

Abstract
Chronic inflammation activates the tryptophan-degrading enzyme IDO, which is well known to impair T cell proliferation. We have previously established that bacille Calmette-Guérin (BCG), an attenuated form of Mycobacterium bovis, is associated with persistent activation of IDO in the brain and chronic depressive-like behavior, but a causative role has not been established. In these experiments we used both pharmacologic and genetic approaches to test the hypothesis that IDO activation is responsible for the development of chronic depression that follows BCG infection. BCG induced TNF-α, IFN-γ, and IDO mRNA steady-state transcripts in the brain as well as the enzyme 3-hydroxyanthranilic acid oxygenase (3-HAO) that lies downstream of IDO and generates the neuroactive metabolite, quinolinic acid. Behaviors characteristic of depression were apparent 1 wk after BCG infection. Pretreatment with the competitive IDO inhibitor 1-methyltryptophan fully blocked BCG-induced depressive-like behaviors. Importantly, IDO-deficient mice were completely resistant to BCG-induced depressive-like behavior but responded normally to BCG induction of proinflammatory cytokines. These results are the first to prove that the BCG-induced persistent activation of IDO is accompanied by the induction of 3-hydroxyanthranilic acid oxygenase and that IDO is required as an initial step for the subsequent development of chronic depressive-like behavior.
Approximately one-third (>2 billion people) of the global population is infected with Mycobacterium tuberculosis (1). Although active tuberculosis will develop in only 10% of these individuals, coinfection with HIV-1 and the ensuing immunodeficiency causes an 800-fold increase in the prevalence of tuberculosis and its accompanying clinical symptoms (2). Bacille Calmette-Guérin (BCG),3 an attenuated strain of Mycobacterium bovis, is a classical intracellular pathogen widely used as a vaccine against tuberculosis (3, 4) in countries other than the United States. A successful protective response against mycobacterial infections is dependent on de novo granuloma formation (5), and infection of mice with BCG is a validated and well-studied model of cellular immunity and granuloma formation (5, 6). Inoculation of BCG i.p. is followed by rapid and persistent mycobacterial dissemination to distant organs, including the spleen, liver, lung, and CNS (7). Mycobacteria express both TLR2- and TLR4-recognized pathogen associated molecular patterns, and macrophages and dendritic cells in draining lymph nodes present mycobacterial Ags to T cells (8). The ensuing chronic production of IFN-γ by CD4+ and CD8+ lymphocytes is critical for intracellular destruction of the bacilli in macrophages (9-12).
In contrast to the short-lived rise in IFN-γ that follows acute exposure to LPS (13), infection with BCG induces a chronic inflammatory response, with plasma IFN-γ remaining elevated for at least 1 mo following exposure (14). Chronic systemic inflammation is now recognized to be involved in nearly all major health issues ranging from infectious, neoplastic, and autoimmune diseases to obesity and cardiovascular disease. Emerging evidence has pointed to the symptom burden (15) experienced by patients suffering from chronic inflammation caused by various conditions, including coinfection with M. tuberculosis and HIV-1 (16). Neuropsychiatric disorders, such as depression and anxiety, are among the most commonly identified comorbidities of both infectious (tuberculosis and HIV) and noncommunicable diseases (cardiovascular disease, rheumatoid arthritis, and diabetes) (17). Chronic infection, such as occurs following exposure to Toxoplasma gondii (18) and BCG (14), leads to increased synthesis of IFN-γ for up to a month following infection. IFN-γ is a potent inducer of the tryptophan-degrading enzyme IDO. This rate-limiting enzyme diverts tryptophan metabolism from serotonin to kynurenine synthesis. The resulting depletion of tryptophan, coupled with the production of downstream toxic metabolites of kynurenine, plays a key role in protection from intracellular and extracellular bacteria, viruses, and parasites (reviewed in Ref. 19).
IDO activation leads to both immunoregulation and neuroregulation. IDO prevents allogenic fetal rejection (20) by suppressing T cell-mediated local inflammatory responses against neonatal Ags (21). Increased IDO enzymatic activity suppresses T cell proliferation by local depletion of available tryptophan (22-25). Both Ag-presenting macrophages (22, 23) and dendritic cells (24, 25) regulate T cell responses at least partially via up-regulation of IDO. Tumor cells also express IDO as a means to evade the immune system. IDO activity contributes to tumorigenic tolerance both by direct inhibition of T cells and by enhancing regulatory T cell-mediated immunosuppression in tumor-draining lymph nodes (26, 27). In addition to its immunoregulatory activity, IDO has been proposed to lie at the interface between chronic inflammatory disease and depression (15). Reduced circulating tryptophan levels and a concomitant increase in the IDO metabolite kynurenine have been reported in patients with chronic inflammatory diseases (28-32) and major depression for nearly two decades (33-36). In degrading tryptophan along the kynurenine pathway, IDO creates several neuroactive kynurenine metabolites and reduces the bio-availability of tryptophan for the synthesis of serotonin, a neurotransmitter that is well known to be involved in mood regulation.
Increased IDO activity has been positively correlated with the severity of depressive scores (37, 38), but there are as yet no data demonstrating a role for IDO activation as a casual factor in mediating the depression associated with chronic inflammation. In this study we used the clinically relevant BCG model of chronic infection to test the hypothesis that long-lived up-regulation of IDO plays a causal role in the development of persistent depressive symptoms. The experiments also tested the possibility that BCG would not only up-regulate IDO but also the downstream enzymes that are responsible for the production of kynurenine metabolites. We report compelling new data using both pharmacological and genetic approaches to clearly demonstrate an essential role for IDO in causing depressive-like behaviors during long-term systemic inflammation induced by BCG infection.
Materials and Methods
Animals and treatments
All experiments were conducted in accordance with guidance for the care and use of laboratory animals from the National Research Council (Washington, D.C.) and with the approval of the Institutional Animal Care and Use Committee of the University of Illinois (Urbana, IL). Pharmacological experiments with 1-methyl tryptophan (1-MT) were performed on 8- to 12-wk-old male Crl:CD1(ICR) mice obtained from Charles River Laboratories. Genetic studies were performed on either B6.129-Indotm1Alm/J (IDO−/−) or C57BL/6J wild-type (WT) controls obtained from The Jackson Laboratory (stock nos.005867 and 000664). Mice were individually housed in standard shoe box cages with wood shavings litter in a temperature- (23°C) and humidity-controlled (45–55%) environment and a 12-h/12-h modified dark-light cycle (light on 10:00 PM-10:00 AM). Food and water were available ad libitum. Mice were individually handled once daily for at least 7 days before the initiation of experiments.
On the day of injection, fresh solutions were prepared by dispersing lyophilized BCG cultures (Sanofi Pasteur) in sterile, endotoxin-free, isotonic saline and then injected i.p. at a dose of 108 CFU per mouse. This dose was selected on the basis of its ability to induce the full spectrum of the acute sickness response that develops into depressive-like behaviors by 7 days and reliably increases peripheral and brain IDO activity, a putative mechanism in the depressive-like behavior induced by immune activation (14, 39). Placebo or 1-MT time-release pellets designed to continuously release 5 mg of a drug per day for 21 days were purchased from Innovative Research of America. This dose was selected on the basis of its ability to fully block the development of depressive-like behavior in LPS-treated mice (13). Pellets were implanted subcutaneously beneath the dorsal skin surface according to the manufacturer's instructions 1 wk before i.p. injection of BCG or saline. Detectible levels of 1-MT were found only in mice implanted with the drug-containing pellet (plasma: 7.87 ± 0.87 μmol/L in 1-MT mice vs undetectable in placebo mice).
Behavioral experiments
All behavioral experiments were performed during the first 4 h of the dark phase of the light cycle.
Locomotor activity
The effects of BCG on locomotor activity were assessed in mice individually placed into a clean, novel cage similar to the home cage but devoid of bedding or litter. The cage was divided into four virtual quadrants, and locomotor activity was measured by counting the number of quadrant entries and rearings over a 5-min period. Counting was done by a trained observer who was blind to the treatments.
Forced swim test
The forced swim test (FST), a standardized test of depressive-like behavior in which depression is inferred from increased duration of immobility, was conducted as described previously (13). The duration of immobility was determined during the test using the mobility function of Observer Basic software (Noldus). Program analysis settings were as follows: sampling rate, 3/s; detection method, subtraction with a low threshold of 20, a high threshold of 255, and a minimum detectable object size of 200 pixels; image filtering, two pixel erosion and dilation; and mobility threshold, 20% with 3 interval averaging.
Tail suspension test
The tail suspension test (TST), a standardized test of depressive-like behavior in which depression is inferred from increased duration of immobility, was conducted as previously described (13) using the Mouse Tail Suspension Package (MED-TSS-MS; Med Associates). Program analysis settings were as follows: integration, on; resolution, 0.1 s; gain, 4; start trigger, 20.
RNA extraction and reverse transcription
Total RNA from whole brain samples was extracted in TRIzol reagent. All reverse transcriptase reactions were conducted in a Stratagene Robocycler Gradient 96 temperature cycler using an Ambion (catalog no. 1710) reverse transcriptase kit according to manufacturer's instructions using 125 ng of total RNA and random decamer primers for each reaction. All RNA samples were reverse transcribed simultaneously to minimize the interassay variation associated with the reverse transcription reaction.
