

Abstract
Oncolytic virotherapy of tumors has shown promising results in both preclinical and clinical studies. Here, we investigated the therapeutic efficacy of a replication-competent vaccinia virus, GLV-1h68, against human pancreatic carcinomas in cell cultures and in nude mice. We found that GLV-1h68 was able to infect, replicate in, and lyse tumor cells in vitro. Virus-mediated marker gene expressions were readily detected. Moreover, s.c. PANC-1 pancreatic tumor xenografts were effectively treated by a single i.v. dose of GLV-1h68. Cancer killing was achieved with minimal toxicity. Viral titer analyses in homogenized organs and PANC-1 tumors showed that the mutant virus resides almost exclusively in the tumors and not in healthy organs. Except mild spleen enlargements, no histopathology changes were observed in any other organs 2 months after virus injection. Surprisingly, s.c. MIA PaCa-2 pancreatic tumors were treated with similar efficiency as PANC-1 tumors, although they differ significantly in sensitivity to viral lysis in cell cultures. When GLV-1h68 oncolytic viral therapy was used together with cisplatin or gemcitabine to treat PANC-1 tumors, the combination therapy resulted in enhanced and accelerated therapeutic results compared with the virus treatment alone. Profiling of proteins related to immune response revealed a significant proinflammatory immune response and marked activation of innate immunity in virus-colonized tumors. In conclusion, the GLV-1h68 strain showed outstanding therapeutic effects and a documented safety profile in mice, with great promise for future clinical development.
Introduction
Pancreatic carcinoma is one of the most deadly cancers. Of the 32,500 patients diagnosed each year with pancreatic cancer (1), nearly 32,000 will die of the disease. Surgery (2), conventional chemotherapy (3), and radiation therapy (4) have had only modest effects on length of survival and have had almost no effect on the mortality of this disease. Effective novel therapies are therefore sorely needed in treatment of pancreatic cancer.
Oncolytic viral therapy is a novel field in cancer treatment that attempts to exploit the lifecycle of viruses to kill cancer cells. In this discipline, a variety of natural and genetically engineered viruses that specifically infect, replicate within, and lyse tumor cells are currently being studied as treatments for cancer, particularly for tumors resistant to conventional therapies. The most commonly studied viruses include several from the adeno, herpes simplex, Newcastle disease, and myxoma viral groups (5). One of the very promising viruses under study is vaccinia virus. This virus is arguably the most successful biological agent in the history of medicine in that vaccination with vaccinia has eradicated smallpox (6). The anticancer activity of vaccinia virus has been noted previously (7-9). A patient with chronic lymphocytic leukemia went into remission after a severe reaction to smallpox vaccination (9). Engineered versions of the vaccinia virus have also been used as agents in immunostimulatory strategies against cancer and have shown promise in early-phase clinical trials (6, 10, 11).
In the current studies, we present preclinical data on a novel vaccinia virus, GLV-1h68 (12). We have described the construction of this recombinant vaccinia virus, which is a replication-competent virus targeted at tumor cells by mutations in F14.5L, J2R (encoding thymidine kinase), and A56R (encoding hemagglutinin) loci. This virus strain also carries marker genes (ruc-gfp) that allow real-time imaging of tumor-specific entry and replication, as well as colonization of the tumors by the virus, through optical imaging (13). Here, we investigated the infection, replication, and lysis capability of GLV-1h68 and related immune profiles after infection and showed efficient oncolytic viral therapy of human pancreatic tumors in nude mice.
Materials and Methods
Cell Lines
All cells were purchased from the American Type Culture Collection. PANC-1 and Hs766T cells were cultured in DMEM supplemented with antibiotic-antimycotic solution (100 units/mL penicillin G, 250 ng/mL amphotericin B, and 100 units/mL streptomycin) and 10% fetal bovine serum (Invitrogen) at 37°C under 5% CO2. MIA PaCa-2 cells were cultured under similar conditions in DMEM but supplemented with 2 mmol/L L-glutamine and 12.5% fetal bovine serum. AsPC-1 and BxPC-3 cells were cultured in RPMI with 2 mmol/L l-glutamine, 10 mmol/L HEPES, 1 mmol/L sodium pyruvate, 4.5 g/L glucose, 1.5 g/L bicarbonate, and 10% fetal bovine serum. CAPAN-2 cells were cultured in DMEM with 4.5 g/L glucose. All cells were incubated at 37°C under 5% CO2.
