand Tyr ladders are the primary binding site for ThT to these PSAMs. Fig. 5c shows a model of ThT binding to the Tyr surfaces. The fact that the 5-YY/LL mutant in which two Tyr were replaced with Ile (YIYIY) did not bind ThT (Fig. 3f) suggests that the aromatic side chains of the Tyr ladder are particularly favorable binding sites for ThT. Though it is possible that conformational dynamics of the side-chains could permit the formation of a ThT-binding site between the ladders, the large impact of the Tyr replacements and the comparatively small impact of the Leu-to-Lys (“-L”) mutation (Fig. 3f) suggests that this is unlikely. The Leu ladder removes charged residues near the ThT-binding site, and it appears to provide tight packing interactions to stabilize the Tyr rings. Taken together the Leu ladder seems to play a secondary role in forming the ThT-binding site. Like the mechanism proposed for ThT-PSAM interactions, the binding pocket of acetylcholinesterase is highly enriched with aromatic side chains, forming π-stacking and hydrophobic interactions with ThT.24 Strikingly, Trp and Tyr residues lining this pocket form a perpendicular edge that is similar to the orthogonal side-chain surface formed by the Tyr ladder of 5-YY/LL (Figs. 5c and 5d). Interactions mediated by aromatic residues may be particularly effective in immobilizing the two aromatic rings of ThT, the prerequisite for fluorescence enhancement.33 Aromatic side chains have been proposed to play the major role for binding of polyphenol inhibitors to fibrils,5 a view consistent with our results for ThT-PSAM interactions.
In addition to their differences in side-chain composition, the degree of β-sheet flatness of the PSAM mutants also correlates with their ability to bind ThT (Figs. 5e and 5f). While we observed that ThT does not bind to the most twisted sheet of 8-EK/KE, it does interact with the somewhat flatter 8-YY/KE, and even more so with almost perfectly flat 5-YY/LL. We have previously shown that the PSAM β-sheet conformation is sensitive to the size and composition of its constituent side chains, with bulky aromatic residues having a pronounced effect in directing flatter β-sheet conformations.27 Therefore, although our results clearly show that flat β-sheets containing cross-strand Tyr surfaces are sufficient for ThT binding, we cannot conclude whether the flat β-sheet surface itself is a requirement for the interaction or rather a by-product of the ladders introduced. Minimally twisted β-sheets are required for the formation of the laminated, cross-β structure common in fibrils.20; 21 It is quite possible that the flatness is critical for high affinity ThT binding, which may explain why ThT does not bind to larger β-sheets in water-soluble proteins that are often heavily twisted.39
The rigidity of the β-sheet scaffold may also be an important factor for high-affinity ThT binding. We have previously shown that a long Tyr cross-strand ladder (8-YY/KE) enhances the rigidity of the PSAM scaffold.27 Such increased rigidity is likely to be shared by 5-YY/LL. A high-level of rigidity may promote the formation of a well-ordered ThT-binding site and reduce the entropic penalty upon ThT binding.33 Further studies are required to dissect the contributions of β-sheet flatness and flexibility to dye binding.
Using PSAMs, we have experimentally identified minimal structural and chemical requirements for ThT binding in the context of anti-parallel peptide self-assembly. The unique ability of PSAMs to accommodate mutations at specific positions without significantly changing the overall structure of the system was critical for this minimalist design of a ThT-binding site. Our strategy should be applicable to characterizing interactions of other ligands with peptide self-assemblies.
The aromatic channel model provides a molecular mechanism by which ThT binds to a variety of β-rich self-assemblies and helps explain ThT’s moderate binding specificity. Because our results establish a minimal structural threshold for high-affinity ThT binding, they may facilitate interpretation of ThT binding data in molecular terms. Knowledge gained from PSAM studies may therefore be useful for better defining the structure of small self-assemblies (oligomers) that are proposed to play an important role in neurotoxicity1; 40 but remain poorly characterized due to their transient nature.
