

Abstract
We previously have described a mouse model for polycystic kidney disease (PKD) caused by either of two mutations, kat or kat2J, that map to the same locus on chromosome 8. The homozygous mutant animals have a latent onset, slowly progressing form of PKD with renal pathology similar to the human autosomal-dominant PKD. In addition, the mutant animals show pleiotropic effects that include facial dysmorphism, dwarfing, male sterility, anemia, and cystic choroid plexus. We previously fine-mapped the kat2J mutation to a genetic distance of 0.28 ± 0.12 centimorgan between D8Mit128 and D8Mit129. To identify the underlying molecular defect in this locus, we constructed an integrated genetic and physical map of the critical region surrounding the kat2J mutation. Cloning and expression analysis of the transcribed sequences from this region identified Nek1, a NIMA (never in mitosis A)-related kinase as a candidate gene. Further analysis of the Nek1 gene from both kat/kat and kat2J/kat2J mutant animals identified a partial internal deletion and a single-base insertion as the molecular basis for these mutations. The complex pleiotropic phenotypes seen in the homozygous mutant animals suggest that the NEK1 protein participates in different signaling pathways to regulate diverse cellular processes. Our findings identify a previously unsuspected role for Nek1 in the kidney and open a new avenue for studying cystogenesis and identifying possible modes of therapy.
Polycystic kidney disease (PKD) is the most common inherited nephropathy in humans (1). PKD is characterized by massive renal enlargement associated with the growth of fluid-filled intrarenal cysts. Genetic studies of inherited PKD in humans and animal models clearly have shown that mutations at multiple loci with different modes of inheritance result in various forms of PKD (2–7). In recent years, major advances have been made toward understanding the genetics of PKD. In humans, the PKD1 and PKD2 loci that cause autosomal-dominant PKD (ADPKD) and the murine orck locus that causes disease pathology similar to the human autosomal-recessive PKD (ARPKD) have been cloned. The PKD1 gene product, polycystin-1, encodes an integral membrane glycoprotein with significant homology to membrane proteins involved in cell—cell and/or cell—matrix interactions (8). The PKD2 gene product, polycystin-2, is also an integral membrane protein with homology to the voltage-activated Ca2+ channel α1E and the Na+ voltage-dependent channels (9). Analysis of the primary sequence of the orck gene product reveals a protein containing 10 copies of an internally repeated, 34-aa (tetratricopeptide) motif that is found in a number of cell-cycle control proteins (10). Although the primary structure has predicted certain roles for these three proteins, their function still remains largely unknown. However, because patients with mutations in either PKD1 or PKD2 have a similar clinical phenotype and recent in vitro studies have shown that polycystin-1 and polycystin-2 interact via their C-terminal cytoplasmic domains, it has been hypothesized that polycystin-1 and polycystin-2 may function in a common signaling pathway (11–13). The available literature also suggests that there is a wide variation in the clinical manifestations seen among patients with ADPKD, suggesting that other protein factors alter the disease severity caused by PKD1 and PKD2 (14, 15). These observations support the hypothesis that polycystin-1, polycystin-2, and those factors that modulate the disease severity among ADPKD patients belong to a common pathway. The identification of these interacting factors is difficult in humans. However, the various animal models of PKD (7, 16) present us with an opportunity to identify these interacting factors not only in the polycystin pathway but also members in other parallel pathways that are important in stabilizing this critical organ.
We recently characterized two independent mutant alleles, kat and kat2J, that cause pleiotropic effects that include facial dysmorphism, dwarfing, male sterility, anemia, cystic choroid plexus, and a progressive polycystic kidney disease (16, 17). The latent onset of cystogenesis and similarity of the renal pathology to human ADPKD make this an attractive animal model for the study of renal cystogenesis. We mapped the mutations to mouse chromosome 8. To generate a physical map and to eventually clone the gene mutated, the kat2J allele was localized to a genetic distance of 0.28 ± 0.12 centimorgan (cM) between D8Mit128 and D8Mit129 (16). During this fine mapping of the kat2J mutation, it was noted that the severity of PKD in the mutant (C57BL/6J-kat2J/+ × CAST/Ei)F2 generation was more variable than that in the parental C57BL/6J strain. This suggested that genetic background or modifier genes alter the clinical manifestations and progression of PKD, thereby further strengthening the kat series of mutations as a model to study PKD. Genome scans using molecular markers revealed modifier loci that affect the severity of PKD, providing an opportunity to identify other interacting proteins (18). Here, by positional cloning, we report the identification of the NIMA (never in mitosis A)-related kinase, Nek1 (19), as the gene that is altered by the kat and kat2J spontaneous mutations.
