

Introduction
As discussed in other chapters in this issue, there is abundant evidence gathered in recent years suggesting that cellular plasma membranes contain small domains of different lipid composition, different degrees of chain ordering, different thickness, and different elastic properties. The small domains are often referred to as “rafts” that reside in the fluid membrane “sea”, and their physical chemical state is termed “liquid ordered’. The liquid ordered state qualitatively consists of lipids with lateral mobility that is close to that of a fluid state but with chain ordering that is greater than that of a fluid state. Over the past 15 years a variety of experimental techniques have been employed to study the properties of rafts or liquid-ordered (Lo) domains in simple model membrane systems. The model membrane systems are generally multilamellar vesicles, monolayers or giant unilamellar vesicle systems. Techniques employed typically include fluorescence microscopy, single particle tracking, differential scanning calorimetry, X–ray diffraction, NMR, and atomic force microscopy. The reviews in this issue by Keller, Longo, Kenworthy, Feigenson, Garwisch, and Kusumi describe these and other experimental methodologies. It is worth noting that there remains a level of controversy in this field, due in part to the possible affect of the measurements on the structure and composition of observed domains. [15, 8] Although there is little serious doubt that structurally segregated domains do form under certain thermodynamic and compositional states in membranes, the actual sizes may be small (≲100 nm in diameter) and their constituent molecules may exchange dynamically with the surrounding membrane. Given the small size and likely dynamical structure of membrane domains it is not surprising that detailed understanding of the intermolecular interactions which drive the formation and stability of these nano–structures is still largely lacking.
Computational modeling provides a set of tools that may be employed to gain insights into the nature of the lateral organization in membranes at a level that is not easily obtained in experiments. At its most basic level, computational modeling of membranes is done at an atomistic level using Molecular Dynamics (MD) or Monte Carlo (MC) methods. Due to the large conformation space of lipid chains, MD is the most commonly used method. While early MD simulations focussed on the properties of single component lipid bilayers, the field has since rapidly expanded to consider bilayers of heterogeneous lipid composition. In most current MD simulations of heterogeneous membranes [see e.g. 71, 69, 61, 63, 12, 23, 58, 65, 57, 55] the focus has been on global properties (such as molecular area) or localized molecular properties (such as order parameters or hydrogen bonding patterns). There have been relatively few simulation or other types of modeling efforts that directly attempt to address issues related to the lateral organization of heterogeneous bilayers. [see e.g. 24, 1, 58] The principal reason for this is the “diffusion problem”. Diffusion of lipids or cholesterol in bilayers is simply too slow (D ≈ 10−12m2/s) which means that the times needed for significant lateral displacement of lipids in a membrane should be in the tens of microseconds to millisecond range, three or more orders of magnitude longer than most current atomistic MD simulations.
In spite of the diffusion problem, atomistic simulations of heterogeneous membranes can be of great importance. Firstly they are, even at short simulation times, indicative of the overlall structural behavior of the membrane such as chain order, molecular area, average electron density, hydrogen bonding patterns, and other properties. Secondly, atomistic simulations provide details of intermolecular interactions that can be used at both qualitative and quantitative levels to build coarse-grained models that can then address the appropriate time and length scales to model lateral organization in membranes. This is the main focus of this review,
This review is organized as follows. First, we will describe a subset of the atomistic simulations that have been carried out on heterogeneous bilayers, beginning with a description of a new united atom force field developed by Chiu et al. [11]. The simulations to be discussed are those that have been used to formulate, as described above, coarse grained models. (The review presented here is not meant to be comprehensive due to the topical content of this issue. For other reviews of simulations of mixed lipid bilayers, see the papers in this issue by Marrink, Tieleman, Niemelă et al., Rŏg et al., and Berkowitz.) We will then turn to a discussion of coarse grained models. Here the focus will be on continuum and mean-field approaches. The paper by Marrink in this issue will describe a different approach to coarse graining simulations of lipid bilayers.
Atomistic MD simulations of mixed lipid bilayers
Force Fields
In molecular dynamics, trajectories of the system are generated starting from an initial state consisting of initial positions and velocities of all the atoms, and integrating Newton’s equations of motion. Critical to the simulation is the specification of the interaction potential energies between atoms. Most MD simulations use a general potential energy that has the following form:
| (1) |
The sum runs over (in order) bonds, bond angles, improper and proper dihedrals, and all pairs of atoms that are on different molecules or are separated by more than four interatomic bonds on the same molecule. The dihedral function in Eq (1) represents the energy of a connected set of four consecutive atoms on a molecule. The set of all parameters in each term in the sum comprise the “force field” for the simulation. In general each distinct pair of atoms or distinct dihedral requires a different force field parameter. Force field parameters are determined using ab initio calculations and simulations involving “model compounds”. Model compounds are molecular fragments that are structurally identical (or nearly so) to portions of lipid molecules. For example, alkane chains are used as model compounds for saturated lipid hydrocarbon chains. Force field parameters are fine tuned using MD simulations of model compounds by fitting simulation data to experimental thermodynamic data such as density and heat of vaporization. For dihedral parameters ab initio computation is used to obtain a potential energy surface that is then fit to a trigonometric function of the form listed in equation 1. The final test of a force field, after development, is of course a comparison of properties of the simulated bilayers with experimental data. Chiu et al. have recently carried out a detailed re-analysis of a united atom (all hydrogens not in a polar environment are grouped with carbons they are bonded to to form a united atom) force field. This force field is an improvement on the GROMACS [36] 43A1 force field, and is labeled 43A1-S3. [11] In Figure 1 we show a comparison of X-ray form factors calculated by simulation [55] and measured experimentally. [29] The data are for three phospholipids, DPPC, POPC, and DOPC. The very high level of agreement between simulation and experiment for direct X-ray experimental data and other data (not shown) such as order parameter profiles, shows that MD force fields, while approximate, are of suficiently high quality that the predictions of MD simulations can be relied upon. They also provide justification for their use in simulations of heterogeneous lipid bilayers.
Figure 1.

Comparison of form factors computed in simulations and X–ray scattering experiments. (a) Pure DPPC bilayer at 323K, (b) Pure POPC bilayer at 303 K, and (c) Pure DOPC bilayer at 303K. [32, 21] Reprinted from ref [55] with permission.
Lipid-Cholesterol atomistic simulations germane to raft studies
In model membrane studies that aim to understand and catalog domains in mixed lipid bilayers the most commonly studied systems are ternary mixtures of one saturated lipid, one mono-or di-unsaturated lipid, and cholesterol. The saturated lipid can be DPPC but di-saturated sphingomyelin, bovine brain sphingomyelin, or egg sphingomyelin are the more biologically relevant choices. For the unsaturated component, DOPC has been widely used, but POPC has greater biological relevance. Due to computational limitations, many simulations are generally designed to study binary mixtures of one lipid and cholesterol. Earlier simulations of lipid-cholesterol systems have been described in reviews. [65, see, e.g.] Of particular more recent interest are the simulations of Zhang et al. [73] They ran MD simulations of mixed bilayers of POPC, 18:0 SM, and 18:1 SM with 34% cholesterol each (all concentrations cited here are molar). The calculated interaction energies between cholesterol and each of the simulated lipids were found to be nearly identical, suggesting that if cholesterol interacts preferentially with one of the lipids, the interaction is driven by entropy rather than energy. Although only one cholesterol concentration was studied, the partial molecular areas could be estimated following a equation due to Hofsäb et al. [23] The linear equation extracts partial areas from volumetric data for the simulation, and from given partial volumes of water and cholesterol. From this relation Zhang et al. find that cholesterol has an area of about 25 Å2 at 34% concentration in all three simulated bilayers. This value is consistent with earlier [12] and also very recent simulations [55] that produced cholesterol areas that are less than the molecular area for crystalline cholesterol (about 38 Å2). However, the value of the area for cholesterol in POPC found by Zhang et al. is significantly larger than that obtained recently by Pandit et a [55] from a series of simulations of POPC with different cholesterol concentrations. We discuss this simulation set and the areas of cholesterol below, but remark here that the discrepancy in the partial area of cholesterol in POPC is likely to be a result of the use of one versus several simulations to extract the area. Other factors that may contribute to different areas include differences in simulation force fields, or simulation cell size differences, In this issue, other recent simulations of lipid - cholesterol bilayers are reviewed by Berkowitz, Rŏg et al., and Niemelă et al.
Here, we focus on recent lipid-cholesterol simulations that have led to new understanding of interactions between cholesterol and phospholipids of varying states of chain saturation. [56, 55] These simulations have provided essential input into coarse grained Mean Field modeling that will be discussed later. The simulations involved binary mixtures of cholesterol with DPPC, POPC, and DOPC, respectively, at cholesterol concentrations between 15 and 40%. The 43A1-S3 force field was used. Simulations were performed on a hydrated POPC [56], DOPC, and DPPC bilayers [55] with cholesterol at 10%,20%,25%, and 33%. Initial configurations for all the mixed bilayers were generated by randomly positioning 100 mixture molecules per leaflet. The simulations with DOPC and DPPC were performed at 303 K and 323 K respectively using the Nose-Hoover scheme. The choice of temperatures was driven by availability of the experimental data for the respective lipids. Relative to the lipid phase transition temperatures, TM, the reduced temperatures ((T − TM )/TM ) were 0.13, 0.20, and 0.03 for DOPC, POPC, and DPPC, respectively.