Real-time RT-PCR
Real-time RT-PCR was performed on an Applied Biosystems Prism 7900 using TaqMan gene expression assays for TNF-α (catalog no. Mm00443258_m1), IL-1β (catalog no. Mm00434228_m1), IFN-γ (catalog no. Mm00801778_m1), IDO (catalog no. Mm00492586_m1), kynurenine 3-hydroxylase (KMO; catalog no. Mm00505511_m1), kynurenine aminotransferase (KAT) II (catalog no. Mm00496169_m1), kynureninase (Kynu; catalog no. Mm00551012_m1), 3-hydroxyanthranilic acid oxygenase (3-HAO; catalog no. Mm00517945_m1), and GAPDH (catalog no. Mm999999_g1) were purchased from Applied Biosystems. Reactions were performed in duplicate according to the manufacturer's instructions using 125 ng of cDNA template for each reaction. Relative quantitative measurement of target gene levels was performed using the ΔΔCt method, where Ct is the threshold concentration. GAPDH was used as the endogenous housekeeping control gene.
HPLC
Plasma tryptophan and kynurenine were analyzed by HPLC using an ESA Coulochem II detector with a 5041 enhanced analytical cell containing a glassy carbon electrode (+600 mV). Mobile phase (pH 4.6) consisted of 75 mM NaH2PO4, 25 μM EDTA (disodium salt), and 100 μl/L triethylamine in acetonitrile:water (6:94; v:v). The chromatograms were integrated and quantified using Dynamax MacIntegrator II software (Rainin Instruments).
Plasma (50 μl) was mixed with a solution of 10% sulfosalicylic acid solution (10 μl) and allowed to precipitate proteins on ice for at least 30 min. Following precipitation, samples were centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was extracted and loaded into a Costar Spin-X centrifuge tube filter (0.22 μM nylon; part no.8169, Corning Incorporated) and centrifuged at 12,000 × g for 6 min at 4°C. For the current experiments, plasma extracts were diluted at 1/50 following the extraction steps described above.
A standard curve was generated daily from concentrated (2 μM) tryptophan, 1-MT, and kynurenine standards dissolved in 0.02 N HClO4 and held at 4°C until a 20-μl volume was injected into the system. Standards were made using a serial dilution technique that made the standards to levels that would encompass the expected levels in the plasma samples. The standard curve was created using the system software and samples were not analyzed unless a linear standard curve with r2 > 0.995 was achieved.
In situ hybridization (ISH)
A separate group of WT C57BL/6J mice was treated with LPS (0.83 mg/kg; i.p.) and decapitated 6 h postinjection during peak IDO up-regulation (40). Brains were snap frozen in 2-methyl butane chilled over dry ice and stored at −70°C until 15-μm thick coronal sections were cryosectioned. Sections were mounted on gelatin-coated slides, dried, and stored at −70°C until processing. Tissue was fixed with 4% formaldehyde solution followed by acetylation with 0.25% acetic anhydride in 0.1 M triethanolamine-HCl (pH 8.0). The tissue was then dehydrated in ethanol and exposed to chloroform to remove lipid.
An antisense RNA probe directed against murine IDO cDNA was generated and incorporated with α-35S-UTP labeling (specific activity > 1000 Ci/mmol). A riboprobe plasmid, pGEM-3Zindo, was created as the template for the probe. Briefly, PCR was used to generate an Indo cDNA product containing flanking AvaI and SalI sites (underlined sequences) with the forward and reverse primer sequences 5′-TACCCGGGATGG CACTCAGTAAAATATCTCCTACAG-3′ and 5′-GTGTGTCGACCT AAGGCCAACTCAGAAGAGCTTTCTCG-3′, respectively. The PCR contained template plasmid pCMV-Sport6 with Indo cDNA (clone identifier 5387937, GenBank accession no BC049931; catalog no. MMM1013–9200196, Open Biosystems), 500 nM dNTPs, 300 nM forward and reverse primer, and 2.5 U of Pfu Ultra polymerase (Stratagene). The thermocycling protocol was as follows: 1) 95°C for 2 min; 2) 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 80 s; and 3) 72°C for 10 min. The resultant PCR product was digested with AvaI and SalI and ligated into a similarly cut pGEM-3Z plasmid (Promega). The Indo gene in the resultant plasmid, pGEM-3Zindo, was verified to be correct by sequencing. The chromosomal location of the Indo gene is chromosome 8, location 8 A2. The Indo gene is 1221 bp and encodes for a 407-aa residue protein. Thus, if completely transcribed, the full-length riboprobe product is ∼1221 ribonucleotides in length. The Indo gene was inserted in the plasmid such that transcription from the T7 promoter generates the sense riboprobe and transcription from the SP6 promoter generates the antisense riboprobe. The radiolabeled antisense probe was diluted (500,000 cpm/section) and applied to the brain tissue sections. Hybridization took place overnight at 55°C in a humidified chamber. The slides were washed with a 20 μg/ml RNase solution followed by 2 × SSC and 0.2 SSC (55 and 60°C) to reduce nonspecific binding. After dehydration in ethanol, the slides were air dried for autoradiography. The slides were placed in x-ray cassettes opposed to film for 7 days and developed in an automatic film developer. After film development the slides were dipped in nuclear track emulsion (NTB-2; Kodak) and exposed for 2 wk followed by development (D-19; Kodak) and counterstaining with cresyl violet.
Statistical analysis
Data (mean ± SEM) were analyzed using a one-way (treatment), two-way (pretreatment × treatment) or a three-way (pretreatment × treatment × time) ANOVA with repeated measurement on the time factor where appropriate, followed by posthoc pairwise multiple comparisons using the Fisher's least significant difference method if the interaction was significant.
Results
1-MT normalizes the plasma kynurenine/tryptophan ratio without affecting IDO expression
To inhibit IDO in vivo (13, 20), mice were implanted s.c. with either a 1-MT-containing time-release pellet or a placebo pellet 7 days before either inoculation with either 108 CFU of BCG or injection of nonpyrogenic physiological saline in a 2 × 2 factorial arrangement of treatments. We measured the kynurenine/tryptophan ratio in the plasma at day 7 posttreatment (Fig. 1A). As we have previously reported following acute exposure to LPS (13), 7 days following BCG inoculation led to a decrease in circulating tryptophan (p < 0.05; raw data not shown) and an increase in the kynurenine/tryptophan ratio (treatment, F1,28 = 7.69; p < 0.01). Pretreatment with 1-MT normalized the plasma kynurenine/tryptophan ratio 7 days after BCG inoculation (pretreatment × treatment, F1,28 = 4.24; p < 0.05). To confirm that the reduction in circulating kynurenine/tryptophan was due to an antagonism of IDO activity rather than a general down-regulation of IDO expression, both peripheral and brain expression levels of IDO were determined using real time RT-PCR. At 7 days postinfection, BCG induced a significant up-regulation of IDO mRNA in both the lung (Fig. 1B) (p < 0.01) and the brain (Fig. 1C) (p < 0.01). However, there was not a significant 1-MT × BCG interaction (p > 0.10) in either of these tissues. Collectively, these findings indicate that in vivo pharmacological inhibition of IDO activity normalizes the persistent, long-term BCG-induced elevation in the kynurenine/tryptophan ratio independently of the expression of steady-state transcripts for this enzyme.
FIGURE 1.
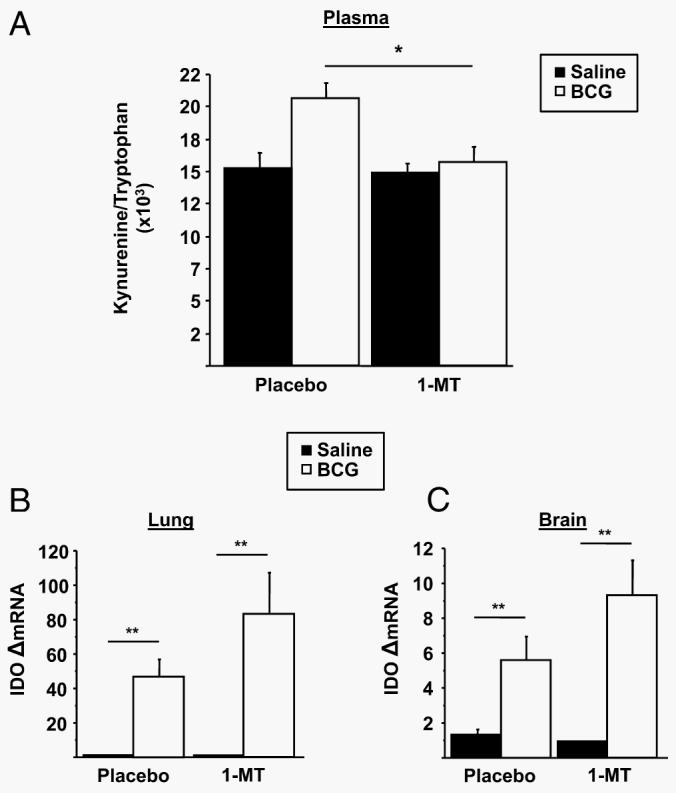
1-MT normalizes the plasma kynurenine/tryptophan ratio but does not impair up-regulation of IDO. A, Immediately following behavioral testing 7 days after BCG infection, mice were sacrificed for blood collection followed by perfusion with ice-cold, heparin-containing PBS and tissue collection. Plasma concentration of kynurenine and tryptophan was measured by HPLC with electrochemical detection. The ratio of kynurenine/tryptophan was determined as an indication of peripheral IDO activity. B and C, Steady-state expression of IDO mRNA transcripts in both the lung (B) and brain (C) were measured by real-time RT-PCR. Data represent means ± SEM (n = 8 mice/group). Bars indicate statistical differences among groups. *, p < 0.05; **, p < 0.01. Average Ct values for placebo- and BCG-treated mice were 25.1 ± 0.9 for the lung and 35.8 ± 2.1 fro the brain.