Materials and Methods
Mice.
The kat and kat2J spontaneous mutations arose at The Jackson Laboratory on RBF/Dn and C57BL/6J inbred strains, respectively. The kat mutation has been transferred to the C57BL/6J background and, like the kat2J mutation, is maintained as a pedigree colony. All parental, control, and mutant stocks are maintained at The Jackson Laboratory, where animal procedures are approved by the Animal Care and Use Committee.
PCR and DNA Sequencing.
Mouse genomic DNA was prepared by using standard techniques (20). DNA was PCR-amplified under the following conditions (3 min at 94°C, 2 min at 55°C, 2 min at 72°C, for a total of 25–35 cycles, followed by a 7-min final extension at 72°C). Primers were: 16335′ (5′-ATAGAACATATTTCTAACCAAAG-3′), 16333′ (5′-TTGTTACCTATTAAGAACTCGAG-3′), Nek125′ (5′-CTCTGAAGCATGTACATGATAG-3′) and Nek123′ (5′-CTTGGTTGGCTCTCTCTACAGC). The PCR products were analyzed on either agarose or polyacrylamide gels. DNA sequencing was performed either manually with the Amplicycle sequencing kit (Perkin–Elmer) or with the ABI model 373A automated DNA sequencer (Applied Biosystems) with M13, SP6, T7, or specific primers.
Cloning of Yeast Artificial Chromosome (YAC) Ends.
Total DNA was prepared from the positive YAC clones by using the standard protocol (21). YAC ends were cloned by inverse PCR (22, 23), with the following modification of primers for the rescue of the left end: VLEFT2, 5′-AGAATTGATCCACAGGACGC-3′, replaced InvL-2 in the first PCR step, and M13REVL2 was replaced by the 18-mer primer LEFT SEQ, 5′-TGGTCGCCATGATCGCGT-3′, in the second step. LEFT SEQ was used to sequence the left-end clone.
Cloning of P1 and Bacterial Artificial Chromosome (BAC) Ends.
P1 and BAC DNAs were purified from Escherichia coli by alkaline lysis (24). The P1 ends were sequenced directly with 21-mer primers complementary to the SP6 and T7 promoter regions of the pAD10SacB11 vector (25). The BAC ends were sequenced with the M13 primers.
DNA Typing.
Single-strand conformation polymorphisms were run on nondenaturing MDE gels (AT Biochem, Malvern, PA) at 6 W for 8 h at 4°C. Simple sequence length polymorphisms were run on nondenaturing 6% polyacrylamide gels at 40 W for 2 h at room temperature.
Direct cDNA Selection.
Hybrid selection protocol (22, 26) was used to obtain cDNA clones for the transcribed sequences contained in the appropriate BAC clones. Poly(A)+ mRNA from adult mouse kidney was used as the starting material for the preparation of cDNA selection products. Total RNA was prepared by using Trizol (GIBCO/BRL), and poly(A)+ mRNA was purified by using Streptavidin Magnesphere Paramagnetic Particles (Promega). cDNA selection products had an average length of 500–600 bp.
Northern Blots and Reverse Transcription–PCR (RT-PCR).
Total RNA and poly(A)+ RNA were prepared from adult mouse testis and kidney, and either 20 μg of total or 1.5 μg of poly(A)+ RNA was run on a 1% agarose/formaldehyde gel. After blotting onto Nytran Plus membrane (Schleicher & Schuell), the blot was probed in Church buffer (7% SDS/0.5 M sodium phosphate, pH 7.5/1 mM EDTA) at 65°C. The final wash was 0.1× SSC/0.1% SDS at 65°C. A 1.8-kb mouse actin probe (Ambion) was labeled (Stratagene) and used as a control for equal RNA loading. For RT-PCR, poly(A)+ RNA (1 μg) was reverse-transcribed with avian myeloblastosis virus reverse transcriptase (Promega). PCRs were run as described above for 25 cycles. The RT products were resolved on agarose gel, purified (Qiagen), and sequenced.