For comparison with experiment, the partial volumes of the constituents were calculated as described in [55] It was found that the volumes of the POPC and DOPC simulations decreased linearly with the cholesterol concentration x, but, volumes for DPPC requires two linear fits in different regions of cholesterol concentrations. This is precisely what was found experimentally by Greenwood et al. [21] Computed partial molecular volumes agree very well with the experimental data.
The most significant finding from the simulations came in the calculations of partial molecular areas from the simulations. Following Edholm and Nagle [14], partial molecular areas were extracted from the relation,
Figure 2 shows plots of a(x)/(1 − x) versus the ratio x/(1 − x) where x is the cholesterol concentration. The partial molecular area of a cholesterol at a particular concentration in a particular simulation is the slope of the appropriate curve in Figure 2 at that concentration. Interestingly, the area curves are not linear in x. In fact for cholesterol concentrations close to 10%, in all the simulations, the partial molecular area of a cholesterol is negative. Negative partial molecular areas are a consequence of the condensation effect of cholesterol on surrounding lipids.[14] At cholesterol concentrations above 20% the curves in Figure 2 are linear and the resulting partial molecular areas of cholesterol and lipids are 23.84, 12.42, 20.84 Å2 for cholesterol, DPPC, POPC, and DOPC respectively. Pandit et al. [55] interpret the minimum in the partial molecular area seen in POPC in terms of the anisotropic interactions between the two faces of cholesterol (rough methylated β face and smooth α face) and the anisotropic structure of the POPC chains. They suggest that an entropic preference of β–face for the unsaturated chain and α–face for the saturated chains. In POPC bilayer both types of chains are present to realize this special arrangement. The anisotropic interaction between cholesterol and lipid chains turns out to have important implications for modeling, as will be discussed below.
Figure 2.

Plot of the a(x)/(1 − x) of the simulated systems versus concentrations of cholesterol(solid line). Light dashed lines represent linear fits to data for non-zero cholesterol. The slope of the fitted lines correspond to the partial molecular areas of the cholesterol and the y–intercept gives the partial molecular area of DPPC, POPC, and DOPC. Reprinted from ref [55] with permission.
Sphingomyelin (SM) is an important component of rafts or domains in outer leaflets of mammalian cell membranes, but until recently sphingolipids have not been studied nearly as extensively as phospholipids. In 2003, Chiu and co-workers ran an MD simulation of a fully hydrated bilayer of (18:0) SM, (SSM) that consisted of 1600 SM molecules and 51200 water molecules, at 50° C. [13] The simulation clearly showed the increased level of lipid chain ordering in SSM, compared to saturated or unsaturated phospholipids, in the area per molecule and the chain order parameter profiles. For the SM bilayer, there is significant intramolecular hydrogen bonding between the phosphate oxygen and the amide hydrogen. There is also significant hydrogen bonding between water and the sphingosine hydroxyls, that occurs on over 75% of the molecules, on average, resulting in a reduction by about one molecule of the number of waters hydrogen bonded to the lipid headgroups.
Simulations of 16:0 sphingomyelin (PSM) were carried out by Niemală et al. [52] and compared to simulations of DPPC. Their analysis of intra- and intermolecular hydrogen bonding, as well as hydrogen bonding with water, are consistent with the above SSM simulations. Additionally they report lifetimes of hydrogen bonds, with the longest times associated with OH and NH groups with phosphate oxygens on the same molecule. The calculated diffusion constant for PSM is about a factor of three smaller than that of DPPC.
More germane to the issue of domain formation, Khelashvili and Scott used MD to examine the effect of cholesterol on SM bilayers in two 18:0 sphingomyelin lipid bilayer simulations, at temperatures of 50°C and 20 °C respectively. [28] The bilayers consisted of 266 SM, 122 CHOL, and 11861 water molecules. Structurally there was little difference between the simulations at the two temperatures in spite of the fact that they spanned the 18:0 SM melting temperature (about 45 °C). This structural similarity across 30° is indicative of a liquid ordered phase that has been induced in SM by cholesterol at about 31% cholesterol concentration. The same level of intramolecular hydrogen bonding on SM molecules was observed in the presence of cholesterol in the simulations. Over the short time frame of the MD simulations, no lateral organizational trends of the cholesterols in the SM were observed at either temperature.
Other simulations of sphingomyelin and cholesterol are recently been carried out by Rog and Pasenkiewicz-Gierula [64], Bhide et al [10], and Zhang et al. [73] Rog and Pasenkiewicz-Gierula simulated SSM at concentrations different from those of Khelashvili and Scott [28], and compared the results with simulations of pentadecanoyl-steroylphoephatidylcholine(15SPC)-cholesterol mixtures. They also considered two different temperatures above and below the SSM phase transition temperature. Similar to Khelashvili and Scott, they found that the properties of the SSM-cholesterol bilayers were very similar at both temperatures. However the 15SPC bilayer, despite strong structural similarity to SSM, remained in a gel phase at 37°C while the SSM bilayer exhibited a liquid-orderd phase. Both bilayers exhibited liquid ordered phases at 50°C. Since sphingolipids are primarily found in outer leaflets of membranes, Bhide et al. [10] have extended SM-cholersterol simulations to an asymmetric bilayer. One leaflet of the bilayer was a mixture of SSM and cholesterol while the other leaflet was a mixture of steroyl-oleoyl phosphatidylserine (SOPS). They found little interaction between the two leaflets, which exhibited structural properties similar to symmetrical bilayers of each lipid, respectively. In recent work of Zhang et al [73, 74], the group studied energetics of cholesterol lipid systems using umbrella sampling techniques. They investigated the change in free energy of insertion of cholesterol in lipid POPC and SM bilayers. In these simulations the free energy cost of pulling cholesterol out of SM bilayer is larger than that of POPC bilayer. [74]
The above set of simulations of binary lipid-cholesterol mixtures form a basis for the analysis, by MD and by coarse graining, of more complex and biologically interesting model membranes. These will be discussed in the following sections.
Ternary mixture atomistic simulations
Ternary mixtures of lipids and cholesterol have been studied in simulation by Pandit et al.[60, 59] and most recently by Niemală et al. [53] In two sets of simulations, Pandit et al. examined the structure of lipid bilayers containing mixtures of 18:0 SM, DOPC, and cholesterol. In the first case, they constructed a large bilayer consisting of 1424 molecules of DOPC, 266 molecules of 18:0 SM, 122 molecules of cholesterol, and 62,561 water molecules. The simulation initially placed all of the SM and cholesterol in a central, pre-formed “domain” that was surrounded by DOPC molecules. Due to the large size, this simulation was only run for about 20 ns. During that time the pre-formed region there was no mixing of SM with DOPC but there was some mixing of cholesterols, that left the SM region and diffused into the DOPC region.
More recently, Niemelă et al. also ran large scale simulations of lipid bilayers consisting of 1024 lipids in 1:1:1 and 2:1:1 POPC:16:0 SM:cholesterol. [53] The simulations were run for 100 ns each, and all of the molecules were initially distributed randomly in the bilayer. This study provided pressure profiles and order profiles that showed the rigidity of the bilayers. The lateral distribution of chain order is indicative of the beginnings of a lateral organization process. Again, the timescales were not sufficient to directly simulate lateral re-organization in the membrane.
A set of ternary mixture simulations of smaller bilayers [58] consisted of 100 DOPC, 100 SM, and 100 cholesterol molecules plus 9600 waters. These simulations were started from random distributions of DOPC, SM, and cholesterol. The simulations were run for 250 ns with the goal of determining the interactions, structural, and dynamical parameters that drive the formation of domains. As a control, a simulation of a binary system consisting of 100 SM plus 100 DOPC, with no cholesterol was run. Figure 3 shows before and after snapshots of the ternary system, and a 200 ns snapshot of the binary system. We have performed the same series of structural calculations on the random mixtures as we did on the single-domain simulation described in the preceding paragraph. One of the important findings of this work, revealed in quantitative analysis of the lateral molecular area correlations, is that, on the 200 ns timescale, the presence of cholesterol enhances the formation of segregated domains rich in either SM or DOPC.
Figure 3.

(left) Snapshots of ternary DOPC-SM-cholesterol left: initial states (after 4 ns of equilibration). next right: after 150 ns, third right: after 200 ns; far right: binary system after 200 ns. Color code is: Black: DOPC, orange: SM, yellow: cholesterol. Reprinted from ref [59] with permission.