Administration of 1-MT prevents development of chronic BCG-induced, depressive-like behaviors
To investigate the effect of IDO inhibition on the development of depressive-like behaviors, mice were subjected to the FST and the TST after the acute behavioral response had clearly resolved, i.e., 7 days after BCG inoculation. Following inoculation, mice were weighed daily and monitored for behavioral signs of sickness or depressive-like behavior before being euthanized for the collection of tissue samples. BCG induced a transient reduction in body weight (time × treatment, F1,196 = 35.51; p < 0.01; Fig. 2A) and food intake (treatment × time, F1,196 = 38.27, p < 0.01) that subsided by days 3–4 and was not affected by 1-MT pretreatment (pretreatment, p > 0.05; Fig. 2B). Similarly, BCG induced a transient reduction in locomotor activity (time × treatment, F1,140 = 43.62; p < 0.01) that was not affected by 1-MT pretreatment (pretreatment, p > 0.05; Fig. 2C).
FIGURE 2.

Inhibition of IDO prevents the development of BCG-induced, depressive-like behaviors. Mice were implanted s.c. with a time-release pellet of 1-MT, a competitive IDO inhibitor, or a placebo pellet. One week after this pretreatment, mice received a single i.p. injection of nonpyrogenic saline or BCG (108 CFU). The concomitant changes in body weight (A) and food intake (B) were measured daily, and the reduction in locomotor activity (C) was measured every other day for 7 days after BCG aministration until BCG-treated mice had returned to baseline levels. One week after BCG inoculation, the duration of immobility in the forced swim test (D) and the tail suspension test (E) was recorded. Data represent means ± SEM (n = 6–9 mice/group). Bars indicate statistical differences among groups. *, p < 0.05; **, p < 0.01 vs same treatment placebo mice at each time point.
We recently reported that BCG causes a significant increase in depressive-like behaviors that can be detected for at least 3 wk following infection (39). In this study we confirmed those findings and significantly extended them by determining the potential role of IDO by pharmacologically blocking IDO activity with 1-MT (Fig. 2, D and E, respectively). Enhanced immobility in the FST that occurred 7 days following exposure to BCG was entirely blocked by pretreatment with 1-MT (pretreatment × treatment, F1,28 = 4.88; p < 0.05) (Fig. 2D). Four hours after the FST, mice subjected to a different behavioral test, the TST, also displayed an increased duration of immobility in response to BCG treatment, which was also inhibited by 1-MT pretreatment (pretreatment × treatment, F1,22 = 4.75; p < 0.05) (Fig. 2E).
Collectively, these results confirm that BCG induces an acute sickness response that resolves and then is followed by the development of chronic and persistent depressive-like behaviors. More importantly, these are the first data to establish that pharmacological inhibition of IDO specifically prevents the long-term expression of depressive-like behavioral responses without altering the acute sickness response to BCG.
The BCG-induced proinflammatory cytokine response is not impaired by IDO inhibition
To demonstrate that pretreatment with 1-MT or placebo did not interfere with the up-regulation of the major proinflammatory cytokines responsible for inducing IDO (41), steady-state mRNA expression of both TNF-α and IFN-γ were measured by real-time RT-PCR in both the lung and the brain tissues of saline- or BCG-inoculated mice. As expected, BCG induced a significant increase in TNF-α mRNA expression in both the lung (treatment; p < 0.01) and brain (treatment; p < 0.01) 7 days postinoculation (Fig. 3, A and B). Similarly, as shown in Fig. 3, C and D, BCG also increased IFN-γ mRNA expression in both the lung (treatment; p < 0.01) and brain (treatment; p < 0.05). However, there was not a significant effect of pretreatment with 1-MT (p > 0.10) or a significant 1-MT × BCG interaction in either the lung or brain tissue for either cytokine (p > 0.10). Together, these findings demonstrate that 1-MT inhibition of IDO activity and BCG-induced depressive-like behaviors is not caused by a blunted up-regulation of the key IDO-inducing proinflammatory cytokines.
FIGURE 3.
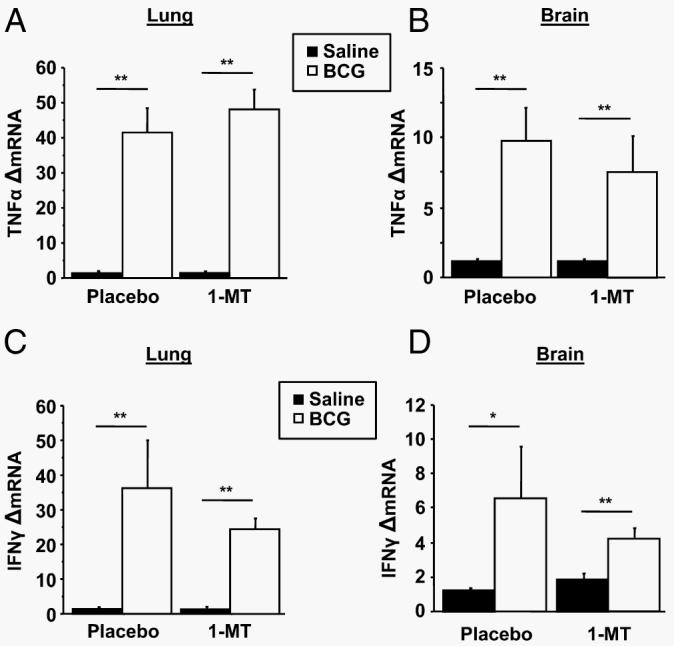
1-MT does not impair BCG-induced up-regulation of proinflammatory cytokines. Steady-state mRNA expression of TNF-α (A and B) and IFN-γ (C and D) were measured by real-time RT-PCR in both the lung and brain tissue 7 days after saline or BCG administration. Data represent means ± SEM (n = 8 mice/group). Bars indicate statistical differences among groups. *, p < 0.05; **, p < 0.01. Average Ct values for placebo- and BCG-treated mice were 27.8 ± 0.4 for lung TNF-α, 32.9 ± 0.5 for brain TNF-α, 30.1 ± 0.7 for lung IFN-γ, and 37.0 ± 1.1 for brain IFN-γ.
Proinflammatory cytokines, but not IDO, are inducible in B6.129-Indotm1Alm/J mice
To confirm the pivotal role of IDO in mediating BCG-induced depressive-like behaviors, a genetic IDO-deficient (IDO−/− mouse model was used. Before being subjected to behavioral testing, the absence of IDO or inducible IDO was confirmed in these mice. To do so, we cloned the full-length 1221-bp murine Indo cDNA, expressed it in a pGEM-3AZindo riboprobe plasmid, and searched for IDO mRNA in the lungs and brain using ISH. Both WT and IDO-deficient mice received a single i.p. injection of non-pyrogenic saline or LPS (0.83 mg/kg) at a dose we have previously shown to potently up-regulate IDO expression both at the periphery and in the brain (13, 40). After sectioning both lung and brain tissues, this IDO riboprobe generated a very low background in saline-treated mice (Fig. 4A). However, this IDO signal was markedly increased following LPS treatment (Fig. 4B). When injected systemically into the IDO−/− mice, this same dose of LPS failed to induce any positive hybridization signal (Fig. 4C). These results were confirmed by real-time RT-PCR in lung tissue from the same mice, whereby LPS induced nearly a 200-fold increase in the steady-state expression of IDO mRNA in the WT mice that was not detectible in their LPS-treated IDO−/− counterparts ( p < 0.01) (Fig. 5). As a positive control for IDO specificity in this study, the LPS-inducible proinflammatory cytokines IL-1β, TNF-α, and IFN-γ were measured in WT and IDO−/− mice. As expected, LPS robustly increased the expression of mRNA for IL-1β (p < 0.01), TNF-α (p < 0.10), and IFN-γ (p < 0.01), but there was no difference between WT and IDO−/− mice (Fig. 5). In the brain, ISH revealed diffuse, moderate hybridization in the striatum of saline-treated mice (Fig. 4D), and LPS induced scattered punctate labeling (Fig. 4E) that was primarily associated with the vasculature (data not shown). Again, no positive hybridization was observed in the brain sections of IDO−/− mice (Fig. 4F). These data are important because they demonstrate that IDO-deficient mice respond normally to systemic inflammation by increasing expression of the critical proinflammatory cytokines even though they fail to express IDO either before or after systemic immune activation.
FIGURE 4.
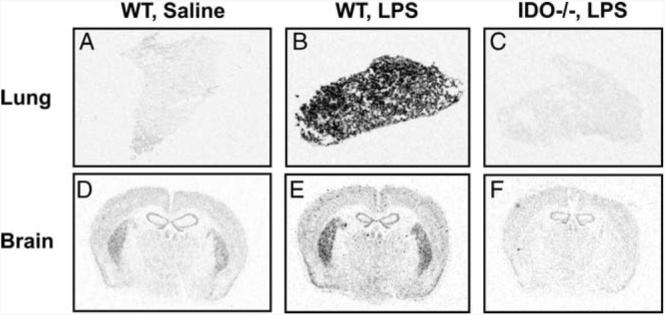
IDO is not inducible in IDO-deficient mice. WT mice were injected with either nonpyrogenic saline or LPS (0.83 mg/kg), whereas age- and sex-matched IDO−/− mice were injected with LPS (0.83 mg/kg). Six hours after injection, mice were sacrificed and tissues were snap frozen for analysis by ISH. Lung (A–C) or brain (D–F) tissue sections were labeled with a full-length 35S-IDO cDNA riboprobe and exposed for 7 days before visualization. ISH images are representative of 4–8 distinct slices.