Southern Blotting.
The mouse genomic DNAs (5 μg) were digested with various restriction enzymes and resolved on a 0.8% agarose gel. After blotting onto Nytran Plus membrane (Schleicher & Schuell), the blot was processed as described above.
Genome Walk.
The mouse Genome Walker kit from CLONTECH was used for the genome walk. The walk was performed according to the manufacturer's protocol. The primer used for the walk was NekGE1 (5′-ATGGAGTGCCTATGCAAGTTCGAGCCAGCTCTAC). All the five libraries in the kit were amplified by using NekGE1 and AP1 primers. The amplified fragment was resolved on a 1.2% agarose gel, purified (Qiagen), and sequenced.
Results
To identify the gene affected by these mutations we generated a physical map of the region between D8Mit128 and D8Mit129. We screened three YAC libraries (27–29) for clones encompassing the closest flanking markers, D8Mit129 and D8Mit128. The ends of these YACs were rescued by inverse PCR and sequenced. Because the ends of the YACs obtained by screening with D8Mit129 were chimeric, repetitive, or oriented in the opposite direction of the intended walk, we walked only from the distal end (D8Mit128). In addition to the YAC clones, we also identified BAC and P1 (25) clones from this region. The ends of every BAC and P1 clone obtained were sequenced, and PCR primers were designed to generate probes to integrate the physical and the genetic map and also to screen the libraries for the next walk. The genomic DNAs from the animals that had recombination events in the interval between D8Mit128 and D8Mit129 were analyzed by single-stranded conformation and restriction fragment length polymorphism. The contig was completed when the walk from D8Mit128 crossed the proximal recombination event (mouse 161). The detailed contig of the region encompassing the kat2J mutation is depicted in Fig. 1.
Figure 1.
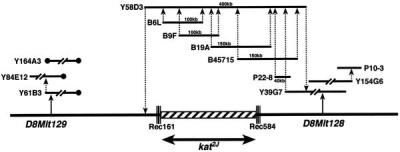
Contig of YACs (Y), BACs (B), and P1s (P) across the kat2J locus on chromosome 8. The triple lines across the chromosome represent the proximal and distal recombination (Rec) events in the animals 584 (mouse with a proximal recombination event) and 161 (mouse with a distal recombination event). The hatched box represents the nonrecombinant region that must contain the kat2J mutation. All the libraries were screened by PCR. The solid circles represent the chimeric ends of the YACs.
A hybrid selection protocol (22, 26) was used to obtain cDNA clones representing the transcribed sequences contained in the contig using BAC clones B6L, B9F, B19A, and B45715 as templates. Because colony hybridization of the hybrid-selected clones with labeled Cot1 DNA showed that only a few of the clones contained repetitive elements, we randomly selected 50 clones for each of the templates used in the hybrid-selection protocol. DNA from each of these clones was sequenced, and blast searched against the various databases. Based on the results of the blast search, 40 cDNA clones were selected for expression analyses. Northern blots of total RNA from the kidneys of 1-month-old mutants (kat/kat and kat2J/kat2J) and age-matched (+/+) controls were probed with the 40 hybrid-selected cDNA clones. Of the 40 clones, only 1 of the cDNA clones, clone 26A, representing Nek1 (19), detected an altered mRNA expression in both mutants. This cDNA clone hybridized to the BAC (B19A), which was used as a template in the cDNA hybrid-selection protocol, showing that the Nek1 cDNA maps to the critical region that encompasses the kat2J mutation. In addition, recently the Nek1 gene was mapped to the same region of mouse chromosome 8 (30), to which we have mapped the kat2J mutation (16). Because the Nek1 gene is known to be expressed in testis and kidney, a Northern blot containing poly(A)+ RNA from these tissues was hybridized with the same Nek1 cDNA clone. Results of this analysis showed a reduction in size of the Nek1 transcripts in both testis and kidney of the kat/kat mutant animal, indicating a possible deletion. The level of expression of the 6.5- and 4.4-kb transcripts in both tissues from kat2J/kat2J was reduced slightly (Fig. 2). These data suggested that the kat and kat2J mutations disrupt the Nek1 gene.
