

Abstract
The accumulation of misfolded protein aggregates is a common feature of numerous neurodegenerative disorders including Alzheimer disease (AD). Here, we examined the effects of different assembly states of amyloid beta (Aβ) on proteasome function. We find that Aβ oligomers, but not monomers, inhibit the proteasome in vitro. In young 3xTg-AD mice, we observed impaired proteasome activity that correlates with the detection of intraneuronal Aβ oligomers. Blocking proteasome function in pre-pathological 3xTg-AD mice with specific inhibitors causes a marked increase in Aβ and tau accumulation, highlighting the adverse consequences of impaired proteasome activity for AD. Lastly, we show that Aβ immunotherapy in the 3xTg-AD mice reduces Aβ oligomers and reverses the deficits in proteasome activity. Taken together, our results indicate that Aβ oligomers impair proteasome activity, contributing to the age-related pathological accumulation of Aβ and tau. These findings provide further evidence that the proteasome represents a viable target for therapeutic intervention in AD.
Keywords: Proteasome, Alzheimer's disease, Amyloid beta, Tau, Transgenic, Oliogmers, Immunotherapy
1. Introduction
Neurodegenerative disorders, such as Alzheimer disease (AD) are characterized by the accumulation of misfolded or aggregated proteins, suggesting that impairments in the ubiquitin-proteasome system may play a pivotal role in its pathogenesis. For example, there is compelling biochemical evidence showing that the chymotrypsin- and PGPH-like proteasome activity are selectively impaired in AD brains (Keller et al., 2000). Recent genetic evidence also suggests a significant association between AD and various single-nucleotide polymorphisms in ubiquilin-1, which is an ubiquitin-like protein with the capacity to interact with both the proteasome and ubiquitin ligases (Bertram et al., 2005). Key aspects of AD pathology have been replicated in the 3xTg-AD mice, which provides a valuable tool for studying the molecular mechanisms by which amyloid beta (Aβ) modulates tau pathology (Oddo et al., 2005, 2003a,b). Work from our lab implicates the proteasome as one pathway linking Aβ and tau pathology. For example, we have shown that clearance of tau following Aβ immunotherapy is dependent upon the proteasome, suggesting that Aβ may interfere with proteasome function, thereby facilitating the development of tau pathology (Oddo et al., 2004). Consequently, understanding the role of the ubiquitin-proteasome system in AD is critical, as its successful manipulation may represent a novel therapeutic avenue for treating this disorder.
Previous studies have examined the effects of Aβ on the ubiquitin-proteasome system. One study demonstrated that Aβ40 does not affect ubiquitin conjugate formation or deubiquitination, but inhibits ubiquitinated protein degradation by the proteasome (Gregori et al., 1995). Recent reports also indicate that extracellular Aβ can enter the cytoplasm and inhibit the proteasome of neurons from the Tg2576 mice (Oh et al., 2005). Furthermore, accumulation of Aβ within Tg2576 primary neurons leads to impairments in proteasome and ubiquitination activities (Almeida et al., 2006). With respect to Aβ degradation itself, the prevailing view is that this process is accomplished primarily by neprilysin (NEP) and insulin-degrading enzyme (IDE) and perhaps other enzymes (Farris et al., 2004; Hama et al., 2001; Shirotani et al., 2001), but the role of the proteasome in Aβ degradation remains unresolved. Although one study concluded that Aβ40 was not degraded by the proteasome (Gregori et al., 1995), other studies have since shown that Aβ42 could be degraded by cell extracts from astrocytes and neurons (Gregori et al., 1995; Lopez Salon et al., 2003).
Proteasome-mediated degradation of tau, the primary component of neurofibrillary tangles, has also been examined using a variety of systems. In SH-SY5Y cells, tau, either stably transfected or non-transfected, was prevented from being degraded by the addition of the proteasome inhibitor lactacystin (David et al., 2002). To further elucidate whether tau degradation was ubiquitin-dependent, non-ubiquitinated recombinant tau was proteolyzed by purified 20S proteasomes in vitro and this proteolysis was also inhibited by lactacystin (David et al., 2002). Also using the inhibitors leupeptin and Z-GPFL-CHO for the trypsin and glutamyl/BrAAP like activities, respectively, it was shown that these are the predominant activities that degrade tau, with the chymotrypsin-like activity having minimal degradative effects (Cardozo and Michaud, 2002). More recently, it has been demonstrated that CHIP (carboxyl terminus of the Hsc70-interacting protein), a protein thought to be involved the process of quality-control of proteins as well as being an E3-ligase, regulates the degradation of tau and may prevent its aggregation (Murata et al., 2003, 2001; Petrucelli et al., 2004; Sahara et al., 2005).
In this study, we further examine the role of the proteasome in the pathogenesis of AD using 3xTg-AD mice. We show that Aβ impairs various key components of the ubiquitin-proteasome system and define the consequences these impairments have on the pathogenic accumulation of Aβ and tau in vitro and in vivo. We find that Aβ oligomers, but not monomers, inhibit the proteasome and that proteasomal activity is impaired in 3xTg-AD mice when Aβ oligomer levels are high. We also demonstrate that inhibiting proteasome function in vivo in the 3xTg-AD mice causes the pathological accumulation of both Aβ and tau. Lastly, removal of intraneuronal Aβ by immunotherapy decreases the accumulation of Aβ oligomers and reverses the deficits in proteasome activity in the 3xTg-AD mice, demonstrating the dependence of these impairments on the presence of Aβ. Taken together, our study implicates the proteasome as a key pathway linking Aβ and tau pathology.