FIGURE 5.
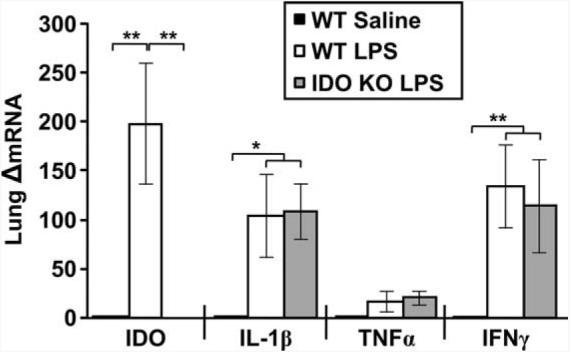
Induction of proinflammatory cytokines is normal in IDO-deficient mice. Lungs from WT and IDO−/− mice were collected from the same mice described in Fig. 4 at 6 h following injection. Steady-state mRNA expression of IDO, IL-1β, TNF-α, and IFN-γ in lung samples was analyzed by real-time RT-PCR. Data represent means ± SEM (n = 2–4 mice/group). Bars indicate statistical differences among groups. *, p < 0.05; **, p < 0.01. Average Ct values for LPS treated mice were 26.5 ± 0.3 for IDO, 21.7 ± 0.5 for IL-1β, 21.8 ± 0.8 for TNF-α, and 27.9 ± 1.4 for IFN-γ. KO, knockout.
IDO-deficient mice exhibit a normal transient sickness response to BCG
To determine the impact of genetic IDO deletion on the transient sickness response to BCG, WT and IDO−/− mice received a single i.p. injection containing either nonpyrogenic saline or BCG (108 CFU) in a 2 × 2 factorial arrangement of treatments. After injection, mice were weighed daily and monitored for behavioral symptoms of sickness. Consistent with pharmacological experiments using 1-MT, BCG induced a transient reduction in body weight (treatment × time, F1,301 = 72.13; p < 0.01) that had returned to saline-treated levels 5–6 days posttreatment (Fig. 6A). However, there was no strain × treatment interaction (p < 0.10). Likewise, BCG induced an acute reduction in locomotor activity (treatment × time, F1,112 = 10.18; p < 0.01) that returned to baseline levels by 3 days after BCG (Fig. 6B) and was independent of strain (strain × treatment; p > 0.10). Collectively, these findings establish that genetic deletion of IDO does not interfere with the short-term sickness response that is induced by BCG infection.
FIGURE 6.
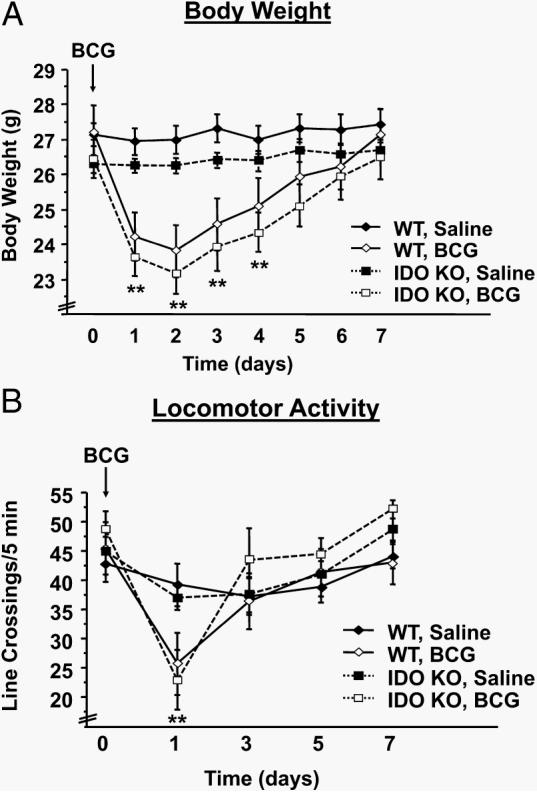
IDO-deficient mice exhibit a typical BCG-induced acute sickness response. A, WT or IDO−/− mice (IDO KO (knockout)) received a single i.p. injection of nonpyrogenic saline or BCG (108 CFU). Body weight change was measured daily for 1 wk following BCG administration. B, Similarly, locomotor activity was monitored every other day for 1 wk. Data represent means ± SEM (n = 12 mice/group). Bars indicate statistical differences among groups. *, p < 0.05; **, p < 0.01 vs same strain of saline-treated mice at each time point.
IDO-deficient mice do not show any BCG-induced elevation in the plasma kynurenine/tryptophan ratio or depressive-like behaviors
To confirm the pharmacological data showing that IDO is a critical mediator of BCG-induced depressive-like behaviors, we tested the hypothesis that the chronic induction of depressive-like behaviors that occurs following BCG inoculation would not occur in mice that were devoid of the IDO gene. In these experiments, the duration of immobility in the FST and TST was assessed in WT and IDO−/− after inoculation with BCG or injection with saline at a later time point of 7 days, after the shorter-term symptoms of sickness dissipated. Mice were euthanized immediately following behavioral testing for determination of the plasma kynurenine/tryptophan ratio. As expected, BCG induced a significant increase in the duration of immobility during the FST that did not occur in IDO−/− mice (strain × treatment, F1,43 = 6.31; p < 0.05) (Fig. 7A). The mice were subjected to the TST 4 h later. Again, BCG-treated WT mice displayed a marked increase in duration of immobility compared with their saline-treated WT counterparts, and BCG failed to elicit an increased immobility in IDO−/− mice (strain × treatment, F1,42 = 8.78; p < 0.01) (Fig. 7B).
FIGURE 7.
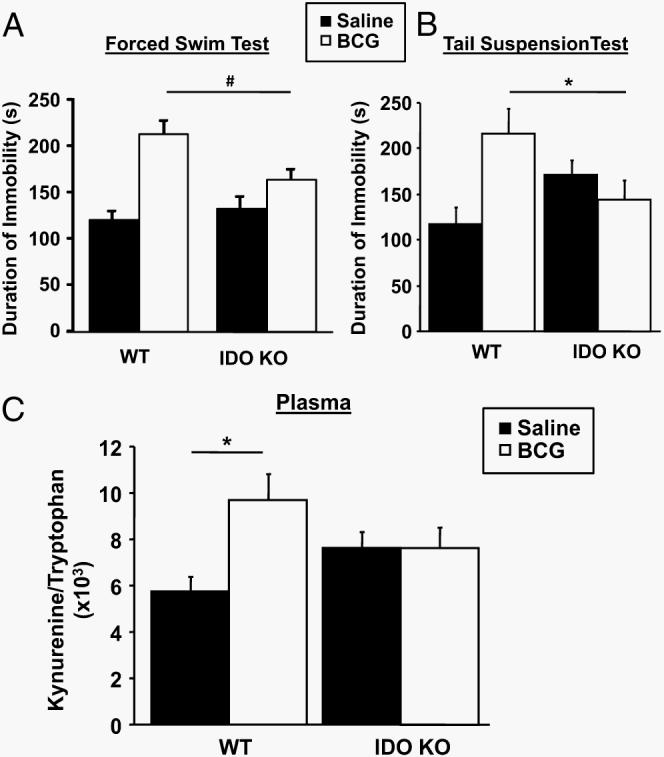
IDO is required for BCG to increase the plasma kynurenine/tryptophan ratio and to precipitate development of depressive-like behaviors. One week after receiving a single i.p. injection of either nonpyrogenic saline or BCG (108 CFU), duration of immobility in the forced swim test (A) and the tail suspension test (B) was measured in WT and IDO−/− (IDO KO (knockout)) mice. Immediately following behavioral testing, mice were euthanized and blood was taken. The plasma kynurenine/tryptophan ratio (C) was determined by HPLC with electrochemical detection as an indication of IDO activity. Data represent means ± SEM (n = 11–12 mice/group). Bars indicate statistical differences among groups. *, p < 0.05; p = 0.06.
Consistent with our pharmacological findings using 1-MT, the development of depressive-like behaviors in BCG-inoculated WT mice was accompanied by a reduction in plasma tryptophan (p < 0.01; raw data not shown) and an increase in the kynurenine/tryptophan ratio (treatment, F1,43 = 5.00; p < 0.05). As hypothesized, this increase in the kynurenine/tryptophan ratio did not occur in the IDO−/− mice that are unable to express IDO either before or after systemic inflammation (strain × treatment, F1,43 = 5.11; p < 0.05) (Fig. 7C). These results with a genetic model of IDO deficiency confirm the pharmacological results and establish an essential role for IDO in mediating the development of depressive-like behaviors following chronic immune activation by M. bovis BCG.