While the 250 ns time of these simulations is still not long enough for significant lateral reorganization to occur, it does provide a large data set of snapshots from which correlations can be calulated. To this end, Pandit et al. [58] calculated two-dimensional radial distribution functions in both distance r in the membrane plane, and the plane polar angle ϕ:
where N(r,φ) is the number of CH1 atoms of lipids in an area element 2πrδrδφ at the point (r,φ) measured from the oxygen of cholesterol. The data are averaged over all cholesterols and over the last 150 ns of the simulation. The results, reproduced in Figure 4, show by two-dimensional radial distribution function density plots, between cholesterol and SM, and cholesterol and DOPC. The orientation of the cholesterol is taken into account., and darker color indicates stronger correlation(larger g(r,ϕ)). Cholesterol is represented by the T figure, with the horizontal appendage as the methylated rough face. The color distribution shows a clear preference of SM for the smooth face of cholesterol. By default a similar calculation for DOPC-cholesterol shows a slight preponderance of DOPC nearer to the rough face of cholesterol molecules.
Figure 4.

(left) Density plot of g(r,ϕ) on the XY plane of the cholesterol body coordinate system at 200 ns. A top view of the cholesterol molecule reference frame is schematically shown as black T. The radial distribution function was calculated between O atoms of cholesterol and the backbone CH1 moiety of SM (and also DOPC). Left: DOPC-cholesterol; right: SM-cholesterol. Reprinted from ref [59] with permission.
The atomistic interactions span times of at most a few hundred ns. To model the lateral organization of membranes, it is necessary to extend the timescales to microseconds and beyond. In this effort it is crucial to the validity of any coarse graining effort that accurate atomistic data be included at a quantitative level. We describe such an effort to make use of the above MD simulation results for construction of a coarse grained model in the following section.
Coarse grained simulations of heterogeneous bilayers
As a consequence of the diffusion-based limitations on atomistic simulations, there are multiple ongoing efforts to devise alternative “coarse grained” simulation models that retain the essential features of atomistic lipids, but that have otherwise severely reduce the number of molecular degrees of freedom, thereby greatly accelerating the simulations. We now describe several coarse graining strategies that have the goal of extending simulations to greater scales of length and time. In each case atomistic detail is sacrificed that may affect the resulting properties of the coarse granied models.
One way to model lipid bilayers on larger scales of length and time is to reduce the number of degrees of freedom per molecule. This can be done by simply reducing the number of atoms in the simulation. For example, one can condense a typical lipid molecule from about 50 (non-hydrogen) atoms to between four and twenty “pseudo-molecules” by combining atoms along the three lipid chains. The pseudo-molecules are constructed as chains of typically 4–10 pseudo-atoms (solvent is also represented by pseudo-molecules). After defining suitable forcefields for the coarse-grained model, the properties of the system are calculated using standard MD or MC. The effect of the reduced number of degrees of freedom is to allow for a time step of ≈ 5 picoseconds in MD, or larger moves in MC. Thus with a coarse grained model for the lipids one can obtain an improvement of about 3–4 orders of magnitude in computational speed [22, 30, 31, 67]. Lipowski and co-workers have studied similar coarse-grained molecular models for bilayers using Monte Carlo (MC) and MD simulation. [19, 18] Marrink et al. have developed a robust forcefield for coarse grained simulations of lipid bilayers [41] allowing for the most comprehensive simulations of lipids in this approximation to date. The model, called the “Martini” model, is described in the paper by Marrink in this issue. The reduction in degrees of freedom and the choice of parameters allow Marrink and co-workers to run MD simulations for over roughly 10 microseconds (μs) (this type of coarse graining changes the time scale so “real” times must be estimated). As a consequence, the model has been used to simulate such events as gel phase formation [43]and hexagonal phase formation. [42]
In an interesting application of coarse graining, Stevens has used this type of coarse graining to show how domains form in and how they match across the two leaflets of a bilayer. [70] In another coarse graining effort, Orsi et al. [54] have developed a coarse-grained model that represents water molecules as sticky dipoles, and lipid hydrocarbon chains as strings of Gay-Berne ellipsoids. This approach has the advantage of including electrostatic interactions explicitly. The model has been applied to the simulation of the self-assembly of a DMPC bilayer, with resulting structure that agrees qualitatively with experiment. The model has not been applied to heterogeneous lipid mixtures at this time.
The pseudoatom approach to coarse graining produces results that are of clear qualitative value in understanding the dynamical interactions of membranes on a large scale. However there is no real connection between the atomic level interactions and the coarse grained model. The parameters for the latter were determined from experimental data rather than from atomistic forces. This approach is most useful for thermodynamic properties, but is not as useful for understanding small scale biological interactions.
A series of modeling efforts aimed at extension of atomistic simulation predictions to biological scales have been proposed by Voth and co-workers. [2, 4, 3, 39, 5, 6, 7] In this approach the lipid bilayer is represented as a continuous elastic membrane. Connection to the atomistic scale is made through calculation of the elastic modulus for the membrane using non-equilibrium MD or dissipative particle dynamics (DPD) methods based on input from the MD for the bulk properties. In these papers Voth and co-workers describe several methods by which they use local bulk elastic properties for their mesoscopic simulations and by which they map mechanical properties from MD simulations onto coarse grained models. The coarse graining in this case is from the atomic scale to a continuous elastic field. The elastic modulus for the membrane is calculated by non-equilibrium MD or DPD simulations. Properties of the elastic field are calculated by continuum simulation methods. For modeling binary lipid-cholesterol mixtures the elasticity field is coupled with a composition field. The composition field evolves in time via the Cahn-Hilliard equation. [37] When applied to a mixture of cholesterol and di-myristoyl phosphatidylcholine (DMPC) the model predicts composition-curvature fluctuations are interpreted as micro-domains rich in cholesterol and ordered DMPC. The work of the Voth group is the only approach to date that links atomistic simulations to mechanical properties of membranes, and the predicted coupling of curvature and lateral organization is an important result that should be tested against experiment. Due to the complex interactions in this model simulation times may for complex systems be limited to the nanosecond scale, the same as atomistic MD, so some of the advantage of this coarse graining may be lost. We also note that the above papers discuss several independent approaches to link atomistic simulations to mechanistic properties of membranes. This underscores the non-uniqueness of such mappings and suggests caution must be applied in the interpretation of properties predicted by coarse-grained models developed in this manner.
A novel approach to coarse graining that makes direct connection with atomistic MD simulations has been developed by Isvestov and Voth. [26, 27] the coarse-graining begins by reducing the number of molecular degrees of freedom, as discussed above in work by others, However the interaction parameters are now obtained from MD simulations by a force-matching process. In this process interaction parameters in the coarse-grained representation are determined my optimally matching bonded and non-bonded interactions between coarse-grained atoms to forces between the centers of mass of their atomistic multi-atom counter parts. The resulting coarse-grained simulations of DMPC yield radial distribution functions that closely match the atomistic RDF’s. Atom distributions also match those of atomistic simulations. This is a complex effort but can provide a data base of interaction potentials for coarse grained simulations that represent atomistic forces, and, when used in simulations, can be considered as extended predictions of atomistic MD.
The major challenge for all coarse-graining modeling is to link the coarse-grined dynamical entities (pseudo-particles or fields) with accurate atomic level detailed properties of the system. This connection is difficult to construct in the pseudoatom coarse-graining strategy because the pseudoatoms’ degrees of freedom cannot be easily decomposed into sums of atomistic degrees of freedom. In order to achieve a predictive level of modeling between atomistic and coarse-grained modeling a direct connection between some property or properties of the MD simulation and the parameters in the coarse-grain model needs to be established.
For the modeling of large scale equilibrium properties of bilayers, it is desirable to devise coarse-grained models that are amenable to statistical mechanical methods. One such set of models was developed by Mouritsen and co-workers. [51, 50]. The models are base on the well known two-dimesional Ising model. The original model allocates one degree of freedom (spin) to each molecule on a lattice, and interactions occur between nearest neighbor pairs only. The new models add additional degrees of freedom and multi-spin interactions, and use either a random lattice [51], or go to an off-lattice version. [50] By adjusting the parameters associated with the additional degrees of freedom, a rich set of possible phase properties are accessible. The model is applied to a description of the comparative effects of cholesterol, ergosterol, and lanosterol on a bilayer in which the lipid is palmitoyl-petroselinoyl phosphatidylcholine (PPetPC), in collaboration with NMR and DSC experiments. [46] Simulation-predicted phase diagrams are in generally good agreement with experimental results in this work. This level of phenomenological modeling can yield insights into how intermolecular interactions lead to observable experimental results. It is difficult, however, to connect the parameters in the Ising-like models with more detailed and physically realistic atomistic interactions calculated in MD simulations.