2. Methods
2.1. Cell-free and homogenized tissue proteasome activity assays
Proteasomal degradation of Aβ was performed by incubating 100 ng of purified human 20S proteasome (Biomol, Plymouth Meeting, PA) with synthetic human Aβ1–40 or Aβ1–42 at a final concentration of 8000 or 800 pg/ml for 16 h at 37 °C (assay buffer: 50 mM Tris, pH 7.5, 25 mM KCl, 10 mM NaCl, 1 mM MgCl2, 0.03% SDS). Aβ was detected by ELISA using end-specific anti-Aβ antibodies against Aβ40 or Aβ42 under non-denaturing conditions. For proteasome activity assays, synthetic human Aβ was added to the give a final concentration of 100,000, 10,000, 1000, or 100 ng/ml (assay buffer: 25 mM HEPES, pH 7.5, 0.5 mM EDTA, 0.05% NP-40, 0.03% SDS). The Aβ was incubated with 100 ng of purified human 20S proteasome for 10 min at 30 °C then 75 μM suc-LLVY-AMC, to assay for chmyotrypsin-like proteasome activity, was added immediately prior to kinetic reading, 37 °C every 1–1.5 min for 60–90 min (excitation 360 nm, emission 460 nm), on the Synergy HT using the KC4 software (BioTek, Winooski, VT). For brain homogenates, 10, 5 or 1 μg of homogenized cortex or hippocampus was applied to the assay instead of the synthetic human Aβ and the purified human 20S proteasome in the same assay buffer lacking SDS.
2.2. Organotypic slices
7-day-old 3xTg-AD pups were taken and euthanized by decapitation. The brains were extracted and the hippocampus dissected out from the cortex and cerebellum. The hippocampus was then sectioned coronally at 400 μM using a microtome. Sections were then suspended in warmed media (50% MEM, 25% Horse serum, 25% Hanks, 0.65 mg/ml d-glucose, and 1% Pen-Strep, pH ∼ 7.2) and then placed on a Millicell Sterilized Culture Plate Insert in a 6-well plate. 1 ml of freshly warmed media was then added to the 6-well plate so the insert and the slices sat directly on the layer of media. The slices were then incubated at 37 °C with 5% CO2 and media replaced every 3 days. After 14 days in culture the slices maintain a uniform transparent appearance indicating a stable culture. Sections were fixed with 4% paraformaldehyde for 1 h and then the slices removed from the plate insert and immunostained as described for the DAB immunohistochemistry protocol below.
2.3. Antibodies
6E10, 1 mg/ml (Signet Laboratories, Dedham, MA), was used to detect APP and Aβ in immunoblots at dilution of 1:1000 and immunohistochemistry at 1:500. HT7 (Innogenetics, Alpharetta, GA) was also used to detect total human tau at 1:1000–2000 for immunoblotting and 1:3000–5000 for immunohistochemistry (detects both phosphorylated and non-phosphorylated tau). The antibody used for the loading control actin was anti-actin at 1:5000 (Sigma–Aldrich Corp., St. Louis, MO). Secondary antibodies used for immunofluorescence were Alexa Fluor 488 goat anti-mouse or Alexa Fluor 488 goat anti-rabbit (Molecular Probes Inc., Eugene, OR). TOTO-3 iodide for nucleic acid staining was also obtained from Molecular Probes.
2.4. Immunoblotting
Tau nucleofected HEK cells were trypsinized, pelleted by centrifugation into 1.5 ml tubes and lysed with 2% SDS. Brain homogenates were prepared in the proteasome activity assay buffer without SDS using a Dounce homogenizer from the cortex or hippocampus of transgenic and non-transgenic mice. Proteins were resolved on 10% Bis–Tris gels using the NuPage system (Invitrogen) and transferred to nitrocellulose membranes usually loading 10–20 μg of protein as determined by the Bio-Rad protein assay dye reagent (Bio-Rad, Hercules, CA). Membranes were incubated for 1 h at room temperature in 5% milk dissolved in TBS-Tween (TBS-T). Membranes were then probed with primary antibodies overnight at 4 °C. The membranes were then washed in TBS-T and probed with the appropriate secondary antibody for 1 h at room temperature. The blots were again washed with TBS-T and visualized using ECL+ and Storm 840 imaging system (Amersham Biosciences, Piscataway, NJ). Blots were quantified using the ImageQuant software package and statistically analyzed by ANOVA or t-test comparison using the Graphpad Prism software.
2.5. Mice and surgical procedures
Male and female 3xTg-AD mice previously characterized were used in this study. Housing and surgical procedures were performed a previously described (Oddo et al., 2003b). Briefly, mice weighing 25–30 g at the time of surgery were anesthetized with 0.6 ml/25 g body weight of avertin (1.3% tribromoethanol, 0.8% amylalcohol) and placed in a stereotactic apparatus (MyNeuroLab, St. Louis, MO) with a mouse adaptor (Oddo et al., 2004, 2003b). 2.5 μl of epoxomicin at a final concentration of 5 mg/ml or DMSO control were injected into the intracerebral ventricle at coordinates relative to the bregma, −2.5 mm posterior, 0.0 mm lateral, and −3.0 mm ventral to the skull with a 10 μl Hamilton syringe (Hamilton Company, Reno, NV) at approximately 1 μl/min. The syringe was removed 2 min after the injection to allow the siRNAs to diffuse. Mice were warmed on heating pads until they recovered from the anesthesia then housed in individual cages until they were sacrificed for tissue processing. All animal procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all appropriate measures were taken to minimize pain and discomfort in experimental animals.