Figure 2.

Northern analyses of Nek1 expression in control and mutant animals. Lanes 1–3 contain poly(A)+ RNA from the testis, and lanes 4–6 contain poly(A)+ RNA from the kidneys of 3-month-old animals. A was probed with a hybrid-selected Nek1 cDNA, clone26A, that represents 1838–2325 nt of the Nek1 transcript (19). B represents the same blot stripped and probed with a 1.8-kb human β-actin cDNA fragment, a sample loading control. Size markers used to estimate kilobases were 28S and 18S RNA.
To determine whether the putative deletion in the kat/kat mutant animals was due either to a splicing defect that resulted in exon(s) skipping or to a deletion in the genomic DNA, we performed Southern blot analyses. A Southern blot containing equal amounts of EcoRI-digested genomic DNA from +/+, kat/+, and kat/kat was hybridized with a cDNA probe representing +918–2040 nt (Fig. 3B) of the Nek1 cDNA. This probe did not detect any fragments in the kat/kat genomic DNA. This result is consistent with a deletion in Nek1 genomic DNA as the cause of the kat mutation. The hybridization of the same blot to the labeled cDNA probes representing +575–1011 nt and +2877–3738 nt of the Nek1 cDNA detected the same genomic fragments in the +/+, kat/+, and kat/kat genomic DNAs (Fig. 3 A and C). This showed that the kat mutation causes an internal deletion of the Nek1 structural gene. To determine the extent of deletion caused by the kat mutation, we analyzed Nek1 transcripts from +/+ and kat/kat mutants by RT-PCR. Primers were designed along the length of the Nek1 cDNA to amplify overlapping fragments, and the RT-PCR products obtained from +/+ and kat/kat mice were sequenced. The sequence analysis of the Nek1 RT-PCR product from the kat/kat mutant animals showed an internal deletion that included residues 791 to 2105 of the Nek1 transcript, confirming that the kat mutation causes a deletion within the Nek1 gene.
Figure 3.

Identification of the kat mutation. Southern analyses of a blot containing EcoRI-restricted genomic DNA from +/+, +/kat, and kat/kat. The blot was probed with cDNA probes that represented 575–1011 nt (A), 918–2040 nt (B), and 2877–3738 nt (C) of the Nek1 transcript.
To determine whether the kat2J mutation also causes a sequence alteration in the Nek1 transcript, we performed RT-PCR on the mRNA obtained from the kat2J/kat2J mutant animals. The sequence analyses of the RT-PCR product from the kat2J/kat2J mutant showed an insertion of a guanosine residue at position +996 nt (Fig. 4A). This insertion causes a frame shift that results in a premature stop codon.
Figure 4.
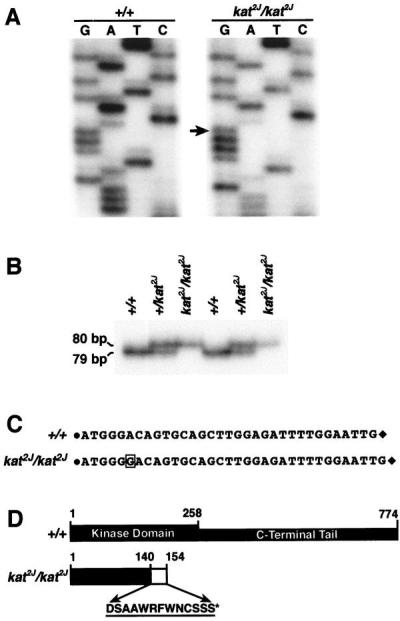
Identification of the kat2J mutation. (A) Sequencing of the RT-PCR product amplified by the primer pair Nek125′ and Nek123′ from +/+ and the KAT2J mutant. The arrow represents the guanosine residue inserted because of the kat2J mutation. The insertion occurs at position +996 nt of the Nek1 transcript. (B) Assay to identify the kat2J mutation in the genomic DNA. Genomic DNAs from known +/+, +/kat2J, and kat2J/kat2J were PCR-amplified with the 16335′ and 16333′ primer pair. The amplified products were resolved on a 6% polyacrylamide gel. The experiment was done on DNAs from two sets of animals. (C) Represents the sequence between the 16335′ and 16333′ primer pair of the genomic PCR fragments obtained from +/+ and kat2J/kat2J animals. The solid circle represents the 16335′ primer, and the solid diamond represents the 16333′ primer. The sequences of the primers are provided in Materials and Methods. The boxed guanosine residue is the insertion resulting from the kat2J mutation. (D) Structural comparison of the wild-type NEK1 protein and the truncated NEK1 protein from the kat2J/kat2J mutant. The amino acids incorporated because of the frame shift are represented by a hatched box and by their single-letter code. The asterisk indicates the position of the premature stop codon.