Viral Construct
The triple-mutant GLV-1h68 virus was constructed as described previously (12). Briefly, GLV-1h68 was generated by inserting three expression cassettes [encoding Renilla luciferase-Aequorea green fluorescent protein (GFP) fusion protein, β-galactosidase (β-gal), and β-glucuronidase] into the F14.5L, J2R, and A56R loci of the LIVP strain viral genome, respectively. After sequencing confirmation of the three inserts, a clone of GLV-1h68 was then amplified in CV-1 cells.
Cell Survival Tests and Viral Proliferation Assays
The cytotoxic effect of viral infection on pancreatic cancer cells was determined by quantifying cytoplasmic lactate dehydrogenase and comparing these levels with levels in untreated control cells grown under identical conditions. Cells (2 × 104) were grown in 12-well flat-bottomed plates and infected with the virus at a virus to tumor cell ratio multiplicities of infection (MOI) of 0.01, 0.1, and 1.0. Cells were left to incubate at 37°C for 1 to 7 days. Every other day beginning on day 1 after infection, cells were washed in PBS and lysed with a 1.35% Triton X-100 solution (% volume/PBS) to release intracellular lactate dehydrogenase. Lactate dehydrogenase was quantified with a Cytotox 96 cytotoxicity assay (Promega), which measures the conversion of a tetrazolium salt into a formazan product. Absorbance was measured at 450 nm by using a microplate reader (EL 312e; Bio-Tek Instruments). Results are expressed as the surviving fraction based on the percentage of the lactate dehydrogenase cells released compared with that of untreated, control cells. All samples were analyzed in triplicate. Standard plaque assays quantified viral replication following infection with GLV-1h68. Supernatants were collected from virally treated cells at 1, 3, 5, and 7 days postinfection. Serial dilutions of supernatants were cultured on confluent layers of CV-1 cells in 12-well plates and titers were determined 72 h later.
Animal Models and In vivo Studies
All mice were cared for and maintained in accordance with animal welfare regulations under an approved protocol by the Institutional Animal Care and Use Committee of LAB Research International (San Diego Science Center).
PANC-1 or MIA PaCa-2 xenograft tumors were developed in 6-8-week-old male nude mice (NCI:Hsd:athymic nude-Foxn1nu; Harlan) by implanting 5 × 106 PANC-1 or MIA PaCa-2 cells s.c. on the right hind leg. Tumor growth was recorded once a week in three dimensions using a digital caliper. Tumor volume was calculated as [(length × width × height) / 2] and reported in mm3. At a specific time point after tumor cell implantation, a single i.v. inoculation of 1 × 106 plaque-forming units (pfu) of GLV-1h68 virus in 100 μL PBS was delivered.
Net body weight change (%) was calculated using the following formula: [(gross weight − tumor weight) − (gross weight at T71 − tumor weight at T71)] / (gross weight at T71 − tumor weight at T71). “T71” means 71 days after tumor cell implantation, which was also the time of virus injection.
After virus injection, mice were killed at different time points for analysis of viral distribution. The tumors and organs were excised, weighted, and homogenized using MagNA Lyser (Roche Diagnostics) at a speed of 6,500 for 30 s. After three cycles of freeze and thaw, samples were sonicated twice for 30 s, and the supernatants were collected by centrifugation at 1,000 × g for 5 min. The viral titers were determined in duplicate by standard plaque assay using CV-1 cells.
Combination Therapy
Cisplatin and GLV-1h68 Combination Treatment
Mice with 32-day-old s.c. PANC-1 tumors were divided into four groups (n = 8 per group). One group was injected with 1 × 106 pfu/mouse of GLV-1h68 virus in the tail vein, and another group was treated with a combination of GLV-1h68 (a single dose of 1 × 106 pfu/mouse in the tail vein) and i.p. injection of 6 mg/kg cisplatin (Sigma; prepared in PBS) once daily on each of days 42 to 46. A third group was treated with consecutive doses of cisplatin only. No treatment was given to the negative control group. Tumor growth was followed by measuring tumor volume (mm3).
Gemcitabine and GLV-1h68 Combination Treatment
Mice with 27-day-old s.c. PANC-1 tumors were divided into four groups (n = 8 per group). One group was injected with 5 × 106 pfu/mouse of GLV-1h68 virus in the femoral vein, and another group was treated with a combination of GLV-1h68 (a single dose of 5 × 106 pfu/mouse in the femoral vein) and i.p. injection of 50 mg/kg gemcitabine (Eli Lilly; prepared in PBS) twice per week for 3 consecutive weeks, starting day 42 of tumor development. A third group was treated with gemcitabine only. No treatment was given to the negative control group. Tumor growth was followed by measuring tumor volume (mm3).