2.6. Immunohistochemistry
As described previously, mice were sacrificed by CO2 asphyxiation and brains were extracted and drop fixed in 4% paraformaldehyde for 48 h (Oddo et al., 2004). A vibratome (Pelco, Redding, CA) was used to prepare 50 μM sections and stored free-floating in PBS with sodium azide. Sections were incubated in 0.3% H2O2 for 30 min to quench endogenous peroxidase activity. Sections were then incubated in 90% formic acid, washed, and incubated overnight at 4 °C with the appropriate primary antibody. Sections were developed with diaminobenzidine (DAB) substrate using the avidin-biotin horseradish peroxidase system (Vector Labs, Burlingame, CA). For immunofluoresence, no peroxide pre-treatment was required and the sections were visualized using the appropriate fluorescent secondary from Molecular Probes. Immunofluoresence was detected using the Bio-Rad 2100 confocal imaging system (Bio-Rad Laboratories, Hercules, CA), quantified using the ImageQuant software package and statistically analyzed by ANOVA or t-test comparison using the Graphpad Prism software.
3. Results
3.1. Aβ oligomers impair the proteasome
To investigate whether different assembly states of Aβ differentially affect proteasome activity, we compared Aβ monomers and oligomers, particularly as recent studies implicate oligomers as the major pathologic species (Walsh et al., 2002a,b; Walsh and Selkoe, 2004). We employed a cell-free proteasome activity assay using model fluorogenic substrates for the three major proteasome activities (LLVY-AMC for the chymotrypsin-like (ChT-L), VGR-AMC for the trypsin-like (T-L), and LLE-AMC for the PGPH/caspase-like (P-L) activity). Aβ42 monomers and oligomers as well as Aβ40 oligomers were incubated with purified human 20S proteasomes and the appropriate fluorogenic substrate for each major proteasome proteolytic activity. The concentrations of Aβ used ranged from 100,000 to 100 ng/ml. We found that oligomeric assemblies of Aβ40 and Aβ42 significantly decreased all three proteasome proteolytic activities in a concentration-dependent manner (Fig. 1A). In contrast, monomeric Aβ40 and Aβ42 exhibited minimal and non-significant alterations of ChT-L, T-L or P-L activity at all concentration except for Aβ40 at 10,000 and 1000 ng/ml for T-L activity only (Fig. 1A). These data indicate that certain assembly states of Aβ preferentially inhibit the proteasome, and it is notable that oligomers of both Aβ40 and Aβ42 yielded comparable results.
Fig. 1.
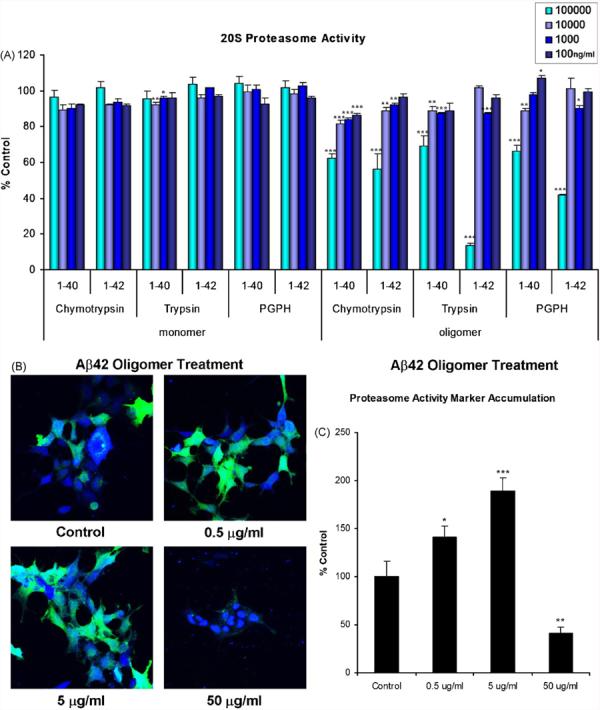
Aβ oligomers but not monomers impair proteasome function in vitro. (A) Purified human 20S proteasome was incubated in the presence of synthetic Aβ42 monomers and oligomers and Aβ40 oligomers. Substrates corresponding to the three major proteasome proteolytic activities, LLVY (chymotrypsin-like), VGR (trypsin-like) and LLE (PGPH-like), were used to monitor these activities via release of the fluorogenic AMC coupled to the C-terminus of these substrates. The rates of AMC release were compared to 20S proteasome activity alone. Aβ42 monomers do not significantly affect the chymotrypsin-like activity of the 20S proteasome (other activities not shown). Aβ40 and Aβ42 oligomers appear to inhibit all the major activities of the 20S proteasome. Concentrations of the Aβ species are in ng/ml. Statistically significant differences were found at 100,000 ng/ml for oligomeric Aβ42 ChT-L: 56.3 ± 0.7%, T-L: 13.4 ± 1.3%, P-L: 41.5 ± 0.7% and Aβ40 ChT-L: 62.3 ± 2.4%, T-L: 69.2 ± 1.1%, P-L: 66.4 ± 3.3% control, 10,000 ng/ml for monomeric Aβ40 T-L: 92.1 ± 1.3% control, oligomeric Aβ42 ChT-L: 89.0 ± 1.6% and Aβ40 ChT-L: 81.8 ± 1.9%, T-L: 88.6 ± 0.9%, P-L: 88.6 ± 1.6% control, 1000 ng/ml for monomeric Aβ40 96.0 ± 0.9% control, oligomeric Aβ42 ChT-L: 92.2 ± 0.9%, T-L: 87.1 ± 0.6%, P-L: 90.0 ± 1.4% and Aβ40 ChT-L: 84.1 ± 0.6%, T-L: 88.8 ± 4.3% control and 100 ng/ml for oligmeric Aβ40 P-L: 107.1 ± 1.3% control. An n of at least 3 was collected for all concentrations and activities. (B, C) HEK cells stably expressing ZsPro were treated with varying doses of oligomeric Aβ42 or were untreated. ZsPro accumulation was dectected by confocal microscopy, and is apparent by emitting green fluorescence. The extent of proteasome dysfunction is indicated by the intensity of the green signal. The blue signal indicates TOTO-iodide nucleic acid staining. Statistically significant differences were found for proteasome activity marker accumulation for all doses. Significance denoted as, *p < 0.05, **p < 0.01, ***p < 0.001. Data shown are representative of at least three treatments per dose.