An interesting and promising statistical mechanical method for the export of atomistic data from MD simulations to coarse grained models has been proposed by Mutrola et al. [48, 47] The method has a base in the Renormalization Group approach to critical phenomena, and was developed initially by Lyubartsev and Lalksonen. [38] In the application to a lipid bilayer, one first defines a simple two-dimensional Hamiltonian function of the form:
| (2) |
where the Kα are coupling constants and the Sα (r) depend on the coordinates of the particles. The sum runs over all interparticle distances, out to a cutoff. Lyubartsev and Lalksonen take Sα (r) to be the pair distribution function(RDF) for the system (times a normalization factor). Then they propose an iterative Monte Carlo scheme that renormalizes the Kα until the Sα (r) approach pre-determined values within a tolerance level. In the application of this method to lipid bilayers, Mutrola et al. [48, 47] calculate atomistic RDF’s between selected pairs of atoms for DPPC-cholesterol mixtures from MD simulations. This process effectively maps an entire leaflet of the bilayer onto a two-dimensional model that can easily me further explored using Monte Carlo simulation. In the most recent version of the method, Murtola et al. [47] use several different RDF’s between different pairs of lipid atoms, and they expand the iterative procedure to include additional thermodynamic constraints. The optimization procedure now becomes multi-dimensional. Multiple solutions are possible, so that the coarse-grained model that results may not be unique. The authors’ resulting coarse grained model predicts, for certain cholesterol concentrations, the formation of localized domains in DPPC-cholesterol bilayers. This approach is a promising procedure and the predictions are amenable to comparison with experimental data.
A different approach to the analysis of lipid conformations in bilayers has been proposed by Murtola et al. [49] that uses self-organizing maps. The maps were able to identify the lateral distribution of lipid chain conformations in an MD simulation, based on the distributions of chain dihedral angle values. This method offers a new and novel approach to understanding lateral organization in membranes.
The above approaches are technically limited to equilibrium, static properties by the Monte Carlo approach (although one can loosely relate MC steps to time steps, the connection is not clear in most cases). In the following section we describe a lattice-based approach that includes dynamical properties of bilayers.
Mean Field theory for mixed lipid bilayers
Equilibrium Mean Field theory
Rather than use a pseudoatomistic approach to coarse graining, one can build a coarse grained model based on selected structural properties of the original molecules. Then methods of statistical mechanics can be employed for the large scale equilibrium and dynamical properties, and the basic building blocks can be related to atomistic simulations.
A method for the statistical mechanical simulations that can address this problem is Langevin Dynamics driven by free energy gradients in a locally defined molecular order. [37, 9, 62] In this type of modeling a system is defined over an area or volume by a localized free energy functional of a suitably defined, localized, order parameter. Minimizing this functional over the order parameter in the absence of perturbants or external driving forces leads to the usual static Ginzburg-Landau theory for phase transitions. Dynamics are introduced via the time-dependent Ginzburg-Landau (TDGL) equation. [37] The key variable that specifies the state of a molecule is in this approach the order parameter. For lipid chains the natural choice is the well known segmental chain order parameter.
In 1974 Marcelja [44] formulated an equilibrium statistical mechanical model for a lipid bilayer using segmental order parameters, and this model can provide the needed connection to atomistic simulations. The model was based on the Maier Saupe model [40] for liquid crystals. In this approach, the inter-atomic interactions that contribute to conformation-dependent intermolecular interactions are reduced to a Hamiltonian based on molecular segmental order parameters. The molecular order parameter is defined as a weighted average of the segmental order parameters over the chain:
| (3) |
where θm is the angle between the plane of the C-H bond vectors for carbon m of the chain at the position r⃗ and the bilayer normal. In this definition the position r⃗ is a two-dimensional chain-averaged position vector on the bilayer plane, but could, in a more detailed model, represent the positions of each carbon on the chain. [66] n is the total number of carbons for which the order parameter is calculated in the chains of the lipids, ns is the number of dihedral angles in a chain, and ntr is the number of dihedrals in trans conformations. In the equilibrium formulation of this model for a uniform bilayer s(r) does not depend on the position r⃗.
The Hamiltonian function is then defined as
| (4) |
where the sum extends over nearest neighbor pairs of lipid chains. The coarse-graining in this model consists of replacing pair interactions between all atoms in the system with effective pair interactions between pairs of chains where the details of the atomistic potentials are replaced by a bilinear energy function that is dependent on the average chain order parameters. Since the chain order parameters are the hallmark of the main lipid chain melting phase transition, this coarse graining approach is similar to many other models for phase behavior in statistical mechanics. [37]
In the Mean Field approximation the Hamiltonian is further reduced to that of a set of independent molecules interacting through an effective average “mean field” with its neighbors:
| (5) |
where < s > is the bilayer-average chain order parameter which does not depend on the molecular position in a homogeneous bilayer.
The free energy of the system in the mean field approximation is
| (6) |
where Ztot is the total partition function of the system, kB is the Boltzmann constant, T is the absolute temperature, and the summation on the right hand side is over all the possible configurations of all the lipid chains. Evaluating F in Self-Consistent Mean Field Theory (SCMFT) reduces the sums in Eq 6 to expressions that can be evaluated explicitly, and self-consistency requires that the theory be closed by the self-consistent equation:
| (7) |
Here, Φ is the mean field at a lipid chain due to the average interactions of all neighboring chains, Eq 8,
| (8) |
and depends on the values of 〈 sj 〉 at the neighboring sites. ν − ci denotes the number of neighbor sites occupied by lipid molecules (this allows for generalization to a number of sites occupied by cholesterols, ci). Equation 7 is a non-linear equation, which can be solved numerically. In the solution procedure, in order to evaluate the partition function Z it is necessary to sum over lipid chain conformations. The terms in the sum over configurations was generated numerically by Marčelja d using a simple three-state (rotational-isomeric) model, but this approach is not unique, as will be discussed below. The resulting model, a phenomenological choice of the coupling constant V0, agrees quantitatively with experimental data for simple lipid chain melting transitions.
At this point a general comment on Mean Field theory is appropriate, as it is often assumed that Mean Field theory is not likely to apply to a system with the conformational degrees of freedom of a lipid bilayer. However, numerous recent applications, some of which are summarized by Matson, and by Muller [20], show that Mean field theory is quite robust and appears to be well suited to modeling complex anisotropic systems, as long as one is not concerned about behavior very close to a second order critical point. In other words, as long as intermolecular correlations in the order parameters are not of extremely long range, Mean Field theory provides an excellent modeling platform.
Schick and co-workers have extended the Marčelja model to lipid bilayers containing lipids having different chain saturation states and cholesterol. [66, 16, 17] For the application of the model to di-unsaturated chains Scoville-Symonds and Schick [66] reduce the effective chain length in the Marčelja model by reducing the number of CH2 molecules that contribute to the average order in chains with double bonds by two. Scoville-Symonds and Schick considered models in which the double bonds had different locations on the chains, and calculated the effect or the locations of the double bonds on the properties of the membrane phase transition temperatures.
In another significant extension of Mean field modeling of equilibrium properties of lipid bilayers, Elliot et al. [16] developed a model that includes details of intermolecular interactions along the membrane normal direction in the interior bilayer. Elliot et al. calculated single lipid phase transitions and, more significantly, phase separation transitions, and binary phase diagrams for the DPPC-DOPC mixtures. [17] The model was applied to DPPC-cholesterol using the same basic model and the SCMFT approach. The phase diagram they calculated was in good agreement with experiment.
The Mean Field models of Schick and co-workers calculate equilibrium thermodynamic properties of the lipid bilayers under consideration. In order to also extract dynamical behavior of mixed lipids, on a larger scale than is possible in MD simulations, self-consistent Mean field theory can be combined with Langevin Dynamics moeling. We describe work in this direction in the following subsection.
MD based Langevin Dynamics and Mean Field theory for mixed lipid bilayers
Another challenge for all coarse-graining modeling is to link the dynamical properties of membranes with a accurate atomic level set of intermolecular forces. For this modeling, the equilibrium methods of the previous section must be extended to allow time-dependent behavior. To address the dynamics question, as noted above, one can make use of a method for simulations that has a basis in self-consistent Mean field theory (SCMFT), and can be run at scales of micrometers and milliseconds, by introducing Langevin Dynamics driven by free energy gradients in a locally defined molecular order. [37, 9, 62] A system is specified by a localized free energy functional of a suitably defined local order parameter, and minimizing the free energy functional over the order parameter leads to the equilibrium picture for phase transitions. Dynamics are introduced by implementing the TDGL formalism. [37] An example of this method to a two-dimensional hard rod system is given by Peng et al. [62] For this application, the basic free energy functional assumed to have the general form [33] shown in Eq 9
| (9) |
where ψ(r⃗) is the order parameter for the model. The order parameter in general for a system is a property that undergoes a sharp change in value at the phase transition of the system. This could be the orientation of spins in a simple ferromagnet, the difference between liquid and vapor phase densities in a simple fluid, or the chain order, Eq 3, in a lipid bilayer. The Landau free energy functional represents a generic Taylor expansion of the free energy and is not sensitive to the details of the system.