We note that the Tyr surfaces observed in the anti-parallel PSAMs may not exist in parallel self-assemblies, where the aromatic side chains in a ladder can form contiguous π-stacking interactions.20 Still, we speculate that certain cross-strand ladder motifs rich in aromatic amino acids may be important for dye binding in parallel self-assemblies. We moreover note that while our results demonstrate cross-strand Tyr ladders are sufficient for recapitulating the ThT binding behavior of fibrils, these particular amino acid motifs are not a universal explanation for amyloid dye-binding interactions. The breadth of ThT binding to cross-strand ladders of various compositions and in distinct structural environments (anti-parallel vs. parallel β-sheets) could be uncovered by an approach analogous to ours presented here.
Finally, the acquisition of a ThT binding function by introducing the Tyr ladder is also interesting from an evolutionary point of view. It demonstrates that a point mutation to Tyr in a short peptide can create a self-assembly with a new binding function. It is notable that Tyr, among the natural amino acids, is particularly suited for producing a binding interface.41 Thus, such simple point mutations may have played an important role in the evolution of primordial peptides from inert to functional self-assemblies.
Materials and Methods
Sample preparation
Mutagenesis, expression and purification of PSAMs were performed as described previously.26 Each protein was analyzed using size-exclusion chromatography (Superdex 75, Amersham). A majority of PSAMs were found to be predominantly monomeric, and thus they were used without further purification. Because of its high affinity to ThT, 5-YY/LL and its point mutants (Fig. 3f) were further purified using size-exclusion chromatography to eliminate potential contamination of oligomers. For crystallization, the ladder mutants were transferred into the OspAsm1 scaffold that contains mutations in the N- and C-terminal globular domains (“caps”) of OspA, which increase crystallization efficiency without perturbing global structure or the SLB region.30 An N-terminal his-tag was removed prior to crystallization. ThT, bis-ANS, BTA-1, and Congo-Red were purchased from Sigma-Aldrich.
Dye Binding
ThT fluorescence measurements were conducted using 90 μL of sample in a black, 96-well NUNC plate using a Tecan Safire 2 plate reader with excitation at 440 nm. Samples were dissolved in 10 mM sodium phosphate buffer pH 7.4 containing 150 mM NaCl. For characterizing low-affinity variants, fluorescence measurements of 10 μM ThT were performed by titrating protein into the solution. For high-affinity mutants, ThT was titrated into a solution containing 10 μM protein. Competition assays with BTA-1 and bis-ANS were conducted using 1 μM protein and 5 μM ThT. BTA-1 was dissolved in DMSO and diluted to a final DMSO concentration of 1% v/v for measurement. Bis-ANS binding was conducted using 10 μM protein, with excitation at 390 nm and detection at 505 nm. Congo Red binding was assessed with an absorbance scan from 300–700 nm using 10 μM protein.
Crystallization and structure determination of the 5-YY/LL PSAM
Crystallization conditions are listed in Table 1. X-ray diffraction data were collected using the Advanced Photon Source at the Argonne National Laboratory. The 5-YY/LL structure was determined by molecular replacement using Phaser 42 with 8-YY/KE (PDB 2OY7) as the search model. Model building and refinement were performed in the same manner as the wild-type (EK) PSAMs.25 Structures were analyzed using CCP4.43 Molecular graphics were generated using PyMOL (http://www.pymol.org).
Circular dichroism spectroscopy
Measurements were performed using an Aviv CD spectrometer using a 1 mm path length cuvette at 30 °C. The spectra of 0.5 mM ThT were measured with and without 0.5 mM of PSAM. A spectrum of each 0.5 mM PSAM was used as a blank. Three separate reads were averaged and then smoothed with a 9-nm window.
Acknowledgments
We thank Dr. Valentina Tereshko for help with x-ray diffraction data collection and analysis. This work was supported in part by NIH grants R01-GM72688, NSF grant CMMI-0709079 and by the University of Chicago Cancer Research Center. MB was supported by NIH grant T90-DK070076 and the Paul K. Richter and Evalyn E. Cobb Richter Memorial Fund. GM/CA CAT has been funded with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357.