The insertion was confirmed by sequencing the kat2J mutation in genomic DNA. Initial attempts to PCR-amplify the wild-type genomic DNA by using primers based on the Nek1 cDNA sequence encompassing the insertion site failed, indicating the presence of intron(s). Because the intron–exon boundaries of the Nek1 gene are not known, we analyzed the Nek1 cDNA sequence for all the possible exon–exon junctions by using the computer program rnaspl (ref. 31; http://dot.imgen.bcm.tmc.edu:9331/seq-search/gene-search.html). The analyses indicated the presence of a number of putative splice sites 5′ to the mutation. We performed a genomic walk toward the intron that was 5′ to the mutation by using a 30-nt primer (NEKGE1). The fragment cloned by this walk was sequenced. The alignment of the genomic and the cDNA sequence of Nek1 allowed us to determine the sequence of the intron 5′ to the mutation. Based on this sequence, a primer pair (16335′ and 16333′) flanking the kat2J insertion was designed and used to amplify genomic fragments from +/+, +/kat2J, and kat2J/kat2J animals. Fig. 4B shows that the fragment generated by the mutant allele is longer than the one generated by the wild-type allele, thereby establishing an assay to distinguish between the kat2J and the wild-type allele. To confirm the identity of the PCR products generated and the nature of the kat2J mutation, we sequenced the fragments generated from +/+ and the kat2J/kat2J animals. Fig. 4C shows the comparison of the sequences, confirming that the kat2J mutation results from an insertion of a guanosine residue in the Nek1 gene. The putative truncated protein would be 154 aa in length, inclusive of the 13 different amino acids incorporated because of the frame shift, and lack the entire C-terminal tail (Fig. 4D). The results presented in this paper show that the two mutations, kat and kat2J, cause an alteration in the Nek1 gene. Because the kat mutation deletes most of the NEK1 kinase domain and both kat and kat2J lead to the same clinical manifestations, we predict that both the mutations result in a null phenotype.
Discussion
Nek1 previously has been cloned and characterized from a mouse erythroleukemia cDNA expression library by using antibodies directed toward phosphotyrosine (19). The Nek1 gene encodes a 774-aa dual specific protein kinase that contains an N-terminal domain (1–258 aa) with homology to the catalytic domain of NIMA, a protein kinase that controls the initiation of mitosis in Aspergillus nidulans. In situ RNA analysis of Nek1 expression in mouse gonads revealed a high level of expression in both male and female germ cells, with a distribution consistent with a role in meiosis. Based on these results, it was suggested that NEK1 is a mammalian relative of the fungal NIMA cell cycle regulator (19, 32). The previous results showing Nek1 expression in the testis combined with the male sterility we see in the kat mutants are in support of a role for NEK1 in spermatogenesis. However, our observation that the homozygous mutant female animals breed suggests that the NEK1 protein may play a different role in male and female germ cells. In situ RNA hybridization analysis has shown high levels of Nek1 expression in distinct regions of the nervous system (33). The facial dysmorphism observed in the kat mutants may be attributed, in part, to the loss of Nek1 function in the cranial ganglia (ninth/tenth inferior complex). We previously have mapped modifier loci that exclusively alter the anemia in the F2 mutants (18), suggesting that in addition to the progressive renal disease being responsible for the anemia, the NEK1 protein may play a direct role in erythropoiesis. Based on the complex phenotype we have described in the kat mutants we propose that NEK1 is a multifunctional protein that participates in multiple pathways regulating different tissue-specific processes. This functional diversity of the NEK1 protein may be due to its involvement in diverse protein–protein interactions and to its dual specific kinase activity.