Imaging
The expression of the Renilla luciferase reporter gene carried by the virus was determined as described previously (14). In vitro imaging of GFP was done using a Zeiss Axiovert 200M inverted stand microscope (Carl Zeiss) and the MetaMorph Imaging System (Universal Imaging). In vivo imaging of GFP in mice was done under a stereo fluorescence macroimaging system (Lightools Research).
β-Gal Expression Assay
Cancer cells were plated at 2 × 104 per well in 12-well flat-bottomed plates. After overnight incubation, the cells were infected with GLV-1h68 at a MOI of 2. At 6, 12, and 24 h postinfection, cells were washed with PBS, fixed with 1% glutaraldehyde for 5 min, and washed three times with PBS. Cells were then stained for 4 h in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (1 mg/mL) in an iron solution of 5 mmol/L K4Fe(CN)6, 5 mmol/L K3Fe(CN)6, and 2 mmol/L MgCl2 and examined by microscopy (×100).
Immune-Related Protein Antigen Profiling
Mice with 32-day-old s.c. PANC-1 tumors were divided into two groups (n = 4 per group). One group was injected i.v. with 5 × 106 pfu/mouse of GLV-1h68 virus. No treatment was given to the negative control group. At 21 and 42 days after virus treatment, two mice from each group were killed. Tumors were excised, weighted, and homogenized using MagNA Lyser at a speed of 6,500 × g for 30 s. Samples were then analyzed for immune-related protein antigen profiling by Multi-Analyte Profiles (Rules Based Medicine) using antibody-linked beads. Results were normalized based on total protein concentration.
Statistical Analysis
In the combination therapy studies of pancreatic tumors with GLV-1h68 and cisplatin and combination therapy with GLV-1h68 and gemcitabine, statistical analyses were done using SPSS 10.0 software. Statistical significance was assessed using a two-tailed, unpaired t test to compare the differences between groups. Differences with P < 0.05 were considered statistically significant.
Results
Viral Infection and Replication in Tumor Cells Were Confirmed through Marker Gene Expressions
To verify the infection and replication of GLV-1h68 in tumor cells, we followed marker gene expression levels in PANC-1 and MIA PaCa-2 cell cultures over time (Fig. 1). Virus-mediated marker gene expressions were analyzed using β-gal staining, fluorescence microscopy, and luciferase luminescence photon counting, respectively. Six hours after virus infection, β-gal expression could already be detected in infected PANC-1 cells (Fig. 1A). The estimated amount of β-gal-positive cells was ∼26%. At 12 h, significant levels of luciferase activities were confirmed in both PANC-1 and MIA PaCa-2 cell cultures based on photon emissions after the addition of coelenterazine (Fig. 1B). At 24 h, even higher levels of luciferase activity were found in both cells, although the signal from PANC-1 cells was much stronger than from MIA PaCa-2 cells. Examining the same culture under a fluorescence microscope, we noticed that not all PANC-1 cells were positive for GFP (Fig. 1C, top). After 48 h, however, we found that almost all PANC-1 cells were positive for GFP (Fig. 1C, bottom). These results indicate that the mutant virus was able to efficiently infect and replicate in both PANC-1 and MIA PaCa-2 cells, although viral infection and/or replication may be more permissive in PANC-1 cells than in MIA PaCa-2 cells.
Figure 1.
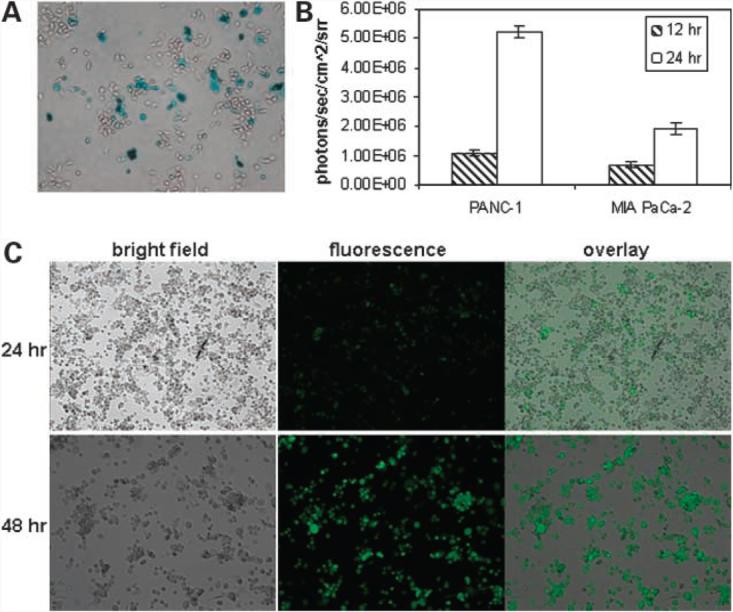
GLV-1h68 virus-mediated marker gene expressions in PANC-1 and MIA PaCa-2 cell cultures at different time points postinfection. A, β-gal staining (6 h) in PANC-1 cells. B, Renilla luciferase-catalyzed light emissions (12 and 24 h) in PANC-1 and MIA PaCa-2 cells. C, GFP expression (top, 24 h; bottom, 48 h) in PANC-1 cells.