BCG increases expression of an enzyme downstream of kynurenine, 3-HAO
IDO not only depletes the microenvironment of tryptophan to impair T cell proliferation, but it also creates kynurenine that is used as a substrate to generate bioactive compounds that regulate both the immune system and CNS (19, 42). We recently demonstrated that systemic injections of kynurenine alone can induce depressive-like behavior independently of any immune stimulation (13). Because kynurenine is unlikely to be active by itself (43), we tested the hypothesis that BCG increases the expression of enzymes that create bioactive kynurenine metabolites. We first confirmed that BCG increases IDO expression 7 days after inoculation in C57BL/6J mice as it does in CD1 mice. In this latter strain of mice, BCG increased IDO transcripts by roughly 9-fold (Fig. 1B). In C57BL/6J mice, an identical inoculation with BCG increased brain IDO mRNA 7 days later by 18.0 ± 5-fold compared with the saline-injected mice (p < 0.05; data not shown).
Kynurenine is degraded by three major enzymes, KAT, KMO, and Kynu (Fig. 8A). Although IDO is the rate-limiting enzyme in this pathway, it is possible that BCG also up-regulates expression of these enzymes, as was recently reported to occur in the brains of LPS-treated rats (44). Seven days following BCG infection, we found little evidence for this possibility. BCG did not affect expression of either KMO or KAT II (Fig. 8B). Although there was a tendency for BCG to increase mRNA of Kynu, this effect was not statistically significant (p = 0.12). We also measured the expression of 3-HAO, which does not directly catabolize kynurenine but is required for the ultimate production of quinolinic acid. This compound is a tryptophan-derived kynurenine metabolite that can act as a neurotoxic N-methyl-d-asparate receptor agonist and lead to the generation of both nitrogen and oxygen free radicals (45). We found that BCG at 7 days following infection increased (p < 0.05) the expression of 3-HAO compared with the saline-injected controls (Fig. 8B). These data indicate that the preferred metabolic fate of IDO-generated kynurenine following chronic infection with BCG is along the pathway that favors the generation of quinolinic acid, a well-recognized neurotoxic and immunosuppressive compound.
FIGURE 8.
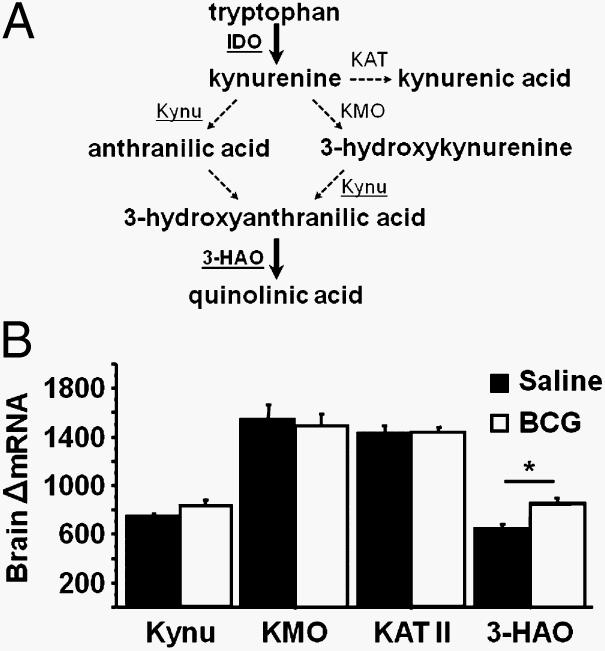
The tryptophan metabolizing enzyme 3-HAO is up-regulated in the brain following infection with BCG. A, Diagram of the major enzymes and their products in the kynurenine pathway of tryptophan metabolism. B, Steady-state mRNA expression of KMO, KAT II, Kynu, and 3-HAO was measured by real-time RT-PCR in brain tissue 7 days following saline or BCG administration. Data represent means ± SEM (n = 8 mice/group). Bars indicate statistical differences among groups. *, p < 0.05. Average Ct values for BCG-treated mice were 29.3 ± 0.2 for KMO, 29.4 ± 0.1 for KAT II, 30.6 ± 0.1 for Kynu, and 30.2 ± 0.1 for 3-HAO.
Discussion
Chronic, low-grade, systemic inflammation characterizes a large number of costly diseases including obesity, heart disease, and AIDS, all of which are associated with major comorbid pathologies (16). Clinical depression is a predominant comorbid symptom that occurs during chronic inflammation. IDO activation has been strongly implicated in clinical depression (43), a concept that was initially based on the shunting of tryptophan from serotonin to kynurenine synthesis in the brain (reviewed in Ref. 15). We recently reported that systemic inflammation induced by a single i.p. injection of LPS augments IDO expression in the brain that peaks within 24 h and declines to near baseline by 36 h (13, 40, 46, 47). This acute injection of LPS induces a short-lasting bout of depressive-like behavior that disappears within 72 h (47). In contrast to LPS, BCG does not induce either central or systemic IDO within 24 h (14, 39, 47). It is not until 7 days after BCG infection that we detected a significant rise in IDO activity and an increase in kynurenine. In sharp contrast to LPS, these changes in IDO are maintained for at least a month following exposure to BCG, which is a classic infectious model of chronic activation of both the acquired and innate immune systems. In the current study we used this BCG model to investigate the potential causative role of IDO as a component of a neuroactive kynurenine pathway that can lead to depressive-like behaviors. This possibility has never been reported in a chronic model of infection and inflammation. The use of IDO knockout mice, which we confirmed to lack IDO following immune activation, was complemented by experiments using pharmacological inhibition of IDO. We report for the first time that IDO is responsible for the long-lasting, depressive-like behavior induced by a chronic infection with BCG.
Mycobacterial granuloma formation is a critical element of the host antimicrobial response, and proinflammatory cytokines like TNF-α (48) and IFN-γ (4, 9, 11) are essential for maintaining granuloma integrity and for mediating protective cellular immunity. Inoculation with BCG to mimic a low level of chronic mycobacterium infection is followed by persistent infection of the lung, spleen, and liver (4, 7). These tissues are rapidly infiltrated with large numbers of IFN-γ-secreting CD4+ T lymphocytes (4). Persistently elevated levels of proinflammatory cytokines, including TNF-α and IFN-γ, can be detected for several weeks following BCG inoculation, and the tryptophan-degrading enzyme IDO is potently activated following inoculation with BCG (14, 39). Proinflammatory cytokines orchestrate not only the cellular immune response but also the communication loops between the immune system and brain, that regulate behavioral functioning of the host (15). Inflammatory mediators and their effects on the metabolism of tryptophan have been proposed to precipitate comorbid depression in a number of chronic inflammatory conditions (29). In the present experiments, we demonstrate for the first time that IDO activation is a pivotal factor in the relationship between chronic inflammation induced by BCG and depression. The three major novel findings in this paper that support this conclusion are as follows: 1) mice with a genetic deletion of IDO do not develop depressive-like behavior in response to BCG (Fig. 7); 2) BCG induction of proinflammatory cytokines is normal in IDO-deficient mice (Fig. 5), indicating that these cytokines lie upstream of IDO activation; and 3) in a model of chronic infection that differs from acute injections of LPS, a competitive IDO inhibitor is effective in blocking the development of depressive-like behavior (Fig. 2).
IDO has been most intensively studied by cellular immunologists for its role in mediating T cell proliferation and local immunotolerance. However, increased IDO activity creates conditions of immunoprivilege that can be both beneficial and deleterious to the host. Specifically, IDO activity supports the persistence of commensal bacteria (49), helps maintain normal pregnancy (20), and promotes the survival of transplants (50). Conversely, IDO activity also increases tumor cell survival and pathogen persistence (26) and can enhance allergic responses/asthma (51) (reviewed in Ref. 52). This immunological paradox is especially apparent in chronic infections such as M. bovis, Leishmania major, or T. gondii where cytokine-induced IDO activation is an important aspect of granuloma formation and pathogen clearance, but at the same time it may act to create an immunosuppressive microenvironment around the granuloma. The paradox extends to the current series of experiments in which the inhibition of IDO actually prevented the depressive-like behavioral responses known to accompany infection. We observed a clear increase in expression and activity of IDO following inoculation with BCG. This increase was coupled to an increase in both peripheral and central expression of TNF-α and IFN-γ. The IFN-γ-inducible tryptophan reduction, mediated by IDO, plays an important role in a number of antimicrobial and antiviral responses, and these immunoregulatory properties are enhanced in the presence of TNF-α (53). Also, IDO expression appears to augment the innate immune response against Mycobacterium avium induced by immunostimulatory DNA (54). Considering these findings, one might have expected that the inhibition of IDO would have made mice more susceptible to BCG infection and exacerbated the concomitant proinflammatory neuroimmune responses. In contrast to this prediction, in the present series of experiments both pharmacological inhibition as well as genetic deletion of IDO normalized the plasma kynurenine/tryptophan ratio following BCG inoculation without altering cytokine gene expression or BCG-induced splenomegaly (data not shown), a physiological correlate of infection. However, it is possible that the relatively short duration of infection (1 wk) was of insufficient duration to observe a defective antimicrobial response.