A coarse-grained model for lipid bilayers based on the TDGL equation and the Landau free energy functional of Eq. 9 was formulated by Shi and Voth. [68] In this approach, Shi and Voth directly obtain parameters for their model from coarse grained MD simulations using the Marrink model mentioned above and in the review by Marrink in this issue. [41] The MD simulations were carried out using large, coarse-grained, bilayers of 1024 DPPC molecules and 1024 molecules of dipalmitoyl phosphatidylethanolamine (DPPE). The Marrink model simulations of this system at 308 K produced leaflets with separated regions of high and low lipid chain order after 480 ns of simulation which is argued to be equivalent to 1.9 μ sec of real time. Although 308 K is below the phase transition temperature of both DPPC and DPPE, the coarse-grained lipids nonetheless evolved to a phase separated state. For the TDGL model an order parameter and a free energy must be defined. For the order parameter, Shi and Voth transform the lipid chain-averaged order parameter:
| (10) |
where Sz is the average order parameter for a coarse-grained chain:
| (11) |
with θi being the angle between the ith C-C bond and the bilayer normal, and NCC the number of such bonds. The TDGL free energy was found by Shi and Voth by analysis of the height fluctuations (“kinks”’) in the phase boundaries in the simulations. as functions of the local distribution of chain order as defined by Eq 10 The result was a free energy functional of the form of Eq 9 with parameters derived from the coarse-grained simulation. The model of Shi and Voth is noteworthy for its use of data from MD simulations. In principle this is a direct method for the extension of MD simulations to much larger time and length scales. However the model has not yet been applied to raft-like ternary mixtures.
To model domain formation in bilayers, Ayton et al. [5] developed a method for coupling Landau-Ginzburg based Mean Field theory with continuum mechanics modeling to predict the evolution of lateral organization and domain formation in giant unilamellar vesicles. In this approach a Landau free energy functional is constructed that contains contributions from the bilayer bending energy, lateral composition inhomogeneities over the bilayer, and a term that couples bendnig to lateral composition. A time-dependent Ginzburg-Landau equation (to be discussed in detail below) or a Cahn-Hilliard equation is used to propagate the system in time. Ayton et al. [5] solved the equations for a spherical geometry to model a large unilamellar vesicle (LUV). the results, for appropriate choices of interaction parameters, bear qualitative resemblance to experimental observations of domains in LUV’s.
We now describe an approach similar to that of Shi and Voth [68] that uses fully atomistic MD simulations for its input, and can be applied to binary and ternary lipid mixtures to obtain predictions that are in excellent accord with experiments. For the application to a mixed lipid bilayer, we have developed a TDGL-based approach that uses the Mean field free energy function calculated explicitly for lipid bilayers by the Marcelja model described above. This approach calculates interactions and chain conformation from libraries obtained from atomistic simulations. This approach has been used to model mixtures of dipalmitoylphosphatidylcholine (DPPC) and cholesterol. [28, 56] It can also be extended to ternary mixtures of raft composition, as we describe below.
The Langevin modeling is based on a Ginzburg-Landau approach to the study of coexisting phases. [37] One defines the order parameter which characterizes the phase transition, ψ, and a free energy functional F(ψ, t). For a lipid bilayer the appropriate choice is the same as used by Marčelja with ψ as the local average chain order parameter field defined in Eq 3. The time evolution of the local order parameter is driven by local free energy gradients and by random thermal fluctuations. These terms are incorporated into the TDGL Equation [37, 62]:
| (12) |
where Γ is an “order parameter mobility”. This equation treats the lipid fluid phase as a continuum. The order parameter ψ(r⃗) is not conserved over the bilayer (i.e. the sum of ψ(r⃗) over all lipids is not fixed). To model a membrane containing cholesterol in the membrane, we take advantage of the rigid fused ring structure of a cholesterol molecule and model the cholesterol molecules as hard rods of appropriate size. Then, in addition to a chain order field, the system contains discrete rods, each of which has an orientation θ and a position r⃗. The variables θi and r⃗i evolve in time according to Langevin equations based on associated gradients in the system free energy F [62]
| (13) |
| (14) |
where M and M′ are mobilities, and ηi and ζi are thermal fluctuations which are derived from an appropriate Gaussian distribution.
In the adaptation of this methodology to a lipid bilayer one first defines and initializes a large two dimensional lattice network of lipid chains plus cholesterols. Typically the lattice should have 105–106 sites. The lipid chain order parameters values are assigned to points on the lattice while the cholesterols, modeled as small hard rods which move continuously over the plane, are given initial random (non-overlapping) center of mass positions and orientations. The cholesterol rod length is set at 0.7 times the lattice constant to match the relative size of a cholesterol molecule relative to lipid chains. In a mean field simulation the first step is to establish the initial distribution of order over the system by solving the self consistent mean field equations for initial state. The initial distribution of order is thereby determined by the lipid-lipid interactions, the temperature, and the lipid-cholesterol interactions in the initial cholesterol distribution. The time is then advanced by one step in which cholesterols move according to Eq 12. Then the self consistent mean field equations with new cholesterol distribution are solved, and the process is repeated by starting the next time step. This procedure allows the lipid order to equilibrate between each incremental change in the locations of the cholesterols, consistent with the vary rapid chain equilibration timescale. As the simulation progresses, the result is a prediction of the time evolution of the lateral organizational structure of the membrane.
The model was applied to DPPC-cholesterol bilayers over a range of cholesterol concentrations and temperatures. [56] In this modeling effort, phenomenology is kept to a bare minimum; the interaction energies between DPPC and cholesterol were calculated from MD simulation data. Further, the sums over chain configurations were carried out using a library of DPPC chain conformations generated by a large MD simulation of a hydrated pure DPPC bilayer. The mobilities used in Eqs 13 and 14 are related to the experimental diffusion constant D by: Mr = D/kbT; Mθ ≈ (10–100) × Mr, and Γ ≈ 10Mr. The dimensionless simulation timestep Δτ is related to the real-time step length , where a is the lattice constant, set in preliminary work at 0.65 nm. This allows for nanosecond or greater real time step sizes with reasonable Δτ for coarse grained simulations. Predictions of the model can for this reason be considered to be extensions of MD simulation predictions. Results are best visualized using two-dimensional color density plots of the lipid order in the systems simulated. The Figure below shows typical for a simulation of DPPC-cholesterol mixtures.
Using this model we can calculate the average order parameter as a function of temperature for various cholesterol concentrations within this model. Figure 5 shows color density plots of the variation of the order parameter field across the simulation cell for three different cholesterol concentrations. with darker colors representing higher chain order. The small lines represent the locations and orientations of cholesterols, and are inevitably surrounded by regions of high relative chain order. In the absence of cholesterol, the model exhibits a first order phase transition at 315 K. The order parameters at temperatures below and above phase transition are similar to the values expected in gel and liquid crystalline phases of DPPC respectively. At concentrations above 25 %, the system has uniform order across all the temperatures investigated here, representing the liquid ordered (β) phase. A closer examination of simulations at other cholesterol concentrations reveals that the phase transition is first order (within numerical resolution) up to about 5% cholesterol concentration, but becomes smoothed, diminishes, and completely vanishes for cholesterol concentrations above 15 %. Similar results are reported by the NMR and DSC experiments of Huang et al. [25] Simulated distributions for order parameters are in very good agreement with this data. [56]
Figure 5.
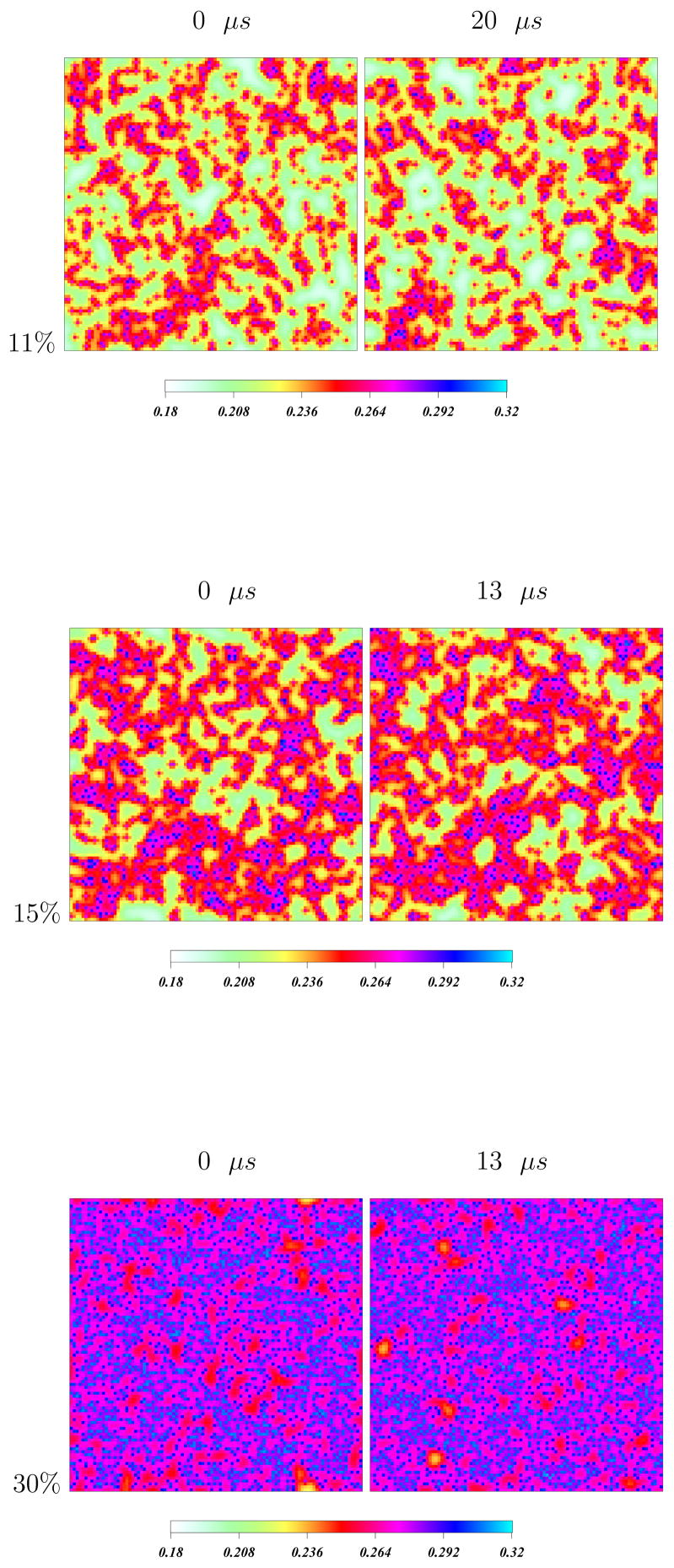
Order parameter density plots of the local DPPC chain order parameter distribution for three different cholesterol concentrations. Times are t = 0(left), and t = 20μs(right). The color scale indicates magnitude of local order parameters on the lattice, with darker colors indicative of greater order. At concentrations between 12–20 % there are regions of high order (blue-red) that are separated by regions of relatively lower order (pale green). Reprinted from ref [57] with permission.