We employed a proteasome activity marker, for in vitro assays, containing the mouse ornithine decarboxylase degradation domain (MODC d410) fused to green fluorescent reef coral protein (Zoanthus sp.), referred to as ZsPro for proteasome sensor (Hoyt et al., 2003). We examined the effects of varying doses of synthetic oligomeric Aβ42 or control buffer exogenously applied to HEK cells stably expressing ZsPro. Although there was some background fluorescence in the control cells, a marked increase in the number of cells and intensity of ZsPro fluorescence following treatment with a low and medium dose (0.5 and 5 μg/ml, respectively) of oligomeric Aβ was evident. High doses (50 μg/ml) caused marked cell loss, as reflected by morphological changes in the cells, including compaction of the cell nucleus (Fig. 1B and C). Taken together, these data demonstrate that Aβ oligomers can directly inhibit the proteasome and that exogenously administered Aβ42 oligomers also inhibit the proteasome in cell culture.
3.2. Age-dependent proteasome impairments in an in vivo model of AD
We next determined if the buildup of Aβ in the brain also impairs the proteasome in the hippocampus of the 3xTg-AD mice as a function of age. As with AD, these mice contain amyloid plaques that are surrounded by ubiquitin-immunopositive neurites (Fig. 2A–F), suggesting a pathological accumulation of ubiquitinated proteins in association with these lesions (Fig. 2G–L). We focused on the hippocampus, as it is one of the earliest structures affected in the 3xTg-AD mice, with the accumulation of intraneuronal Aβ appearing as the first neuropathological change. Proteasome activity in hippocampal homogenates from 3xTg-AD mice were compared to age-matched NonTg mice using the proteasome activity substrates LLVY-AMC for ChT-L, VGR-AMC for T-L, and LLE-AMC for P-L. Between 3 and 12 months of age, all three proteolytic activities were impaired in the 3xTg-AD mice (Fig. 3A–C). There were also no differences in the levels of proteasome β-subunits (data not shown). Curiously, the inhibition at 12 months was less than that at 9 months, which actually correlates with the shift in Aβ pathology from largely intraneuronal to extracellular during this time window (Oddo et al., 2006a). Furthermore, this age range (9–12 months) also corresponds to the time when the tau pathology begins to manifest, and may perhaps be due in part to the Aβ-induced impairments in the proteasome. Interestingly, by 15 months of age, there no longer appears to be a significant difference in proteasome activity in the hippocampus between the 3xTg-AD and NonTg mice (Fig. 3A–C).
Fig. 2.
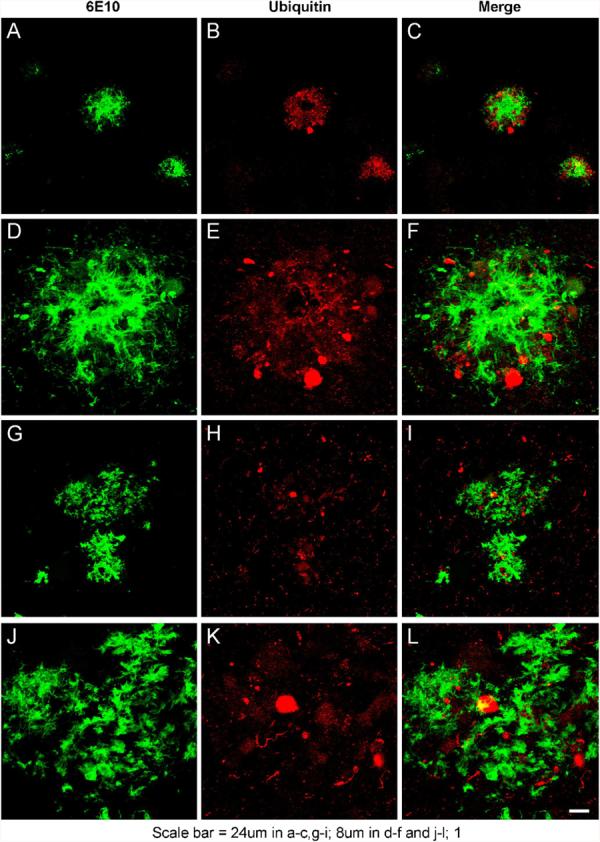
Ubiquitinated proteins are found in association with amyloid plaques in 3xTg-AD mouse model of AD and AD brains. To examine if the 3xTg-AD mouse model generated amyloid plaques associated with ubiquitinated proteins 3xTg-AD brain sections were probed for (A, D, G, J) anti-Aβ (6E10) and (B, E, H, K) anti-ubiquitin (C, F, I, L) merged signals. (A–F) 3xTg-AD plaques. (G–L) Human AD plaques. Scale bar for the various panels are as noted.
Fig. 3.