Footnotes
Accession Code
The coordinates and structure factors of the 5-YY/LL PSAM have been deposited in the Protein Data Bank with an accession code 3EC5.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–99. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–90. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.LeVine H., 3rd Multiple ligand binding sites on A beta(1–40) fibrils. Amyloid. 2005;12:5–14. doi: 10.1080/13506120500032295. [DOI] [PubMed] [Google Scholar]
- 4.Klunk WE, Debnath ML, Koros AM, Pettegrew JW. Chrysamine-G, a lipophilic analogue of Congo red, inhibits A beta-induced toxicity in PC12 cells. Life Sci. 1998;63:1807–14. doi: 10.1016/s0024-3205(98)00454-8. [DOI] [PubMed] [Google Scholar]
- 5.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 6.Cohen T, Frydman-Marom A, Rechter M, Gazit E. Inhibition of amyloid fibril formation and cytotoxicity by hydroxyindole derivatives. Biochemistry. 2006;45:4727–35. doi: 10.1021/bi051525c. [DOI] [PubMed] [Google Scholar]
- 7.Findeis MA. Approaches to discovery and characterization of inhibitors of amyloid beta-peptide polymerization. Biochim Biophys Acta. 2000;1502:76–84. doi: 10.1016/s0925-4439(00)00034-x. [DOI] [PubMed] [Google Scholar]
- 8.Vassar PS, Culling CF. Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol. 1959;68:487–98. [PubMed] [Google Scholar]
- 9.LeVine H., 3rd Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–10. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeVine H., 3rd Stopped-flow kinetics reveal multiple phases of thioflavin T binding to Alzheimer beta (1–40) amyloid fibrils. Arch Biochem Biophys. 1997;342:306–16. doi: 10.1006/abbi.1997.0137. [DOI] [PubMed] [Google Scholar]
- 11.Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem. 2003;278:16462–5. doi: 10.1074/jbc.C300049200. [DOI] [PubMed] [Google Scholar]
- 12.Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S. Mechanism of thioflavin T binding to amyloid fibrils. J Struct Biol. 2005;151:229–38. doi: 10.1016/j.jsb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Dzwolak W, Pecul M. Chiral bias of amyloid fibrils revealed by the twisted conformation of Thioflavin T: an induced circular dichroism/DFT study. FEBS Lett. 2005;579:6601–3. doi: 10.1016/j.febslet.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 14.Loksztejn A, Dzwolak W. Chiral bifurcation in aggregating insulin: an induced circular dichroism study. J Mol Biol. 2008;379:9–16. doi: 10.1016/j.jmb.2008.03.057. [DOI] [PubMed] [Google Scholar]
- 15.Krebs MR, Bromley EH, Donald AM. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J Struct Biol. 2005;149:30–7. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Wu C, Wang Z, Lei H, Zhang W, Duan Y. Dual binding modes of Congo red to amyloid protofibril surface observed in molecular dynamics simulations. J Am Chem Soc. 2007;129:1225–32. doi: 10.1021/ja0662772. [DOI] [PubMed] [Google Scholar]
- 17.Sabate R, Lascu I, Saupe SJ. On the binding of Thioflavin-T to HET-s amyloid fibrils assembled at pH 2. J Struct Biol. 2008 doi: 10.1016/j.jsb.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Nelson R, Eisenberg D. Recent atomic models of amyloid fibril structure. Curr Opin Struct Biol. 2006;16:260–5. doi: 10.1016/j.sbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Tsai HH, Reches M, Tsai CJ, Gunasekaran K, Gazit E, Nussinov R. Energy landscape of amyloidogenic peptide oligomerization by parallel-tempering molecular dynamics simulation: significant role of Asn ladder. Proc Natl Acad Sci U S A. 2005;102:8174–9. doi: 10.1073/pnas.0408653102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 22.Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A. 2005;102:315–20. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato T, Kienlen-Campard P, Ahmed M, Liu W, Li H, Elliott JI, Aimoto S, Constantinescu SN, Octave JN, Smith SO. Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry. 2006;45:5503–16. doi: 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harel M, Sonoda LK, Silman I, Sussman JL, Rosenberry TL. Crystal Structure of Thioflavin T Bound to the Peripheral Site of Torpedo californica Acetylcholinesterase Reveals How Thioflavin T Acts as a Sensitive Fluorescent Reporter of Ligand Binding to the Acylation Site. J Am Chem Soc. 2008 doi: 10.1021/ja7109822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makabe K, McElheny D, Tereshko V, Hilyard A, Gawlak G, Yan S, Koide A, Koide S. Atomic structures of peptide self-assembly mimics. Proc Natl Acad Sci U S A. 2006;103:17753–8. doi: 10.1073/pnas.0606690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan S, Gawlak G, Smith J, Silver L, Koide A, Koide S. Conformational heterogeneity of an equilibrium folding intermediate quantified and mapped by scanning mutagenesis. J Mol Biol. 2004;338:811–25. doi: 10.1016/j.jmb.2004.02.063. [DOI] [PubMed] [Google Scholar]
- 27.Biancalana M, Makabe K, Koide A, Koide S. Aromatic Cross-Strand Ladders Control the Structure and Stability of β-rich Peptide Self-Assembly Mimics. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohnishi S, Koide A, Koide S. Solution conformation and amyloid-like fibril formation of a polar peptide derived from a beta-hairpin in the OspA single-layer beta-sheet. J Mol Biol. 2000;301:477–89. doi: 10.1006/jmbi.2000.3980. [DOI] [PubMed] [Google Scholar]
- 29.Ohnishi S, Koide A, Koide S. The roles of turn formation and cross-strand interactions in fibrillization of peptides derived from the OspA single-layer beta-sheet. Protein Sci. 2001;10:2083–92. doi: 10.1110/ps.15901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makabe K, Tereshko V, Gawlak G, Yan S, Koide S. Atomic-resolution crystal structure of Borrelia burgdorferi outer surface protein A via surface engineering. Protein Sci. 2006;15:1907–14. doi: 10.1110/ps.062246706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khurana R, Ionescu-Zanetti C, Pope M, Li J, Nielson L, Ramirez-Alvarado M, Regan L, Fink AL, Carter SA. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys J. 2003;85:1135–44. doi: 10.1016/S0006-3495(03)74550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazit E. Mechanisms of amyloid fibril self-assembly and inhibition. Model short peptides as a key research tool. Febs J. 2005;272:5971–8. doi: 10.1111/j.1742-4658.2005.05022.x. [DOI] [PubMed] [Google Scholar]
- 33.Stsiapura VI, Maskevich AA, Kuzmitsky VA, Turoverov KK, Kuznetsova IM. Computational study of thioflavin T torsional relaxation in the excited state. J Phys Chem A. 2007;111:4829–35. doi: 10.1021/jp070590o. [DOI] [PubMed] [Google Scholar]
- 34.Ryu J, Kanapathipillai M, Lentzen G, Park CB. Inhibition of beta-amyloid peptide aggregation and neurotoxicity by alpha-d-mannosylglycerate, a natural extremolyte. Peptides. 2008;29:578–84. doi: 10.1016/j.peptides.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 35.Meng F, Marek P, Potter KJ, Verchere CB, Raleigh DP. Rifampicin Does Not Prevent Amyloid Fibril Formation by Human Islet Amyloid Polypeptide but Does Inhibit Fibril Thioflavin-T Interactions: Implications for Mechanistic Studies of beta-Cell Death. Biochemistry. 2008 doi: 10.1021/bi702518m. [DOI] [PubMed] [Google Scholar]
- 36.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo JT, Wetzel R, Xu Y. Molecular modeling of the core of Abeta amyloid fibrils. Proteins. 2004;57:357–64. doi: 10.1002/prot.20222. [DOI] [PubMed] [Google Scholar]
- 38.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002;99:5591–5. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A. 2007;104:6632–7. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collaborative-Computational-Project. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallography D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]