GLV-1h68 Efficiently Lyses Different Pancreatic Tumor Cell Lines in Culture
To determine the lytic activity of GLV-1h68 in infected tumor cells, we infected six human pancreatic tumor cell lines (BxPC-3, CAPAN-2, PANC-1, AsPC-1, MIA PaCa-2, and Hs766T) with the virus at different MOI (Fig. 2A). Oncolytic capability of the virus was examined based on cell survival rate 5 days postinfection. At all MOI levels, significant differences in cell survival were observed among different cell lines. At MOI of 0.01 and 0.1, AsPC-1, MIA PaCa-2, and Hs766T cells seemed to be insensitive to viral lyses and the majority of cells survived treatment. On the other hand, the majority of BxPC-3, CAPAN-2, and PANC-1 cells did not survive the viral infection at these MOI. At MOI of 1, almost all cells of BxPC-3, CAPAN-2, and PANC-1 were killed. The majority of AsPC-1 cells died as well. In contrast, most MIA PaCa-2 and Hs766T cells survived the viral treatment. Therefore, GLV-1h68 was able to more efficiently lyse BxPC-3, CAPAN-2, and PANC-1 cells than MIA PaCa-2 and Hs766T cells in culture. PANC-1 cells infected with GLV-1h68 showed apoptosis in ∼50% of cells 24 h after virus infection (data not shown). At 72 h postinfection, the majority of dying cells were found to be necrotic, and <10% of cells were apoptotic.
Figure 2.

Tumor cell survival and viral titer analysis in pancreatic tumor cell cultures after GLV-1h68 infection at different MOI. A, summary of percentage of cell survival at different MOI 5 d postinfection. +++, most cells survived; −, most cells were dead. B, percentage of cell survival (left, ◆, 0.01 MOI; ■, 0.1 MOI; ▲, 1 MOI) and profile of viral proliferation in PANC-1 cells (right, 0.01 MOI) over the course of 1 wk postinfection. C, percentage of survival of MIA PaCa-2 cells over the course of 1 wk postinfection. ◆, 0.01 MOI; ■, 0.1 MOI; ▲, 1 MOI.
To compare the dynamics of virus-mediated cell lysis, we followed the cell survival rate daily for 1 week (Fig. 2B, left, and 2C). Starting 2 days after virus infection at MOI of 1, the number of surviving PANC-1 cells decreased ∼20% daily for 5 consecutive days (Fig. 2B, left). At MOI of 0.1, the cell survival decreased ∼10% daily for 4 consecutive days starting 3 days postinfection. In contrast, an overall 20% decrease of MIA PaCa-2 cell survival rate was observed only at 1 week postinfection at MOI of 1 (Fig. 2C). Viral proliferation analysis in PANC-1 cells following initial infection of 1 MOI showed 3 logs of viral titer increase in the supernatant during the first week postinfection (Fig. 2B, right). Interestingly, viral titer in the supernatant plateaued between days 3 and 5 before increasing again, which may suggest that initial lysis of cells at day 3 was followed by reinfection and further lysis by day 7. These data correlated well with the cell survival curve (Fig. 2B, left), which showed the sharpest decrease in cell survival between days 3 and 5.