Genetic and biochemical analyses have implicated three fundamental mechanisms in renal cystogenesis: abnormal cellular proliferation and differentiation, anomalies in the extracellular matrix and basement membrane, and, finally, a net trans-epithelial fluid secretion (directed toward the cyst lumen) because of faulty signaling (34, 35). We propose that in the kidney, the NEK1 protein belongs to a signaling pathway that eventually regulates transport functions. It will be important to determine whether the NEK1 protein participates in a pathway that either directly regulates the function of certain ion transporters such as the Na+-K+-2Cl− cotransporter (BSC2), whose activity is regulated by phosphorylation (36), or indirectly by promoting the cellular proliferation/differentiation of the developing tubular cells. Previous studies on the transport mechanisms operative in epithelial cells of the lung, intestine, and kidney have shown that the immature epithelial cells in these tissues secrete Cl− ion and fluid, whereas absorption prevails in terminally differentiated cells (37). Hence, if the NEK1 protein does participate in the pathway that is involved in cellular proliferation/differentiation, then the inactivation of this pathway in the kat mutants may impair terminal differentiation of the renal epithelial cells, which, as a result, show altered transport functions.
It has been speculated that polycystin-1 and polycystin-2 control protooncogenes and cellular programs directing cell cycle progression and cellular differentiation. Consistent with this, studies have shown that polycystin-1 and polycystin-2 may participate in multiple signaling pathways involving protein kinase C (PKC) α and ɛ that converge on the activation of AP-1, a transcription factor that regulates different cellular programs such as proliferation, differentiation, and apoptosis. However, the signaling pathways that lead to the activation of either PKC α or ɛ by the polycystins have not been identified (38, 39). It is enticing to speculate that the NEK1 protein is a member of the polycystin-1- and polycystin-2-induced signaling pathway that leads to the activation of the transcription factor AP-1 or some other factor that is triggered by the combined activation of PKC α and ɛ. Thus, identification of proteins that interact with the NEK1 protein will help us substantiate or refute this speculation and provide insights into the pathway to which NEK1 belongs.
Our previous mapping of modifier loci provides an added avenue for understanding how the loss of Nek1 function causes renal cystogenesis (18). These putative factors may modulate the outcome of the pathway to which the NEK1 protein belongs or they may be members of a salvage pathway that becomes operative as a compensatory mechanism in response to the loss of Nek1 function. For the characterization and identification of these modifier loci, we currently are generating congenic strains. This type of genetic study cannot be done in humans. It is, therefore, exciting to postulate that the future identification of the pathway to which NEK1 belongs and the factors that modulate the renal disease caused by the loss of Nek1 function may allow advances in the understanding and therapeutic intervention of human PKD.
Acknowledgments
This work is dedicated to the memory of Dr. Edward H. Birkenmeier, who passed away on July 27, 1996. We thank Brenda Sprague for technical assistance, Drs. Richard M. Baldarelli, Susan L. Ackerman, and Verity Letts for discussions, technical suggestions, and review of a preliminary version of this manuscript, and The Jackson Laboratory Mouse Mutant Resource for providing mice. This work was supported by National Institutes of Health Grant DK49634 (E.H.B/J.B.), the Center for Innovation in Biomedical Technology (E.H.B.), the Polycystic Kidney Research foundation (E.H.B.), and Grant CA34196 (Institutional Core Grant). The Jackson Laboratory is fully accredited by the American Association for the Accreditation of Laboratory Animal Care.