In order to quantify the existence of regions within the model field of different levels of chain order, one can again examine the distribution of chain over the order parameter field order by a binning procedure. For certain temperature and cholesterol concentrations, coexisting regions of different order are found, as revealed by a bimodality in the distribution. This property can be used to identify different regions of a “phase diagram”. Figure 6 shows distributions of order parameters at 5 %, 25 %, and 30 % of chol concentration at various temperatures. The distributions show peaks around the order parameter values corresponding to gel and liquid crystalline order. For certain values of concentrations and temperatures, these distributions show two peaks indicating coexistence of two “phases”, e.g., at 50 C and 25 % cholesterol concentration the system clearly has multiple peaks in order parameter distribution.
Figure 6.

(a) Average order parameter as a function of temperature for various chol concentrations. Comparison of the average order parameter in the model with the order parameter of the sixth carbon atom reported by Huang et al. for (b) 10 % chol, (c) 15 % chol, (d) 20 % chol, and (e) 30 % chol. Reprinted with permission from ref [57]
By examining all distributions at various temperatures and cholesterol concentrations, a plot can be drawn that outlines the regions in which the distribution of order parameters is bimodal as revealed in Figure 7. This figure is fully consistent with the phase diagram proposed by Vist and Davis. [72] In particular, both model and experiment show non-overlapping regions having chain order at the same cholesterol concentrations and temperatures. At temperatures below 315 K, coexisting regions of gel and intermediate order (gel + β region). At temperatures above 315 K, there are non-overlapping regions of intermediate and low levels of order (Lα + β region). The agreement between the model diagram and the Vist-Davis diagram is shown in Figure 7, and is significant in that no parameters in the model were set phenomenologically to fit the experimental plot.
Figure 7.

(left) Computed phase diagram from the model, (right) Plots of specific heat as a function of temperature for several cholesterol concentrations. Reprinted from ref [57] with permission.
The model was tested further against experiment by computing the specific heat as function of temperature, a property that is well established experimentally but beyond the scope of MD calculations. It was found [57] that the heat capacities at all temperatures agree well with experiment. [25] Another significant prediction of this model is that there is no first order phase separation transition in this model. That is, changes in the lateral organization are continuous. This model prediction is independently supported by recent re-analysis of NMR data by McConnell and Radhakrishnan. [45]
The SCMFT approach is readily generalizable to ternary and higher mixtures of lipids. In order to model the a mixed lipid bilayer, a new field that describes the local molecular composition of the bilayer is introduced on the lattice at each site r⃗, e.g. for sphingomyelin (SM) and dioleylphosphatidylcholine (DOPC) one has:
| (15) |
where ϕx represents the concentration of species x, at the point r⃗ and Φ is then the local difference in concentration. One can construct a Free Energy functional based on the Marčelja model which includes the interactions of all of the lipids in the mixture, all using maximum input from simulations. In addition to propagating cholesterols through the model plane, we now also propagate SM and DOPC molecules over the bilayer plane, in this case using a Cahn-Hilliard equation (in order to conserve the total number of atoms, this equation must be used instead of the Langevin equation) [37]. The functional contains coupling terms between different molecules which will be calculated from simulations, and will be used to predict the lateral organization of membranes on the scale of the TDGL method. Figure 8 shows calculated ordering in mixtures of SM-DOPC-cholesterol obtained using a preliminary estimate of the salient coupling constants from MD simulations, and after an LD-SCMFT run of 100 μ s.
Figure 8.

Ordering in ternary mixtures of SM, DOPC, and cholesterol. Left: 1:1 mixture of DOPC and SM with 10% cholesterol. Right: 1:1 mixture of DOPC and SM with 20% cholesterol. Darker color indicates higher order, and therefore a greater concentration of SM, while small segments represent cholesterol molecules.
It is evident that cholesterol induces the formation of regions of increased order in the field. In this model increased order is correlated with an enhanced concentration of sphingomyelin. By a quantitative examination of the distribution of order over the field, it is possible to map out a ternary phase diagram for this mixture. An alternative approach for the determination of phase boundaries is to map locations of anomalies in the heat capacity on a ternary diagram. Figure 9 shows a preliminary construction of a ternary diagram based on heat capacity. Further work is in progress on this and other ternary phase diagrams.
Figure 9.
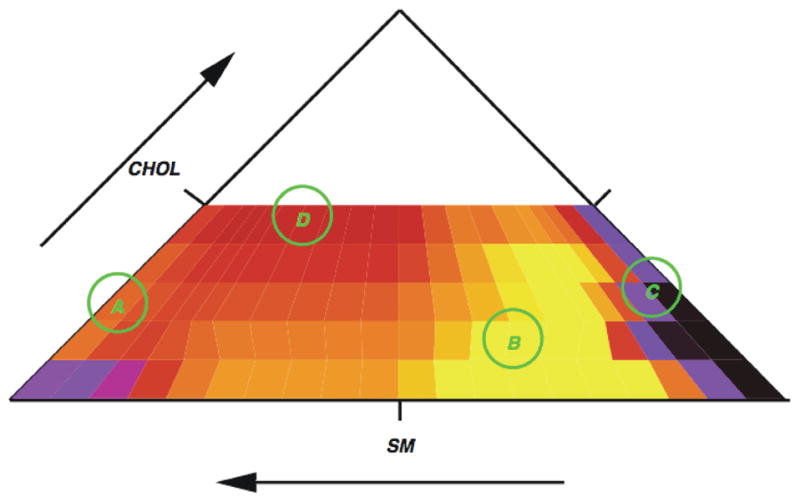
Preliminary partial ternary phase diagram for DOPC/SM/cholesterol based on heat capacity. The color represents heat capacity with red being the largest. On this diagram there are only four data points (circled) at this time so the plot is meant as a preliminary example only. The data are all for a temperature of 303K.
The next level of coarse graining beyond what has been discussed above is continuum modeling, where it may be possible to simulate more complex biological membranes and tissues. In this light we note the continuum modeling of Lin and Brown. [34, 35] Lin and Brown describe a simulation procedure for elastic thin membrane sheets that are coupled to a surrounding solvent. A Langevin dynamical model for the dynamics of the sheet is simulated in Fourier space. In an interesting application, the dynamics are simulated with the membrane sheet locally confined by a random cytoskeletal matrix in an inhomogeneous fluid environment.
Discussion
In this paper we have focussed on the use of atomistic MD simulation as a base for coarse-grained models that can be applied to the study of the conditions under which domains can form in lipid bilayers. These conditions are not directly revealed by atomistic simulations due to the slow rate of lateral diffusion of molecules in bilayers. However, the interatomic and intermolecular interactions and correlations that drive lateral organization are in principle contained in MD simulations. This means that coarse grained models that are based on atomistic data become, by extension, predictions of the MD models. Future work by our lab and others should include the theoretical prediction of ternary mixtures of DOPC/SM/cholesterol, and POPC/SM/cholesterol. Comparison of the predictions with experiment will serve to refine the modeling work. Some possible future goals are:
The inclusion of mechanical properties like elasticity fields (as done by Voth et al. and Lin and Brown, described above) with ordering properties.