Rather than exacerbate the neuroimmune response, inhibition of IDO had no impact on acute sickness responses and completely inhibited the development of depressive-like behaviors. The lack of effect of IDO inhibition on BCG-induced acute sickness is likely due to the fact that these clinical signs are directly induced by proinflammatory cytokines acting in the brain (15),whereas BCG-induced depression requires cytokine-induced IDO activation. The lack of effect of 1-MT on the signs associated with BCG infection besides depression suggests that IDO does not have a predominantly antimicrobial or neuroimmune role, at least during the initial week following infection with BCG. The recent discovery of an IDO-like protein, IDO-2, provides an alternative mechanism by which these seemingly incompatible IDO-mediated events could occur. IDO-2 is expressed in both mice and humans, and the gene is located adjacent to IDO on chromosome 8 (55, 56). The two enzymes are structurally similar and each serves as the first enzyme degrading tryptophan along the kynurenine pathway (56). Although studies aimed at identifying the biological function of IDO-2 are still in their infancy, emerging data indicates that several key differences exist in tissue distribution and regulation (see Ref. 57 for review). Of particular relevance to the present study is the finding that the l- and d-stereoisomers of 1-MT, the small molecule IDO inhibitor used in this study, have differing inhibitory efficacies for the IDO and IDO2. Although there is some disagreement in the literature regarding this topic (58-60), d-1-MT appears to preferentially inhibit IDO-2 in vivo (61, 62). The present study reports that 1-MT prevents the development of depressive-like behaviors in response to chronic inflammation; however, the 1-MT time-release pellets used were manufactured using a racemic mixture of both the l- and d-stereoisomers. Therefore, although the pharmacologic experiment presented in Fig. 4 clearly implicates IDO as a pivotal pathogenic contributor, the data presented in Fig. 7 using IDO−/− mice unequivocally establish IDO, not IDO-2, as the enzyme responsible for mediating this neuroimmune process. These new data represent an important early step in the emerging characterization of the immunological and pathophysiological roles of these two related enzymes.
The role of IDO in mediating comorbid depressive disorders has recently garnered substantial attention. Perhaps the most persuasive clinical evidence for a causal relationship between inflammation and depression mediated by IDO is the development of comorbid depressive disorders in patients receiving cytokine immunotherapy for the treatment of certain cancers or hepatitis. In the context of several studies showing an association between IDO activation and neuropsychiatric disorders, Capuron and colleagues demonstrated a significant positive correlation between depressive symptoms and a depletion in tryptophan in patients undergoing cytokine immunotherapy (37). In that study, renal cell carcinoma patients without preexisting depressive disorders were monitored over the first 4 wk of cytokine therapy. Depression scores increased and serum tryptophan decreased in all patients during this time period. Moreover, the magnitude of these effects was highly correlated and specific for tryptophan, as no other large neutral amino acid was affected (37). Subsequently, Wichers et al. further substantiated the clinical relevance of IDO in mediating inflammation-induced depression, demonstrating that increased kynurenine/tryptophan ratios were associated with higher depression scores (38). The current experimental findings showing that depressive-like behaviors are associated with an increased kynurenine/tryptophan ratio support these clinical findings. Importantly, we were able to demonstrate a causal effect of IDO in mediating this relationship by using both pharmacological and genetic approaches.
Proinflammatory cytokines induced by pathogen-associated molecular patterns such as BCG, bacterial LPS, or even some types of vaccination (63) are well known to induce clinical signs of sickness both in humans and in laboratory animals (15). However, because many aspects of acute sickness are similar to signs of depression, it has been difficult to disentangle the two conditions in laboratory animals. For example, decreased motor activity and reduced food consumption can give false positive results in tests of depressive-like behavior that aim at measuring either resignation (e.g., TST and FST) (64) or preference for a rewarding aliment (e.g., sucrose preference test) (65). For this reason, it is important to measure depressive-like behavior when sickness is minimal. In the model of acute inflammation induced by LPS (66), we have shown that LPS increases the duration of immobility in the TST and FST and decreased sucrose preference when these tests were completed 24 h after LPS induction, at a time when general locomotor activity and food consumption have returned to normal. However, acute induction of depressive-like behavior is far from ideal because it is based on the consequences of a transient (<24 h) activation of the innate immune system, whereas inflammation-associated depression develops clinically on a background of chronic, persistent inflammation.
The BCG model of chronic immune activation appears to accurately recapitulate the etiology of cytokine-induced depression in conditions of chronic inflammation. More importantly, the infectious disease model is particularly relevant to patients afflicted with tuberculosis because depression is an often-overlooked comorbidity in this disease. In two separate studies of tuberculosis patients entering treatment, nearly 50% met the diagnostic criteria for depression (67, 68). Moreover, most of these patients experienced improvements in their depressive symptoms during the course of treatment (68). Depression is a predominant factor for patient nonadherence and most likely represents a substantial impediment to successful treatment, contributing to the growing problem of major depressive disorders that occur in tuberculosis patients.
A role of IDO in the pathogenesis of tuberculosis-associated depression has never been reported, although early studies recognized altered tryptophan metabolism in tuberculosis patients that was characterized by significantly elevated urinary excretion of 3-hydroxykynurenine (69) and xanthurenic acid (70). Interestingly, IDO is the rate-limiting enzyme for the formation of both of these metabolites, and successful completion of antituberculosis cycloserine treatment has been reported to normalize xanthurenic acid levels (70). Besides reproducing the main features of tuberculosis, inoculation of mice with BCG has the advantage of inducing a sustained stimulation of IFN-γ and TNF-α, which are the main cytokines responsible for the activation of IDO (39). Although we have previously demonstrated that acute immune activation using the bacterial mimetic LPS also induces IDO-dependent depressive-like behavior, Fujigaki et al. have demonstrated LPS-induced IDO expression to be IFN-γ independent (41). In that report, LPS induced IDO in both IFN-γ-deficient and IFN-γ-immunoneutralized mice. Using an in vivo primate model of neuro-AIDS, Burudi and colleagues demonstrated that IFN-γ was the primary cytokine responsible for inducing IDO expression in myeloid cells within the brains of SIV-infected primates (71). Therefore, IDO up-regulation within the context of chronic inflammatory disease appears to be largely dependent on IFN-γ. Some in vitro studies also have demonstrated a synergistic effect of TNF-α and IFN-γ acting via NF-κB in up-regulating IDO gene expression (72, 73). This may be a result of activation of the TLR-MyD88 signaling pathway, serving to prime infected macrophages to the subsequent T cell-derived IFN-γ (74).
IDO is a pivotal metabolic enzyme in inflammation-induced neuroimmune responses because it is the rate-limiting enzyme that metabolizes the essential amino acid tryptophan along the kynurenine pathway. Tryptophan is the biosynthetic precursor of the neurotransmitter serotonin that is well known to be important in the regulation of mood. Increased degradation of tryptophan along the kynurenine pathway could potentially deplete the bioavailability of this amino acid precursor for serotonin synthesis. In the present study, BCG did not induce significant changes in brain serotonin and its major metabolite, 5-hydroxyindole-3-acetic acid, when compared with control mice (data not shown).
It is likely that events other than a potential reduction in serotonin are responsible for the depressive-like behaviors induced by BCG. For example, metabolism of tryptophan by IDO results in the generation of several neuroactive kynurenine metabolites, including kynurenic acid, 3-hydroxykynurenine, anthranilic acid, and quinolinic acid, which primarily affect glutamatergic neurotransmission. Support for this possibility was demonstrated years ago by the laboratory of Schwarcz et al. (75-77). Their experiments clearly established that kynurenine is degraded in nonactivated glial cultures and brain slices. Recently, acute treatment of rats with LPS was shown to cause an up-regulation of both IDO and KMO transcripts in the brain, and the authors noted the relative abundance of downstream kynurenine pathway enzymes in relation to IDO (44). In accord with these data, in this study we were able to detect substantial transcripts for the three enzymes that catabolize kynurenine, KAT II, Kynu, and KMO, in the brains of saline-injected mice (Fig. 8). Expression of none of these three enzymes was significantly increased by inoculation with BCG. However, we were able to detect a significant BCG-induced increase in 3-HAO, which leads to the production of quinolinic acid. Guillemin and colleagues (78) used fetal cultures of human tissue treated with IFN-γ to demonstrate that human microglia actively produce quinolinic acid. Our new data significantly extend the recent results in both humans (78) and rats (44), because they are the first to suggest that this increase in quinolinic acid is likely to be mediated by enhanced expression of 3-HAO. More importantly, we speculate that the balance of the putative protective kynurenine metabolite, kynurenic acid, to that of the neurotoxic metabolite, quinolinic acid, is reduced in the brains of mice infected with BCG. If so, these results might form the basis for targeting the quinolinic acid arm of kynurenine metabolism, including 3-HAO, in depressive-like behaviors that occur in this BCG-induced chronic inflammation model.
A limitation of the present experiments is that their design prevented a determination of the relative importance of peripheral vs central IDO activity. That is because in mice treated with 1-MT, the inhibitor was detected in all tissues analyzed, including the brain. This fact, coupled with the data from IDO-deficient mice, clearly indicates that global inhibition of BCG-induced IDO up-regulation is sufficient to prevent the development of depressive-like behaviors. Although IDO is significantly up-regulated in both the brain and lung of BCG-infected mice, IDO expression is several orders of magnitude higher in the lung. This phenomenon is clearly illustrated in the present studies whereby ISH is strikingly more prominent in the lungs of stimulated WT mice. Moreover, both tryptophan and kynurenine from the periphery are readily transported into the brain. Therefore, determination of whether the inhibition of central IDO activity is sufficient or even necessary for development of BCG-associated depressive-like behavior still requires additional experiments aimed at directly testing this hypothesis.