Abbreviations
- PKD
polycystic kidney disease
- ADPKD
autosomal-dominant polycystic kidney disease
- YAC
yeast artificial chromosome
- BAC
bacterial artificial chromosome
- RT-PCR
reverse transcription–PCR
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Grantham J J, Geiser J L, Evan A P. Kidney Int. 1987;31:1145–1152. doi: 10.1038/ki.1987.121. [DOI] [PubMed] [Google Scholar]
- 2.Reeders S T, Breuing M H, Davies K E, Nicholls D R, Jarman A J, Higgs D R, Pearson P L, Weatherall D J. Nature (London) 1985;317:542–544. doi: 10.1038/317542a0. [DOI] [PubMed] [Google Scholar]
- 3.Peters D J M, Spruit L, Saris J, Ravine D, Sandkuijl L A, Fossdal R, Boersma J, van Eijk R, Norby S, Constantinou-Deltas C D, et al. Nat Genet. 1993;5:359–362. doi: 10.1038/ng1293-359. [DOI] [PubMed] [Google Scholar]
- 4.Kimberling W J, Kumar S, Gabow P A, Kenyon J B, Connolly C J, Somlo S. Genomics. 1993;18:467–472. doi: 10.1016/s0888-7543(11)80001-7. [DOI] [PubMed] [Google Scholar]
- 5.Daoust M C, Reynolds D M, Bichet D G, Somlo S. Genomics. 1995;25:733–736. doi: 10.1016/0888-7543(95)80020-m. [DOI] [PubMed] [Google Scholar]
- 6.Zerres K, Mucher G, Bachner L, Deschennes G, Eggerman T, Kaariainen H, Knapp M, Lennert T, Misselwitz J, von Muhlendahl K E, et al. Nat Genet. 1994;7:429–432. doi: 10.1038/ng0794-429. [DOI] [PubMed] [Google Scholar]
- 7.Nazeen A. BioEssays. 1995;17:703–712. [Google Scholar]
- 8.Harris P C and the European Polycystic Kidney Disease Consortium. Cell. 1994;77:881–894. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 9.Mochizuki T, Wu G, Hayashi T, Xenophontos S L, Veldhuisen B, Saris J J, Reynolds D M, Cai Y, Gabow P A, Pierides A, et al. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 10.Moyer J H, Lee-Tischler M J, Kwon H-Y, Schrick J J, Avner E D, Sweeney W E, Godfrey V L, Cacheiro N L A, Wilkinson J E, Woychik R P. Science. 1994;264:1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 11.Ravine D, Walker R G, Gibson R N, Forrest S M, Richards R I, Friend K, Sheffield L J, Kincaid-Smith P, Danks D M. Lancet. 1992;340:1330–1333. doi: 10.1016/0140-6736(92)92503-8. [DOI] [PubMed] [Google Scholar]
- 12.Qian F, Germino F J, Cai Y, Zhang X, Somlo S, Germino G G. Nat Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 13.Tsiokas L, Kim E, Arnould T, Sukhatme V P, Walz G. Proc Natl Acad Sci USA. 1997;94:6965–6970. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milutinovic J, Rust P F, Fialkow P J, Agodoa L Y, Phillips L A, Rudd T G, Sutherland S. Am J Kidney Dis. 1992;19:465–472. doi: 10.1016/s0272-6386(12)80956-5. [DOI] [PubMed] [Google Scholar]
- 15.Peral B, Ong A C M, Millan J L S, Gamble V, Rees L, Harris P C. Hum Mol Genet. 1996;5:539–542. doi: 10.1093/hmg/5.4.539. [DOI] [PubMed] [Google Scholar]
- 16.Janaswami P M, Birkenmeier E H, Cook S A, Rowe L B, Bronson R T, Davisson M T. Genomics. 1997;40:101–107. doi: 10.1006/geno.1996.4567. [DOI] [PubMed] [Google Scholar]
- 17.Vogler C, Homan S, Pung A, Thorpe C, Barker J, Birkenmeier E H, Upadhya P. Am J Nephrol. 1999;10:2591–2598. doi: 10.1681/ASN.V10122534. [DOI] [PubMed] [Google Scholar]