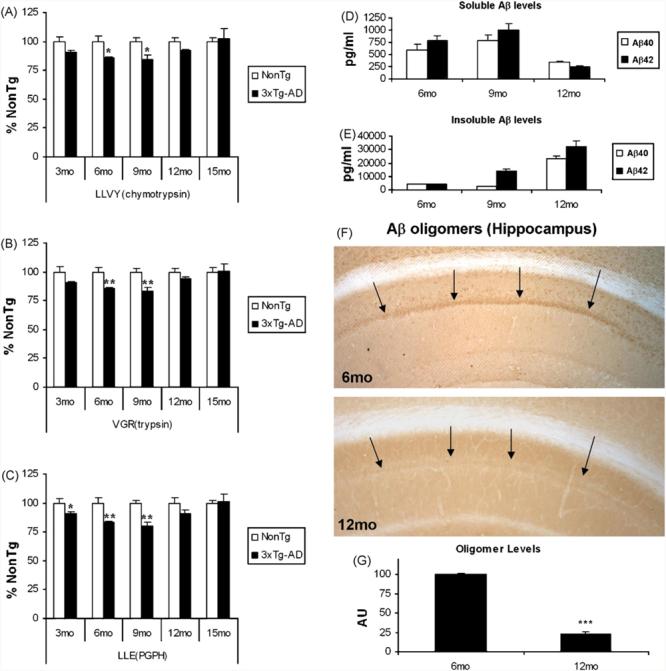
Age-dependent impairments in proteasome activity in the hippocampus of 3xTg-AD mice. (A–C) Homogenates were prepared from the hippocampus of 3xTg-AD and age-matched NonTg mice. These homogenates were assay for chymotrypsin-like, trypsin-like and PGPH-like proteolytic activities. (A) ChT-L proteasome activity was impaired in the 3xTg-AD mice at all ages except for 15 months with significant impairments at 6 (85.7 ± 0.6% NonTg), 9 (84.0 ± 3.9% NonTg) and 24 (81.3 ± 2.1% NonTg) months. (B) T-L proteasome activity was impaired in the 3xTg-AD mice at all ages except for 15 months with significant impairments at 6 (85.7 ± 0.8% NonTg), 9 (83.1 ± 3.2 % NonTg) and 24 (83.6 ± 2.6 % NonTg) months. (C) ChT-L proteasome activity was impaired in the 3xTg-AD mice at all ages except for 15 months with significant impairments at 3 (90.9 ± 1.1% NonTg), 6 (83.4 ± 0.8% NonTg), 9 (80.1 ± 4.0% NonTg) and 24 (82.1 ± 2.5% NonTg) months. An n of at least 3 was collected for all ages and activities. (D) Soluble Aβ40 and Aβ42 levels were assayed by ELISA from 6-, 9- and 12-month-old 3xTg-AD mice brain homogenates. Soluble Aβ levels appear to increase between 6 and 9 months but decrease between 9 and 12 months. (E) Insoluble Aβ40 and Aβ42 levels were assayed by ELISA from 6-, 9- and 12-month-old 3xTg-AD mice brain homogenates. Insoluble Aβ levels appear to increase steadily between 6 and 12 months of age. (F, G) Aβ oligomers are readily detected at 6 months of age in the 3xTg-AD hippocampus but by 12 months are still present but are markedly more difficult to visualize. The anti-Aβ oligomer antibody A-11 was used for immunohistochemistry and the data shown are representative of at least three animals per age. Significance denoted as, *p < 0.05, **p < 0.01, ***p < 0.001.
We were intrigued by the diminished disparity in proteasome activity between the 3xTg-AD and NonTg mice between 9 and 12 months of age, and postulated that changes in soluble and insoluble Aβ levels may underlie this effect. Accordingly, we measured their levels by ELISA. At the earlier time points (6 and 9 months) when significant proteasome inhibition was observed, soluble Aβ levels were readily detectable and appear to be increasing (Fig. 3A). Notably, soluble Aβ levels decreased between 9 and 12 months (Fig. 3D), whereas insoluble Aβ40 and especially Aβ42 levels increased during this time frame, likely due to the emergence of Aβ plaques (Fig. 3E). In addition, intraneuronal Aβ oligomers are readily apparent at 6 months, whereas they are much less evident at 12 months (Fig. 3F and G), although we did not investigate the presence of extracellular oligomers which may help to seed plaques. It is tempting to speculate that as the distribution of Aβ shifts between the soluble and insoluble forms, the repression of the proteasome is relieved, and activity recovers by 15 months. Based on our observations, we conclude that intraneuronal, soluble Aβ oligomers mediate the early inhibition of the three major proteasome activities in the 3xTg-AD mice.
3.3. In vivo inhibition of the proteasome induces Aβ and tau accumulation
As our data and those of others suggest that Aβ inhibits proteasome activity directly, we next determined if the proteasome was also involved in the degradation and clearance of Aβ. We monitored the ability of purified human 20S proteasomes to degrade Aβ40 and Aβ42. Two concentrations of Aβ40 or Aβ42 (8000 and 800 pg/ml) were incubated in the presence or absence of purified human 20S proteasome for 16 h at 37 °C, and levels of Aβ were subsequently measured by ELISA using end-specific antibodies to Aβ40 or Aβ42. Although a portion of the Aβ was not recovered (likely due to sticking to the tubes, etc.), ∼50% of both isoforms of Aβ was degraded relative to the no proteasome control for both concentrations (Fig. 4A). These data demonstrate that the 20S proteasome is capable of degrading Aβ40 and Aβ42 and suggest that the previously described enzymatic activities (e.g., IDE, NEP) are not the sole means for clearing this peptide.
Fig. 4.