The inclusion of other lipid species, including inositols and gangliosides
the inclusion of membrane peptides and transmembrane protein domains
Acknowledgments
HLS is supported by NIH Grant UIUC/NIH 2006-139-1 to the National Center for Biomimetic Nanoconductors, E. Jakobsson, Director
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson TG, McConnell HM. Condensed complexes and the calorimetry of cholesterol–phospholipid bilayers. Biophys J. 2001;81(5):2774–2785. doi: 10.1016/S0006-3495(01)75920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayton G, Bardenhagen S, Sulsky D, Voth GA. Interfacing continuum and molecular dynamics: an application to lipid bilayers. J Chem Phys. 2001;114:6913–6924. [Google Scholar]
- 3.Ayton G, Smondyrev AM, Bardenhagen S, McMurtry P, Voth GA. Interfacing molecular dynamics and macro-scale simulations for lipid bilayer vesicles. Biophys J. 2002a;83:1026–1036. doi: 10.1016/S0006-3495(02)75228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayton G, Smondyrev AM, Bardenhagen S, McMurtry P, Voth GA. Calculating the bulk modulus for a lipid bilayer with non-equilibrium molecular dynamics simulation. Biophys J. 2002b;82:1226–1238. doi: 10.1016/S0006-3495(02)75479-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayton GS, MacWhirter JL, McMurtry P, Voth GA. Coupling field theory with continuum mechanics: A simulation of domain formation in giant unilamellar vesicles. Biophys J. 2005;88(50):38553869. doi: 10.1529/biophysj.105.059436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayton GS, MacWhirter JL, Voth GA. A second generation mesoscopic lipid bilayer model: connections to field-theory descriptions of membranes and nonlocal hydrodynamics. J Chem Phys. 2006;124(50):64906. doi: 10.1063/1.2165194. [DOI] [PubMed] [Google Scholar]
- 7.Ayton GS, Noid WG, Voth GA. Multiscale modeling of biomolecular systems: in serial and in parallel. Curr Op Struct Biol. 2007;17(50):192–198. doi: 10.1016/j.sbi.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Ayuyan A, Cohen F. Lipid peroxides promote large rafts: Effects of excitation of probes in fluorescence microscopy and electrochemical reactions during vesicle formation. Biophys J. 2007;91(9):2127–2183. doi: 10.1529/biophysj.106.087387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balazs A, Ginsburg VV, Qiu F, Peng G, Jasnow D. Multi-scale model for binary mixtures containing nanoscopic particles. J Phys Chem B. 2000;104:3411–3422. [Google Scholar]
- 10.Bhide S, Zhang Z, Berkowitz M. Molecular dynamics simulations of spos and sphingomyelin bilayers containing cholesterol. Biophys J. 2007;92:1284–1295. doi: 10.1529/biophysj.106.096214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu S, Pandti S, Scott HL, Jakobsson E. An improved united atom force field for simulation of mixed lipid bilayers. J Phys Chem B. 2008 doi: 10.1021/jp807056c. submitted. [DOI] [PubMed] [Google Scholar]
- 12.Chiu SW, Jakobsson E, Mashl RJ, Scott HL. Cholesterol–induced modifications in lipid bilayers: A simulation study. Biophys J. 2002;83(4):1842–1853. doi: 10.1016/S0006-3495(02)73949-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiu SW, Vasudevan S, Jakobsson E, Mashl RJ, Scott HL. Structure of sphingomyelin bilayers: A simulation study. Biophys J. 2003;85(6):3624–3635. doi: 10.1016/S0006-3495(03)74780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edholm O, Nagle JF. Areas of molecules in membranes consisting of mixtures. Biophys J. 2005;89(3):1827–1832. doi: 10.1529/biophysj.105.064329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edidin M. The state of lipid rafts: From model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 16.Elliott R, Katsov K, Szleifer I, Schick M. Phase separation of saturated and mono-unsaturated lipids as determined from a microscopic model. J Chem Phys. 2005;122:044904. doi: 10.1063/1.1836753. [DOI] [PubMed] [Google Scholar]
- 17.Elliott R, Szleifer I, Schick M. Phase diagram of a ternary mixture of cholesterol and saturated and unsaturated lipids calculated from a microscopic model. Phys Rev Lett. 2006;96:098101. doi: 10.1103/PhysRevLett.96.098101. [DOI] [PubMed] [Google Scholar]
- 18.Goetz R, Gompper G, Lipowski R. Mobility and elasticity or self-assembled membranes. Phys Rev Lett. 1999;82:221–224. [Google Scholar]
- 19.Goetz R, Lipowski R. Computer simulations of bilayer membranes: self-assembpy and interfacial tension. J Chem Phys. 1998;108:7397–7409. [Google Scholar]
- 20.Gompper G, Schick M. Soft Matter Volume 1: Polymer Melts and Mixtures. Wiley-VCH; 2006. [Google Scholar]
- 21.Greenwood AI, Tristram-Nagle S, Nagle JF. Partial molecular volumes of lipids and cholesterol. Chemistry and Physics of Lipids. 2006;143:1–10. doi: 10.1016/j.chemphyslip.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groot RD, Rabone KL. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants. Biophys J. 2001;81:725–736. doi: 10.1016/S0006-3495(01)75737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofsäb C, Lindahl E, Edholm O. Molecular dynamics simulations of phospholipid bilayers with cholesterol. Biophys J. 2003;84(4):2192–2206. doi: 10.1016/S0006-3495(03)75025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Swanson JE, Dibble ARG, Hinderliter AK, Feigenson GW. Nonideal mixing of Phosphatidylserine and Phosphatidylcholine in fluid lamellar phase. Biophys J. 1993a;64:413–425. doi: 10.1016/S0006-3495(93)81382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang TH, Less CWB, Das Gupta SK, Blume A, Griffin RG. A 13C and 2H nuclear magnetic resonance study of phosphatidylcholine/cholesterol interactions: Characterization of liquid–gel phases. Biochem. 1993b;32(48):13277–13287. doi: 10.1021/bi00211a041. [DOI] [PubMed] [Google Scholar]
- 26.Izvekov S, Voth GA. A multiscale coarse-graining method for biomolecular systems. J Phys Chem B. 2006a;109(3):2469–2473. doi: 10.1021/jp044629q. [DOI] [PubMed] [Google Scholar]
- 27.Izvekov S, Voth GA. Multiscale coarse-graining of mixed phospholipid/cholesterol bilayers. J Chem Theory Comput. 2006b;2(3):637–648. doi: 10.1021/ct050300c. [DOI] [PubMed] [Google Scholar]
- 28.Khelashvili GA, Pandit SA, Scott HL. Self-consistent mean field model based on molecular dynamics: Application to lipid-cholesterol bilayers. J Chem Phys. 2005;123:034910. doi: 10.1063/1.1943412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klauda JB, Kucerka N, Brooks BR, Pastor RW, Nagle JF. Simulation-based methods for interpreting x-ray data from lipid bilayers. Biophys J. 2006;90(8):2796–2807. doi: 10.1529/biophysj.105.075697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kranenburg M, Ventrouli M, Smit B. Molecular simulations of mesoscopic bilayer phases. Phys Rev E. 2003a;67:60901-1–3. doi: 10.1103/PhysRevE.67.060901. [DOI] [PubMed] [Google Scholar]
- 31.Kranenburg M, Ventrouli M, Smit B. Phase behavior and induced interdigitation in bilayers studied with dissipative particle dynamics. J Phys Chem B. 2003b;107:11491–11501. [Google Scholar]
- 32.Kucerka N, Tristram-Nagle S, Nagle JF. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J Membr Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 33.Landau LD, Lifshitz EM. Statistical Physics 3rd Edition Part 1; Course of Theoretical Physics. 2 Vol. 5. Pergamon Press; 1980. [Google Scholar]
- 34.Lin C-LL, Brown FLH. Brownian dynamics in fourier space: Menbrane simulations over long length and time scales. Phys Rev Lett. 2005;93(3):25600-1–25600-4. doi: 10.1103/PhysRevLett.93.256001. [DOI] [PubMed] [Google Scholar]
- 35.Lin CLL, Brown FLH. Simulating membrane dynamics in nonhomogeneous hydrodynamic environments. J Chem Theory Comput. 2006;2(3):472–483. doi: 10.1021/ct050293s. [DOI] [PubMed] [Google Scholar]
- 36.Lindahl E, Hess B, van der Spoel D. Gromacs 3.0: A package for molecular simulation and trajectory analysis. J Mol Mod. 2001;7:306–317. [Google Scholar]
- 37.Lubensky T, Chaikin P. Principles of Condensed Matter Physics. Cambridge University Press; 1995. [Google Scholar]
- 38.Lyubartsev AP, Laaksonen A. Calculation of effective interaction potentials from radial distribution functions: A reverse monte carlo approach. Phys Rev E. 1995;52(4):3730–3737. doi: 10.1103/physreve.52.3730. [DOI] [PubMed] [Google Scholar]