In conclusion, results of the present experiments demonstrate unequivocally that the neuroimmune aspects of the BCG-induced proinflammatory response represent a unique model for studying the molecular mechanisms that mediate cytokine-induced depressive symptoms. Although activation of IDO by proinflammatory cytokines has been speculated to mediate the precipitation of depressive disorders during chronic inflammation, its causative role had yet to be thoroughly investigated in a clinically relevant model. In the present experiments, we demonstrate that both pharmacological inhibition with a competitive inhibitor of IDO, 1-MT, and genetic deletion of IDO results in a specific inhibition of BCG-induced depressive-like behaviors without affecting the acute sickness response or the up-regulation of proinflammatory cytokines. Indeed, 1-MT does not appear to be toxic in either rats or dogs (79), nor does it affect pregnancy in mice (20). Potential toxicity issues in humans will soon be determined, because 1-MT is currently being developed for use as a vaccine adjuvant and combination chemotherapeutic agent. Similarly, the present results point to the possible role of neuroactive kynurenine metabolites along the pathway that forms quinolinic acid in the development of depression. These findings offer strong support for the notion that targeting the activation of IDO, or targeting IDO itself, might represent a viable therapeutic strategy for patients suffering from chronic inflammation-associated depression.
Footnotes
This work was supported by National Institutes of Health Grants R01 AG 029573 (to K.W.K.), R01 MH 71349 (to R.D.), and R01 MH 079829 (to R.D.).
Abbreviations used in this paper: BCG, bacille Calmette-Guérin; Ct, threshold cycle; FST, forced swim test; 3-HAO, 3-hydroxyanthranilic acid oxygenase; ISH, in situ hybridization; KAT, kynurenine aminotransferase; KMO, kynurenine 3-hydroxylase; Kynu, kynureninase; 1-MT, 1-methyl tryptophan; TST, tail suspension test; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.World Health Organization/Stop TB Partnership . Tuberculosis Facts. World Health Organization; Geneva, Switzerland: 2008. [Google Scholar]
- 2.Kaufmann SH, McMichael AJ. Annulling a dangerous liaison: vaccination strategies against AIDS and tuberculosis. Nat. Med. 2005;11(4 Suppl):S33–S44. doi: 10.1038/nm1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer TF. Preventing tuberculosis with Bacillus Calmette-Guerin vaccine: a meta-analysis of the literature. Clin. Infect. Dis. 2000;31(Suppl 3):S64–S67. doi: 10.1086/314072. [DOI] [PubMed] [Google Scholar]
- 4.Mittrucker HW, Steinhoff U, Kohler A, Krause M, Lazar D, Mex P, Miekley D, Kaufmann SH. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc. Natl. Acad. Sci. USA. 2007;104:12434–12439. doi: 10.1073/pnas.0703510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–284. doi: 10.1016/j.immuni.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufmann SH, Ladel CH, Flesch IE. T cells and cytokines in intracellular bacterial infections: experiences with Mycobacterium bovis BCG. Ciba Found. Symp. 1995;195:123–132. doi: 10.1002/9780470514849.ch9. discussion 132–126. [DOI] [PubMed] [Google Scholar]
- 7.Tsenova L, Bergtold A, Freedman VH, Young RA, Kaplan G. Tumor necrosis factor α is a determinant of pathogenesis and disease progression in mycobacterial infection in the central nervous system. Proc. Natl. Acad. Sci. USA. 1999;96:5657–5662. doi: 10.1073/pnas.96.10.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heldwein KA, Fenton MJ. The role of Toll-like receptors in immunity against mycobacterial infection. Microbes Infect. 2002;4:937–944. doi: 10.1016/s1286-4579(02)01611-8. [DOI] [PubMed] [Google Scholar]
- 9.Chackerian AA, Perera TV, Behar SM. Gamma interferon-producing CD4+ T lymphocytes in the lung correlate with resistance to infection with Mycobacterium tuberculosis. Infect. Immun. 2001;69:2666–2674. doi: 10.1128/IAI.69.4.2666-2674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamath AB, Woodworth J, Xiong X, Taylor C, Weng Y, Behar SM. Cytolytic CD8+ T cells recognizing CFP10 are recruited to the lung after Mycobacterium tuberculosis infection. J. Exp. Med. 2004;200:1479–1489. doi: 10.1084/jem.20041690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 2001;193:271–280. doi: 10.1084/jem.193.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serbina NV, Flynn JL. Early emergence of CD8+ T cells primed for production of type 1 cytokines in the lungs of Mycobacterium tuberculosis-infected mice. Infect. Immun. 1999;67:3980–3988. doi: 10.1128/iai.67.8.3980-3988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry. doi: 10.1038/sj.mp.4002148. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreau M, Lestage J, Verrier D, Mormede C, Kelley KW, Dantzer R, Castanon N. Bacille Calmette-Guerin inoculation induces chronic activation of peripheral and brain indoleamine 2,3-dioxygenase in mice. J. Infect. Dis. 2005;192:537–544. doi: 10.1086/431603. [DOI] [PubMed] [Google Scholar]
- 15.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopnisky KL, Bao J, Lin YW. Neurobiology of HIV, psychiatric and substance abuse comorbidity research: workshop report. Brain Behav. Immun. 2007;21:428–441. doi: 10.1016/j.bbi.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet. 2007;370:851–858. doi: 10.1016/S0140-6736(07)61415-9. [DOI] [PubMed] [Google Scholar]
- 18.Fujigaki S, Saito K, Takemura M, Maekawa N, Yamada Y, Wada H, Seishima M. l-tryptophan-l-kynurenine pathway metabolism accelerated by Toxoplasma gondii infection is abolished in gamma interferon-gene-deficient mice: cross-regulation between inducible nitric oxide synthase and indoleamine-2,3-dioxygenase. Infect. Immun. 2002;70:779–786. doi: 10.1128/iai.70.2.779-786.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacKenzie CR, Heseler K, Muller A, Daubener W. Role of indoleamine 2,3-dioxygenase in antimicrobial defence and immuno-regulation: tryptophan depletion versus production of toxic kynurenines. Curr. Drug Metab. 2007;8:237–244. doi: 10.2174/138920007780362518. [DOI] [PubMed] [Google Scholar]
- 20.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 21.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn H. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat. Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 22.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 26.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuchs D, Moller AA, Reibnegger G, Stockle E, Werner ER, Wachter H. Decreased serum tryptophan in patients with HIV-1 infection correlates with increased serum neopterin and with neurologic/psychiatric symptoms. J. Acquir. Immune Defic. Syndr. 1990;3:873–876. [PubMed] [Google Scholar]
- 29.Schrocksnadel K, Wirleitner B, Winkler C, Fuchs D. Monitoring tryptophan metabolism in chronic immune activation. Clin. Chim. Acta. 2006;364:82–90. doi: 10.1016/j.cca.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 30.Schroecksnadel K, Kaser S, Ledochowski M, Neurauter G, Mur E, Herold M, Fuchs D. Increased degradation of tryptophan in blood of patients with rheumatoid arthritis. J. Rheumatol. 2003;30:1935–1939. [PubMed] [Google Scholar]
- 31.Widner B, Sepp N, Kowald E, Ortner U, Wirleitner B, Fritsch P, Baier-Bitterlich G, Fuchs D. Enhanced tryptophan degradation in systemic lupus erythematosus. Immunobiology. 2000;201:621–630. doi: 10.1016/S0171-2985(00)80079-0. [DOI] [PubMed] [Google Scholar]
- 32.Wirleitner B, Rudzite V, Neurauter G, Murr C, Kalnins U, Erglis A, Trusinskis K, Fuchs D. Immune activation and degradation of tryptophan in coronary heart disease. Eur. J. Clin. Invest. 2003;33:550–554. doi: 10.1046/j.1365-2362.2003.01186.x. [DOI] [PubMed] [Google Scholar]
- 33.Smith RS. The macrophage theory of depression. Med. Hypotheses. 1991;35:298–306. doi: 10.1016/0306-9877(91)90272-z. [DOI] [PubMed] [Google Scholar]
- 34.Maes M, Smith R, Scharpe S. The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinology. 1995;20:111–116. doi: 10.1016/0306-4530(94)00066-j. [DOI] [PubMed] [Google Scholar]
- 35.Maes M, Verkerk R, Bonaccorso S, Ombelet W, Bosmans E, Scharpe S. Depressive and anxiety symptoms in the early puerperium are related to increased degradation of tryptophan into kynurenine, a phenomenon which is related to immune activation. Life Sci. 2002;71:1837–1848. doi: 10.1016/s0024-3205(02)01853-2. [DOI] [PubMed] [Google Scholar]
- 36.Widner B, Laich A, Sperner-Unterweger B, Ledochowski M, Fuchs D. Neopterin production, tryptophan degradation, and mental depression- what is the link? Brain Behav. Immun. 2002;16:590–595. doi: 10.1016/s0889-1591(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 37.Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatry. 2002;7:468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- 38.Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M. IDO and interferon-α-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry. 2005;10:538–544. doi: 10.1038/sj.mp.4001600. [DOI] [PubMed] [Google Scholar]