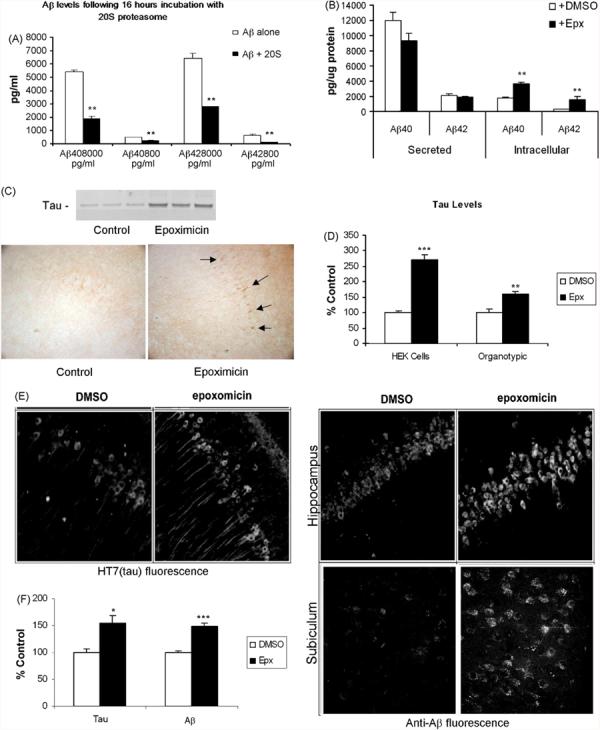
Inhibition of proteasome activity exacerbates the accumulation of AD proteins Aβ and tau. (A) Monomeric Aβ40 or Aβ42 incubated for 16 h at 37 °C with or without 20S proteasome subunit. Sandwich ELISA measurements show that Aβ40 and 42 levels are reduced by ∼50% when incubated with the 20S subunit, indicating that monomeric Aβ40 or Aβ42 can be substrates for the proteasome. An n of at least 3 was collected for all isoforms and concentrations. (B) Mouse neuronal cell line N2A cells were treated for 12 h with the proteasome inhibitor epoxomicin (1 μM). Cell lysates and media were collected and Aβ40 and Aβ42 levels measured by sandwich ELISA. Consistent with (A) intracellular Aβ40 and Aβ42 levels were significantly increased by over 100% whereas secreted Aβ was unaffected. This indicates that Aβ40 and Aβ42 are degraded by the proteasome in vitro and that blocking it increases Aβ levels inside the cell. An n of at least 3 was collected for all isoforms and locations. (C) HEK293 cells were transfected with full-length tau (2N/4R) plasmid and incubated with epoxomicin (10 μM, 12 h) and the lysates probed with HT7. Full-length tau steady state levels were increased, consistent with previous studies demonstrating proteasomal degradation of tau protein. Furthermore, incubation of organotypic hippocampal slice cultures from 3xTg-AD pups in 10 μM epoxomicin (24 h), or vehicle (DMSO), results in increased accumulation of tau protein in cell bodies, as highlighted. (D) Significant increases in tau levels were detected following epoxomicin treatment. Data shown are representative of at least three different transfections or treated slices. (E) 4-mo 3xTg-AD mice were injected intrahippocampally with epoxomicin (2.5 μg), or vehicle, and sacrificed 72 h later. Sections were stained for tau, with HT7, or Aβ, with 6E10 or Aβ specific antibodies, and imaged using fluorescence. Sections shown indicate the CA1 region of the hippocampus. Epoxomicin-treated mice displayed increased tau and Aβ staining, indicating that proteasomal impairment leads to accumulation of both hallmark pathologies of AD, consistent with our in vitro data. (F) Significant increases in tau and Aβ levels were detected following epoxomicin treatment. Significance denoted as, *p < 0.05, **p < 0.01, ***p < 0.001. Data shown are representative of at least four treated animals per group.
To further explore the issue of Aβ degradation by the proteasome, N2A cells were treated with the proteasome inhibitor epoxomicin (1 μM) for 12 h. Cell lysates and media were subsequently collected and Aβ40 and Aβ42 levels measured by sandwich ELISA. We found that intracellular Aβ40 and Aβ42 levels were significantly increased by over 100%, whereas secreted Aβ was unaffected (Fig. 3B). This finding agrees with the results shown in Fig. 4A and indicates that Aβ40 and Aβ42 are degraded by the proteasome in vitro and that selectively blocking proteasome activity increases intracellular Aβ levels.
Tau is generally thought to be a target for proteasome degradation but some recent data challenge this assertion (Cardozo and Michaud, 2002; David et al., 2002; Feuillette et al., 2005; Petrucelli et al., 2004). One report suggests that application of the protein synthesis blocker, cyclohexamide, increases tau levels, which are then further augmented by the application of a proteasome inhibitor, whereas application of the proteasome inhibitor alone actually resulted in a decrease in tau levels (Feuillette et al., 2005). We examined the effects of proteasome inhibition on tau degradation by transiently transfecting HEK cells with wild-type human tau (2N/4R) and treating a subset of these cells with 10 μM epoxomicin for 12 h, as previous work indicated that the half-life of tau in cells is about 12–14 h (David et al., 2002). Compared to the untreated cells, the epoxomicin treatment resulted in a significant increase in the steady-state level of full-length tau (Fig. 3C and D). Taken together, our results suggest that the proteasome degrades both Aβ and tau.
Given the above results, we hypothesized that impairments to the proteasome may result in higher levels of both Aβ and tau. Consequently, we evaluated the effects of proteasome inhibition on organotypic hippocampal slice cultures from the 3xTg-AD mice treated for 24 h with epoxomicin or DMSO vehicle control. Immunostaining for human tau revealed a marked increase in intraneuronal tau staining in the epoxomicin-treated slices compared to DMSO control treatment (Fig. 4C and D). Finally, to assess the effects of in vivo proteasome inhibition on AD relevant proteins, epoxomicin was stereotactically injected into the cerebral ventricle of ∼4-month-old 3xTg-AD mice (n = 4). Seventy-two hours post-surgery, we found that intraneuronal Aβ immunofluorescence was significantly increased in the epoxomicin-injected mice and total human tau was also increased in CA1 neurons in the epoxomicin-injected mice compared to DMSO control injected mice (Fig. 4E and F). Notably, the levels of APP and its relevant C-terminal fragments were not significantly altered by the epoxomicin treatment (data not shown), suggesting that this effect is not due to an inadvertent increase in production of APP or altered APP processing. Therefore, using two different approaches (organotypic slices and sterotactic injection in animals), our studies show that inhibiting proteasome function leads to the intracellular accumulation of tau and Aβ.