- 18.Upadhya P, Churchill G, Birkenmeier E H, Barker J E, Frankel W N. Genomics. 1999;58:129–137. doi: 10.1006/geno.1999.5830. [DOI] [PubMed] [Google Scholar]
- 19.Letwin K, Mizzen L, Motro B, Ben-David Y, Bernstein A, Pawson T. EMBO J. 1992;10:3521–3531. doi: 10.1002/j.1460-2075.1992.tb05435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor B A, Rowe L, Grieco D A. Genomics. 1993;16:380–394. doi: 10.1006/geno.1993.1201. [DOI] [PubMed] [Google Scholar]
- 21.Treco D A. Preparation of Yeast DNA. New York: Wiley; 1991. [Google Scholar]
- 22.Segre J A, Nemhauser J L, Taylor B A, Nadeau J H, Landert E S. Genomics. 1995;28:549–559. doi: 10.1006/geno.1995.1187. [DOI] [PubMed] [Google Scholar]
- 23.Haldi M, Perrot V, Saumier M, Desai T, Cohen D, Cherif D, Ward D, Lander E S. Genomics. 1994;3:478–484. doi: 10.1006/geno.1994.1656. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 25.Pierce J C, Sternberg N, Sauer B. Mamm Genome. 1992;3:550–558. doi: 10.1007/BF00350620. [DOI] [PubMed] [Google Scholar]
- 26.Morgan J G, Dolganov G M, Robbins S E, Hinton L M, Lovett M. Nucleic Acids Res. 1992;20:5173–5179. doi: 10.1093/nar/20.19.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusumi K, Smith J S, Koos D S, Lander S. Mamm Genome. 1993;4:321–322. doi: 10.1007/BF00360591. [DOI] [PubMed] [Google Scholar]
- 28.Larin Z, Monaco D B, Lehrach H. Proc Natl Acad Sci USA. 1991;88:4123–4127. doi: 10.1073/pnas.88.10.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chartier F L, Keer J T, Sutcliffe M J, Henriques D A, Mileham P, Brown S D M. Nat Genet. 1992;1:132–136. doi: 10.1038/ng0592-132. [DOI] [PubMed] [Google Scholar]
- 30.Chen A, Yanai A, Arama E, Kilfin G, Motro B. Gene. 1999;234:127–137. doi: 10.1016/s0378-1119(99)00165-1. [DOI] [PubMed] [Google Scholar]
- 31.Solovyev V V, Salamov A A, Lawrence C B. Nucleic Acids Res. 1994;22:5156–5163. doi: 10.1093/nar/22.24.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fry A M, Nigg E A. Methods Enzymol. 1997;283:270–282. doi: 10.1016/s0076-6879(97)83022-4. [DOI] [PubMed] [Google Scholar]
- 33.Arama E, Yanai A, Kilfin G, Bernstein A, Motro B. Oncogene. 1998;10:1813–1823. doi: 10.1038/sj.onc.1201710. [DOI] [PubMed] [Google Scholar]
- 34.Devuyst O, Beauwens R. Adv Nephrol. 1998;28:439–479. [PubMed] [Google Scholar]
- 35.Murcia N S, Sweeney W E, Jr, Ellis D A. Kidney Int. 1999;55:1187–1197. doi: 10.1046/j.1523-1755.1999.00370.x. [DOI] [PubMed] [Google Scholar]
- 36.Tanimura A, Kurihara K, Reshkin S J, Turner R J. J Biol Chem. 1995;270:25252–25258. doi: 10.1074/jbc.270.42.25252. [DOI] [PubMed] [Google Scholar]
- 37.Sullivan L P, Wallace D P, Grantham J J. Physiol Rev. 1998;78:1165–1190. doi: 10.1152/physrev.1998.78.4.1165. [DOI] [PubMed] [Google Scholar]
- 38.Arnould T, Sellin L, Benzing T, Tsiokas L, Cohen T H, Kim E, Walz G. Mol Cell Biol. 1999;19:3423–3434. doi: 10.1128/mcb.19.5.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnould T, Kim E, Tsikas L, Jochimsen F, Gruning W, Chang J D, Walz G. J Biol Chem. 1998;273:6013–6018. doi: 10.1074/jbc.273.11.6013. [DOI] [PubMed] [Google Scholar]