A Single Systemic Dose of GLV-1h68 Regresses Pancreatic Tumor Xenografts in Nude Mice
The therapeutic efficacy of GLV-1h68 virus against tumors in vivo was examined. Approximately 2 weeks after virus injection, tumor-specific replication of the virus was examined under a stereofluorescence microscope. Spots of GFP expression were seen on the surface of the tumors (Fig. 3A). One month after virus injection, extensive GFP signal was observed throughout the tumor surface but nowhere else on the body. To quantitatively assess the tumor therapy efficacy, we monitored the tumor volumes in virus-treated mice over a period of 9 months. An example of tumor size change in a single mouse over time is illustrated in Fig. 3A. The time points shown were 2 weeks, 3 weeks, 1 months, and 9 months after virus injection. At the end of the observation period, the PANC-1 tumor xenograft in each mouse had dramatically reduced in size, so that only a small whitish remnant of tissue was visible at each original tumor site (Fig. 3A, d). Histologic analysis showed that these whitish tissues, which are often filled with thick fluid, consisted of dead tumor cells and stroma (data not shown).Tumor volumes for mice in the control group were monitored for 2 months before the mice were killed due to the excessive tumor burden.
Figure 3.
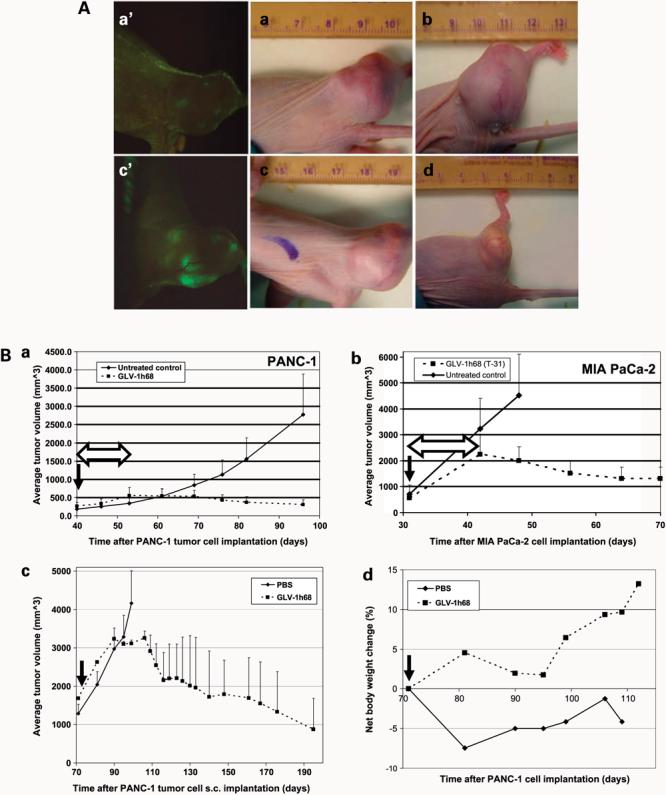
A, real-time imaging of GLV-1h68 viral therapy of s.c. PANC-1 tumors in nude mice. When the tumors reached ∼250 mm3 in size, a single dose (1 × 106 pfu) of the virus was delivered systemically through the i.v. route. No treatment was given in the control group. Tumor sizes of the same mouse as an example at different time points after virus treatment: (a) 2 wk, (b) 3 wk, (c) 1 mo, and (d) 9 mo. Virus-mediated GFP expression in tumors at 2 wk (a′) and 1 mo (c′) postinfection. Note the “whitish” appearance of the residue tumor in d. The major unit of rulers is 1 cm (a-d). B, systemic vaccinia viral therapy of s.c. PANC-1 and MIA PaCa-2 tumors in nude mice. All virus-treated animals received a single i.v. dose of 1 × 106 pfu of GLV-1h68. Median tumor volumes (a and b). Comparison of virus therapy results in PANC-1 and MIA PaCa-2 tumor models (n = 8 per group). T-31, 31 d after tumor cell s.c. implantation into mice. c, virotherapy in nude mice with large PANC-1 tumors (n = 8 per group). d, net animal weight change (%) during the therapy (c). Solid arrows, time points of virus injection; double-headed open arrows, time of tumor regression after virus injection.