- 39.Moreau M, Andre C, O'Connor JC, Dumich SA, Woods JA, Kelley KW, Dantzer R, Lestage J, Castanon N. Inoculation of Bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behavior. Brain Behav. Immun. 2008;22:1087–1095. doi: 10.1016/j.bbi.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andre C, O'Connor JC, Kelley KW, Lestage J, Dantzer R, Castanon N. Spatio-temporal differences in the profile of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration. J. Neuroimmunol. 2008;200:90–99. doi: 10.1016/j.jneuroim.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, Wada H, Noma A, Seishima M. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-γ-independent mechanism. Eur. J. Immunol. 2001;31:2313–2318. doi: 10.1002/1521-4141(200108)31:8<2313::aid-immu2313>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 42.Schroecksnadel K, Zangerle R, Bellmann-Weiler R, Garimorth K, Weiss G, Fuchs D. Indoleamine-2, 3-dioxygenase and other interferon-γ-mediated pathways in patients with human immunodeficiency virus infection. Curr. Drug Metab. 2007;8:225–236. doi: 10.2174/138920007780362608. [DOI] [PubMed] [Google Scholar]
- 43.Kohl C, Sperner-Unterweger B. IDO and clinical conditions associated with depressive symptoms. Curr. Drug Metab. 2007;8:283–287. doi: 10.2174/138920007780362572. [DOI] [PubMed] [Google Scholar]
- 44.Connor TJ, Starr N, O'Sullivan JB, Harkin A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-γ? Neurosci. Lett. 2008;441:29–34. doi: 10.1016/j.neulet.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Fallarino F, Gizzi S, Mosci P, Grohmann U, Puccetti P. Tryptophan catabolism in IDO+ plasmacytoid dendritic cells. Curr. Drug Metab. 2007;8:209–216. doi: 10.2174/138920007780362581. [DOI] [PubMed] [Google Scholar]
- 46.Lestage J, Verrier D, Palin K, Dantzer R. The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain. Behav. Immun. 2002;16:596–601. doi: 10.1016/s0889-1591(02)00014-4. [DOI] [PubMed] [Google Scholar]
- 47.Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O'Connor J, Castanon N, Kelley KW, Dantzer R, Johnson RW. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–740. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- 49.Harrington L, Srikanth CV, Antony R, Rhee SJ, Mellor AL, Shi HN, Cherayil BJ. Deficiency of indoleamine 2,3-dioxygenase enhances commensal-induced antibody responses and protects against Citrobacter rodentium-induced colitis. Infect. Immun. 2008;76:3045–3053. doi: 10.1128/IAI.00193-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brandacher G, Margreiter R, Fuchs D. Implications of IFN-γ-mediated tryptophan catabolism on solid organ transplantation. Curr. Drug Metab. 2007;8:273–282. doi: 10.2174/138920007780362536. [DOI] [PubMed] [Google Scholar]
- 51.Xu H, Oriss TB, Fei M, Henry AC, Melgert BN, Chen L, Mellor AL, Munn DH, Irvin CG, Ray P, Ray A. Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc. Natl. Acad. Sci. USA. 2008;105:6690–6695. doi: 10.1073/pnas.0708809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat. Rev. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- 53.Heseler K, Spekker K, Schmidt SK, MacKenzie CR, Daubener W. Antimicrobial and immunoregulatory effects mediated by human lung cells: role of IFN-γ-induced tryptophan degradation. FEMS Immunol. Med. Microbiol. 2008;52:273–281. doi: 10.1111/j.1574-695X.2007.00374.x. [DOI] [PubMed] [Google Scholar]
- 54.Hayashi T, Rao SP, Takabayashi K, Van Uden JH, Kornbluth RS, Baird SM, Taylor MW, Carson DA, Catanzaro A, Raz E. Enhancement of innate immunity against Mycobacterium avium infection by immunostimulatory DNA is mediated by indoleamine 2,3-dioxygenase. Infect. Immun. 2001;69:6156–6164. doi: 10.1128/IAI.69.10.6156-6164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ball HJ, Sanchez-Perez A, Weiser S, Austin CJ, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, Hunt NH. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396:203–213. doi: 10.1016/j.gene.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 56.Ball HJ, Yuasa HJ, Austin CJ, Weiser S, Hunt NH. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int. J. Biochem. Cell Biol. doi: 10.1016/j.biocel.2008.01.005. In press. [DOI] [PubMed] [Google Scholar]
- 57.Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–3900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 58.Lob S, Konigsrainer A. Is IDO a key enzyme bridging the gap between tumor escape and tolerance induction? Langenbecks Arch. Surg. 2008;393:995–1003. doi: 10.1007/s00423-007-0245-7. [DOI] [PubMed] [Google Scholar]
- 59.Lob S, Konigsrainer A, Schafer R, Rammensee HG, Opelz G, Terness P. Levo- but not dextro-1-methyl tryptophan abrogates the IDO activity of human dendritic cells. Blood. 2008;111:2152–2154. doi: 10.1182/blood-2007-10-116111. [DOI] [PubMed] [Google Scholar]
- 60.Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, Terness P. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 2009;58:153–157. doi: 10.1007/s00262-008-0513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 62.Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound d-1-methyl-tryptophan. Cancer Res. 2007;67:7082–7087. doi: 10.1158/0008-5472.CAN-07-1872. [DOI] [PubMed] [Google Scholar]
- 63.Wright CE, Strike PC, Brydon L, Steptoe A. Acute inflammation and negative mood: mediation by cytokine activation. Brain Behav. Immun. 2005;19:345–350. doi: 10.1016/j.bbi.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 64.Dunn AJ, Swiergiel AH. Effects of interleukin-1 and endotoxin in the forced swim and tail suspension tests in mice. Pharmacol. Biochem. Behav. 2005;81:688–693. doi: 10.1016/j.pbb.2005.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dunn AJ, Swiergiel AH, Zhang H, Quan N. Reduced ingestion of sweetened milk induced by interleukin-1 and lipopolysaccharide is associated with induction of cyclooxygenase-2 in brain endothelia. Neuroimmunomodulation. 2006;13:96–104. doi: 10.1159/000096291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frenois F, Moreau M, O'Connor J, Lawson M, Micon C, Lestage J, Kelley KW, Dantzer R, Castanon N. Lipopolysaccharide induces delayed FosB/ΔFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology. 2007;32:516–531. doi: 10.1016/j.psyneuen.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Husain MO, Dearman SP, Chaudhry IB, Rizvi N, Waheed W. The relationship between anxiety, depression and illness perception in tuberculosis patients in Pakistan. Clin. Pract. Epidemol. Ment. Health. 2008;4:4. doi: 10.1186/1745-0179-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vega P, Sweetland A, Acha J, Castillo H, Guerra D, Smith Fawzi MC, Shin S. Psychiatric issues in the management of patients with multidrug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2004;8:749–759. [PubMed] [Google Scholar]
- 69.Makino K, Satoh K, Fujiki T, Kawaguchi K. Relation of 3-hydroxykynurenine to the Ehrlich diazo reaction of urine in severe tuberculosis. Nature. 1952;170:977–978. doi: 10.1038/170977a0. [DOI] [PubMed] [Google Scholar]
- 70.Nair S, Maguire W, Baron H, Imbruce R. The effect of cycloserine on pyridoxine-dependent metabolism in tuberculosis. J. Clin. Pharmacol. 1976;16:439–443. doi: 10.1002/j.1552-4604.1976.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 71.Burudi EM, Marcondes MC, Watry DD, Zandonatti M, Taffe MA, Fox HS. Regulation of indoleamine 2,3-dioxygenase expression in simian immunodeficiency virus-infected monkey brains. J. Virol. 2002;76:12233–12241. doi: 10.1128/JVI.76.23.12233-12241.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson CM, Hale PT, Carlin JM. NF-κB activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-γ and tumor necrosis factor-α. Cytokine. 2006;35:53–61. doi: 10.1016/j.cyto.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 73.Robinson CM, Shirey KA, Carlin JM. Synergistic transcriptional activation of indoleamine dioxygenase by IFN-γ and tumor necrosis factor-α. J. Interferon Cytokine Res. 2003;23:413–421. doi: 10.1089/107999003322277829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi S, Nathan C, Schnappinger D, Drenkow J, Fuortes M, Block E, Ding A, Gingeras TR, Schoolnik G, Akira S, et al. MyD88 primes macrophages for full-scale activation by interferon-γ yet mediates few responses to Mycobacterium tuberculosis. J. Exp. Med. 2003;198:987–997. doi: 10.1084/jem.20030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Speciale C, Hares K, Schwarcz R, Brookes N. High-affinity uptake of l-kynurenine by a Na+-independent transporter of neutral amino acids in astrocytes. J. Neurosci. 1989;9:2066–2072. doi: 10.1523/JNEUROSCI.09-06-02066.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Speciale C, Schwarcz R. Uptake of kynurenine into rat brain slices. J. Neurochem. 1990;54:156–163. doi: 10.1111/j.1471-4159.1990.tb13296.x. [DOI] [PubMed] [Google Scholar]
- 77.Turski WA, Gramsbergen JB, Traitler H, Schwarcz R. Rat brain slices produce and liberate kynurenic acid upon exposure to L-kynurenine. J. Neurochem. 1989;52:1629–1636. doi: 10.1111/j.1471-4159.1989.tb09218.x. [DOI] [PubMed] [Google Scholar]
- 78.Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. 2005;49:15–23. doi: 10.1002/glia.20090. [DOI] [PubMed] [Google Scholar]
- 79.Jia L, Schweikart K, Tomaszewski J, Page JG, Noker PE, Buhrow SA, Reid JM, Ames MM, Munn DH. Toxicology and pharmacokinetics of 1-methyl-d-tryptophan: absence of toxicity due to saturating absorption. Food Chem. Toxicol. 2008;46:203–211. doi: 10.1016/j.fct.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]