3.4. Aβ immunotherapy reverses deficits in the proteasome system
Lastly, we chose to determine whether reducing intraneuronal Aβ in vivo using Aβ immunotherapy would restore proteasome function. We administered either PBS or the anti-Aβ antibody 1560 into the cerebral ventricle of 6-month-old 3xTg-AD mice. In the mice that received the Aβ antibody, intraneuronal Aβ was significantly reduced in the hippocampal neurons, as depicted by the reduced immunofluorescence in the subiculum and CA1 subfield (Fig. 5A and B). The immunotherapy also decreased Aβ oligomer levels (Fig. 5C). Tau levels were lower as well, consistent with our previous finding that clearance of intraneuronal Aβ lead to clearance of tau (Oddo et al., 2004). Using fluorogenic activity assays, homogenates from the hippocampus of mice receiving Aβ immunotherapy revealed significant increases in all three of the major proteolytic activities of the proteasome compared to the PBS-treated mice (Fig. 5D). No differences were found in the catalytic proteasome β-subunit levels (data not shown). Taken together, these results demonstrate that Aβ oligomers mediate proteasome dysfunction in vivo.
Fig. 5.

Aβ immunotherapy reverses proteasome dysfunction. (A, B) The Aβ antibody 1560 was administered to 6-month-old 3xTg-AD mice and the mice were sacrificed 3 days later. Aβ levels were markedly decreased in the hippocampus including the subiculum and CA1. Consistent with data previously published from our lab, tau levels appear to begin to decrease as well. (C) Levels of Aβ oligomers within the hippocampus were significantly reduced following Aβ immunotherapy as detected by dot blot with the oligomeric-sensitive antibody A11. (D) Homogenates from the hippocampus were assayed for the three major proteasome activities as well as for Aβ oligomer levels. Aβ immunotherapy significantly rescued the proteasome impairment as all three proteolytic activities were enhanced. Quantification revealed that Aβ oligomer levels were markedly decreased in the hippocampus. Significance denoted as, *p < 0.05, **p < 0.01, ***p < 0.001. Data shown are representative of at least four treated animals per group.
4. Discussion
Increasing evidence highlights impairments in the UPS as being involved in AD, as well as a number of other neurodegenerative diseases (Adori et al., 2005; de Pril et al., 2004; Layfield et al., 2005; Ross and Pickart, 2004; Song et al., 2003; van Leeuwen et al., 1998). Furthermore, a recent study has shown that deficiency in ubiquitin carboxyl-terminal esterase L1, a ubiquitin hydrolase, cause LTP and behavioral deficits in APP overexpressing mice (Gong et al., 2006) highlighting the importance of UPS impairments in AD mouse models. Our interest in UPS impairments in the 3xTg-AD mouse model of AD originated from experiments showing that Aβ appeared to modulate tau, and that its clearance via Aβ immunotherapy was dependent upon a functioning proteasome (Oddo et al., 2004). As the link between Aβ and tau pathologies in AD still needs to be fully elucidated, we hypothesized that one pathway by which Aβ may affect tau levels is by interfering with the UPS, preventing tau from being degraded and thus leading to its accumulation and eventual phosphorylation.
As Aβ has been previously shown to inhibit the proteasome, we investigated whether the assembly state of Aβ was critical to this effect. We found that oligomeric Aβ species were most effective, particularly at low doses. In contrast, relatively high concentrations of synthetic Aβ were required to show proteasome inhibition in the past, perhaps because the preparations were largely devoid of oligomeric species (Gregori et al., 1995). This concept is corroborated by studies of other neurodegenerative disorders in which protein aggregates are a prominent feature. For example, α-synuclein oligomers and filaments appear to confer proteasome inhibition in Parkinson disease (Lindersson et al., 2004). In spinocerebellar ataxia type 1 (SCA1), ataxin-1 with expanded polyglutamine tracts, known to be a critical factor for the aggregation of polyQ peptides, was also shown to impair proteasome function (Khare et al., 2005; Park et al., 2005). Thus, our finding that Aβ oligomers inhibit the proteasome, the first such description to our knowledge, is consistent with the idea that proteasome dysfunction is a key component of neurodegenerative disorders where the principal aggregated proteins (Aβ, α-synuclein, polyQ peptides) that characterize these diseases mediate proteasomal inhibition. As Aβ oligomers are the subject of intense study for their ability to inhibit LTP, cause neurotoxicity, and alter neuronal processes and synaptic compartments, our data provide evidence for another mechanism by which these oligomers are pathogenic (Klein et al., 2004; Takahashi et al., 2004; Walsh et al., 2002a,b).
In the 3xTg-AD mice, we have shown that proteasome dysfunction occurs relatively early in the progression of pathological events observed in these mice. Between 6 and 12 months of age, a critical window in which intraneuronal Aβ accumulates, Aβ mediated cognitive behavioral changes occur, Aβ plaques initially emerge and pathological alterations of tau commence (Billings et al., 2005; Oddo et al., 2004, 2005, 2003a,b, 2006b) It is very possible that proteasome inhibition may contribute to the development of these pathologies, as we have demonstrated that its inhibition leads to the accumulation of Aβ and tau, two proteins that are themselves degraded by the proteasome. Taken together, our data suggest that future therapeutic interventions into AD, as well as other protein aggregates related neurodegenerative disorders, may include the manipulation of proteasome function.