- 39.MacWhirter JL, Ayton GS, Voth GA. Coupling field theory with mesoscopic dynamical simulations of multicomponent lipid bilayers. Biophys J. 2004;87(50):3242–3263. doi: 10.1529/biophysj.104.045716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maier W, Saupe A. Z Naturforsch. 1959;A14:882–889. [Google Scholar]
- 41.Marrink SJ, de Vries AH, Mark AE. Coarse grained model for semiquantitative lipid simulations. J Phys Chem B. 2004;108:750–760. [Google Scholar]
- 42.Marrink SJ, Mark AE. Molecular view of hexabonal phase formation in phospholipid bilayers. Biophys J. 2005;12(87):3894–3900. doi: 10.1529/biophysj.104.048710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marrink SJ, Risselada J, Mark AE. Simulation of gel phase formation and melting in lipid bilayers using a coarse grained model. Chem Phys Lipids. 2005;135:223–244. doi: 10.1016/j.chemphyslip.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Marčelja S. Chain ordering in liquid crystals ii. the structure of bilayer membranes. Biochim Biophys Act. 1974;367:156–176. doi: 10.1016/0005-2736(74)90040-6. [DOI] [PubMed] [Google Scholar]
- 45.McConnell H, Radhakrishnan A. Theory of the deuterium NMR of sterol– phospholipid membranes. Proc Natl Acad Sci USA. 2006;103(5):1184–1189. doi: 10.1073/pnas.0510514103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miao L, Nielsen M, Thewalt J, Ipsen JH, Bloom M, Zuckermann MJ, Mouritsen OG. From lanosterol to cholesterol: Structural evolution and differential effects on lipid bilayers. Biophys J. 2002;82(3):1429–1444. doi: 10.1016/S0006-3495(02)75497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murtola T, Falck E, Kartunen M, Vattulainen I. Coarse-grained model for phospholipid/cholesterol bilayer employing inverse monte carlo with thermodynamic constraints. J Chem Phys. 2007a;126(18):075101-1–075101-14. doi: 10.1063/1.2646614. [DOI] [PubMed] [Google Scholar]
- 48.Murtola T, Falck E, Patra M, Kartunen M, Vattulainen I. Coarse-grained model for phospholipid/cholesterol bilayer. J Chem Phys. 2004;121(18):9157–9165. doi: 10.1063/1.1803537. [DOI] [PubMed] [Google Scholar]
- 49.Murtola T, Kupiainen M, Falck E, Vattulainen I. Conformational analysis of lipid molecules by self-organizing maps. J Chem Phys. 2007b;126(18):054707-1–054707-17. doi: 10.1063/1.2429066. [DOI] [PubMed] [Google Scholar]
- 50.Nielsen M, Miao L, Ipsen JH, Zuckerman M, Mouritsen O. Off-lattice models for the phase equilibria of lipid-cholesterol layers. Phys Rev E. 1999;59(5):5790–5803. doi: 10.1103/physreve.59.5790. [DOI] [PubMed] [Google Scholar]
- 51.Nielsen M, Miao L, Ipsen JH, Mouritsen O, Zuckerman M. Random-lattice models and simulation algorithms for phase equilibria in two-dimensional condensed systems of particles with coupled internal and translational degrees of freedom. Phys Rev E. 1996;54(6):6889–6905. doi: 10.1103/physreve.54.6889. [DOI] [PubMed] [Google Scholar]
- 52.Niemală PT, Hivŏnen MY, Vattulainen I. Structure and dynamics od sphingomyelin bilayer: Insight gained through systematic comparison to phosphatidylcholine. Biophys J. 2004;87(11):2976–2989. doi: 10.1529/biophysj.104.048702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niemelă PS, Ollila S, Hyvŏnen M, Kartunen M, Vattulainen I. Assessing the nature of lipid raft membranes. PLoS Computational Biology. 2007;3(2):0304–0312. doi: 10.1371/journal.pcbi.0030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orsi M, Haubertin D, Sanderson W, Essex J. A quantitative coarse-grain model for lipid bilayers. J Phys Chem B. 2008;112(112):472–483. doi: 10.1021/jp076139e. [DOI] [PubMed] [Google Scholar]
- 55.Pandit S, Chiu S, Jakobsson E, Grama A, Scott H. Cholesterol packing around lipids with saturated and unsaturated chains: A simulation study. Langmuir. 2008;24:6858–6865. doi: 10.1021/la8004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pandit S, Chiu SW, Jakobsson E, Grama A, Scott HL. Cholesterol surrogates: a comparison of cholesterol and 16:0 ceramide in popc bilayers. Biophys J. 2007a;92:920–927. doi: 10.1529/biophysj.106.095034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pandit S, Khelashvili G, Jakobsson E, Grama A, Scott HL. Lateral organization in lipid-cholesterol mixed bilayers. Biophys J. 2007b;92:440–447. doi: 10.1529/biophysj.106.093864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pandit SA, Bostick DL, Berkowitz ML. Complexation of phosphatidylcholine lipids with cholesterol. Biophys J. 2004a;86(3):1345–1356. doi: 10.1016/S0006-3495(04)74206-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pandit SA, Jakobsson E, Scott HL. Simulation of the early stages of nano-domain formation in mixed bilayers of sphingomyelin, cholesterol, and dioleylphosphatidylcholine. Biophys J. 2004b;87(5):3312–3322. doi: 10.1529/biophysj.104.046078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pandit SA, Vasudevan S, Chiu SW, Mashl RJ, Jakobsson E, Scott HL. Sphingomyelin–cholestrol domains in phospholipid membranes: Atomistic simulation. Biophys J. 2004c;87(2):1092–1100. doi: 10.1529/biophysj.104.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pasenkiewicz-Gierula M, Rŏg T, Kitamura K, Kusumi A. Cholesterol effects on the phosphatidylcholine bilayer polar region: A molecular simulation study. Biophys J. 2000;78(3):1376–1389. doi: 10.1016/S0006-3495(00)76691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng G, Qiu F, Ginzburg VV, Jasnow D, Bakazs A. Forming supramolecular networks from nonoscale rods in binary, phase-separating mixtures. Science. 2000;288:1802–1804. doi: 10.1126/science.288.5472.1802. [DOI] [PubMed] [Google Scholar]
- 63.Rŏg T, Pasenkiewicz-Gierula M. Cholesterol effects on the phosphatidylcholine bilayer nonpolar region: A molecular simulation study. Biophys J. 2001;81(4):2190–2202. doi: 10.1016/S0006-3495(01)75867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rŏg T, Pasenkiewicz-Gierula M. Cholesterol - sphingomyelin interactions: A molecular dynamics simulation study. Biophys J. 2006;91(11):3756–3767. doi: 10.1529/biophysj.106.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scott HL. Modeling the lipid component of membranes. Curr Op Struct Biol. 2002;12(4):495–502. doi: 10.1016/s0959-440x(02)00353-6. [DOI] [PubMed] [Google Scholar]
- 66.Scoville-Simonds M, Schick M. Theory of the effect of unsaturation on the main chain transition. Phys Rev E. 2003;67:098101. doi: 10.1103/PhysRevE.67.011911. [DOI] [PubMed] [Google Scholar]
- 67.Shelley J, Shelley MY, Reeder R, Ipsen JH, Bandyopadhyay S, Klein MW. A coarse grain model for phospholipid simulations. J Phys Chem B. 2001;105(3):4464–4470. [Google Scholar]
- 68.Shi Q, Voth GA. Multi-scale modeling of phase separation in mixed lipid bilayers. Biophys J. 2005;89(127):2385–2394. doi: 10.1529/biophysj.105.063784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smondyrev AM, Berkowitz ML. Structure of dipalmitoylphosphatidylcholine/cholesterol bilayer at low and high cholesterol concentrations: Molecular dynamics simulation. Biophys J. 1999;77:2075–2089. doi: 10.1016/S0006-3495(99)77049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stevens M. Complementary matching of domain formation within lipid bilayers. J A C S. 2005;12(127):15330–15331. doi: 10.1021/ja043611q. [DOI] [PubMed] [Google Scholar]
- 71.Tu K, Klein ML, Tobias DJ. Constant–pressure molecular dynamics investigation of cholesterol effects in a dipalmitoylphosphatidylcholine bilayer. Biophys J. 1998;75:2147–2156. doi: 10.1016/S0006-3495(98)77657-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vist MR, Davis JH. Phase equilibria of cholesterol/dipalmitoylphosphatidylcholine mixtures: 2H nuclear magnetic resonance and differential scanning calorimetry. Biochem. 1990;29(2):451–464. doi: 10.1021/bi00454a021. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Z, Bhide S, Berkowitz M. Molecular dynamics simulations of lipid bilayers containing mixtures of sphingomyelin with cholesterol and phosphatidylcholine with cholesterol. J Phys Chem B. 2007;111:12888–12897. doi: 10.1021/jp074037i. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Z, Lu L, Berkowitz M. Energetics of cholesterol transfer between lipid bilayers. J Phys Chem B. 2008;112:3807–3811. doi: 10.1021/jp077735b. [DOI] [PubMed] [Google Scholar]