Acknowledgements
This work was supported in part by grants from Alzheimer's Association and the National Institute of Health AG0212982 to FML. We give thanks to Drs. Charles G. Glabe and Rakez Kayed for the generous gift of amyloid beta oligomers and the A11 antibody.
Footnotes
Disclosure statement
The authors disclose that they have no actual or potential conflicts of interest including any financial, personal or other relationships with other people or organizations within 3 years of beginning the work submitted that could inappropriately influence (bias) their work. All necessary approvals pertaining to animal experimentation were sought and obtained, in accord with the University of California, Irvine's regulations.
References
- Adori C, Kovacs GG, Low P, Molnar K, Gorbea C, Fellinger E, Budka H, Mayer RJ, Laszlo L. The ubiquitin-proteasome system in Creutzfeldt-Jakob and Alzheimer disease: intracellular redistribution of components correlates with neuronal vulnerability. Neurobiol. Dis. 2005;19(3):427–435. doi: 10.1016/j.nbd.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 2006;26(16):4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE. Family-based association between Alzheimer's disease and variants in UBQLN1. N. Engl. J. Med. 2005;352(9):884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Cardozo C, Michaud C. Proteasome-mediated degradation of tau proteins occurs independently of the chymotrypsin-like activity by a nonprocessive pathway. Arch. Biochem. Biophys. 2002;408(1):103–110. doi: 10.1016/s0003-9861(02)00493-9. [DOI] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG. Proteasomal degradation of tau protein. J. Neurochem. 2002;83(1):176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- de Pril R, Fischer DF, Maat-Schieman ML, Hobo B, de Vos RA, Brunt ER, Hol EM, Roos RA, van Leeuwen FW. Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum. Mol. Genet. 2004;13(16):1803–1813. doi: 10.1093/hmg/ddh188. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, Eckman CB, Tanzi RE, Selkoe DJ. Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am. J. Pathol. 2004;164(4):1425–1434. doi: 10.1016/s0002-9440(10)63229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillette S, Blard O, Lecourtois M, Frebourg T, Campion D, Dumanchin C. Tau is not normally degraded by the proteasome. J. Neurosci. Res. 2005;80(3):400–405. doi: 10.1002/jnr.20414. [DOI] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126(4):775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D. Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 1995;270(34):19702–19708. doi: 10.1074/jbc.270.34.19702. [DOI] [PubMed] [Google Scholar]
- Hama E, Shirotani K, Masumoto H, Sekine-Aizawa Y, Aizawa H, Saido TC. Clearance of extracellular and cell-associated amyloid beta peptide through viral expression of neprilysin in primary neurons. J. Biochem. (Tokyo) 2001;130(6):721–726. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Zhang M, Coffino P. Ubiquitin-independent mechanisms of mouse ornithine decarboxylase degradation are conserved between mammalian and fungal cells. J. Biol. Chem. 2003;278(14):12135–12143. doi: 10.1074/jbc.M211802200. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J. Neurochem. 2000;75(1):436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Khare SD, Ding F, Gwanmesia KN, Dokholyan NV. Molecular origin of polyglutamine aggregation in neurodegenerative diseases. PLoS Comput. Biol. 2005;1(3):230–235. doi: 10.1371/journal.pcbi.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr., Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol. Aging. 2004;25(5):569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Layfield R, Lowe J, Bedford L. The ubiquitin-proteasome system and neurodegenerative disorders. Essays Biochem. 2005;41:157–171. doi: 10.1042/EB0410157. [DOI] [PubMed] [Google Scholar]
- Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 2004;279(13):12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- Lopez Salon M, Pasquini L, Besio Moreno M, Pasquini JM, Soto E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003;180(2):131–143. doi: 10.1016/s0014-4886(02)00060-2. [DOI] [PubMed] [Google Scholar]
- Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int. J. Biochem. Cell Biol. 2003;35(5):572–578. doi: 10.1016/s1357-2725(02)00394-1. [DOI] [PubMed] [Google Scholar]
- Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2(12):1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Green KN, Liang K, Tran L, Chen Y, Leslie FM, LaFerla FM. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 2005;102(8):3046–3051. doi: 10.1073/pnas.0408500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol. Aging. 2003a;24(8):1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, Laferla FM. A Dynamic Relationship between Intracellular and Extracellular Pools of A{beta} Am. J. Pathol. 2006a;168(1):184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, Laferla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease: a link between Abeta and tau pathology. J. Biol. Chem. 2006b;281(3):1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 2005;126(12):1292–1299. doi: 10.1016/j.mad.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Park Y, Hong S, Kim SJ, Kang S. Proteasome function is inhibited by polyglutamine-expanded ataxin-1, the SCA1 gene product. Mol. Cells. 2005;19(1):23–30. [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004;13(7):703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Ross CA, Pickart CM. The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14(12):703–711. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005;94(5):1254–1263. doi: 10.1111/j.1471-4159.2005.03272.x. [DOI] [PubMed] [Google Scholar]
- Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, Kiryu-Seo S, Kiyama H, Iwata H, Tomita T, Iwatsubo T, Saido TC. Neprilysin degrades both amyloid beta peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 2001;276(24):21895–21901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- Song S, Kim SY, Hong YM, Jo DG, Lee JY, Shim SM, Chung CW, Seo SJ, Yoo YJ, Koh JY, Lee MC, Yates AJ, Ichijo H, Jung YK. Essential role of E2-25K/Hip-2 in mediating amyloid-beta neurotoxicity. Mol. Cell. 2003;12(3):553–563. doi: 10.1016/j.molcel.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 2004;24(14):3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science (New York, NY) 1998;279(5348):242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002a;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002b;30(4):552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett. 2004;11(3):213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]