To determine the kinetics of GLV-1h68 virus distribution in different organs and tumors, we killed three groups of virus-injected tumorous female and/or male mice at 2 weeks, 1 month, and 85 days postinfection. The results of a viral titer analysis using homogenized tissues are presented as pfu/organ or tumor (Table 1). At 2 weeks and 1 month, mild levels of virus were present in lungs and spleens. In a few animals, the virus was also found in the kidneys, ovaries, and liver at 2 weeks. Overall, the viral titers in the tumors were 3 to 8 and 6 to 8 logs higher than the titers found in all the other organs combined at 2 weeks and 1 month, respectively. The median viral titer in tumors increased ∼1 log from 2 weeks to 1 month. At 85 days, 4 of 5 animals did not show viral infection in any organs, except in tumors. The fifth animal had a mild level of virus presence in the lungs. Overall, the viral titer in the tumors was 3 to 7 logs higher than the titer found in all the other organs combined. The lower viral titer in tumors at 85 days, compared with at 2 weeks and 1 month, may be the result of tumor size shrinkage. Histopathology analyses were done on excised organs, including brain, lung, heart, stomach, kidney, liver, spleen, colon, bladder, and testes, 2 months after virus injection. Except mild spleen enlargements in some animals, no any other histopathology changes were found (data not shown). These results showed that the GLV-1h68 virus has an outstanding infection and replication capability and specificity in tumors and that it is safe for systemic delivery.
Table 1.
Distribution of GLV-1h68 virus in female and/or male nude mice with s.c. PANC-1 tumors 2 wk (A), 1 mo (B), and 85 d (C) after i.v. viral injection
| (A) 2 wk | ||||||||
|---|---|---|---|---|---|---|---|---|
| Viral titer in tissue samples (pfu/organ or tumor) |
||||||||
| Female | Male | |||||||
| Animal no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Kidneys | 0 | 0 | 79 | 0 | 0 | 0 | 0 | 0 |
| Spleen | 37 | 447 | 584 | 331 | 68 | 229 | 212 | 274 |
| Ovaries/testes | 0 | 1.6 × 104 | 1.5 × 104 | 0 | 0 | 0 | 0 | 0 |
| Liver | 0 | 820 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lungs | 0 | 222 | 67 | 224 | 38 | 630 | 203 | 427 |
| Brain | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tumor | 5.7 × 107 | 3.9 × 108 | 1.7 × 108 | 3.2 × 108 | 5.1 × 107 | 4.3 × 107 | 6.4 × 107 | 3.3 × 107 |
| (B) 1 mo | ||||||
| Viral titer in tissue samples (pfu/organ or tumor) |
||||||
| Female | Male | |||||
| Animal no. | 1 | 2 | 3 | 4 | 5 | 6 |
| Kidneys | 0 | 0 | 0 | 0 | 0 | 0 |
| Spleen | 0 | 527 | 339 | 0 | 0 | 88 |
| Ovaries/testes | 0 | 0 | 0 | 0 | 0 | 0 |
| Liver | 0 | 0 | 0 | 0 | 0 | 0 |
| Lungs | 178 | 185 | 493 | 0 | 0 | 710 |
| Brain | 0 | 0 | 0 | 0 | 0 | 0 |
| Tumor | 3.8 × 108 | 8.8 × 108 | 5.3 × 108 | 6.0 × 108 | 3.3 × 108 | 9.9 × 108 |
| (C) 85 d | |||||
| Viral titer in tissue samples (pfu/organ or tumor) |
|||||
| Male | |||||
| Animal no. | 1 | 2 | 3 | 4 | 5 |
| Kidneys | 0 | 0 | 0 | 5 | 0 |
| Spleen | 0 | 0 | 0 | 2 | 0 |
| Bladder | 0 | 0 | 0 | 5 | 0 |
| Testes | 7 | 0 | 0 | 9 | 0 |
| Liver | 0 | 0 | 0 | 0 | 0 |
| Heart | 0 | 0 | 0 | 50 | 0 |
| Lungs | 4 | 0 | 0 | 311 | 0 |
| Brain | 0 | 0 | 0 | 1 | 15 |
| Tumor | 9.6 × 104 | 7.4 × 105 | 3.6 × 107 | 1.5 × 105 | 5.3 × 103 |
To further characterize the oncolytic potency of the systemically delivered GLV-1h68 virus in vivo, we tested the virus in nude mice with s.c. MIA PaCa-2 tumors. The therapy results are compared with those from PANC-1 model (Fig. 3B). The MIA PaCa-2 tumors grew at much faster rate, ∼3- to 4-fold the rate of PANC-1 tumors. Surprisingly, we found that, regardless of tumor growth rate, both tumors could be eliminated. We also found that, in both tumor models, tumor regression started at ∼12 days after virus injection (Fig. 3B, double-headed open arrows). These findings do not correspond to cell culture lysis data, in which PANC-1 cells seemed to be much more prone to viral lysis than MIA PaCa-2 cells.
To determine whether the initial tumor size at the time of virus delivery would affect the outcome of therapy, we delayed the time of virus injection (1 × 106 pfu) to 71 days after PANC-1 cell implantation, when the tumors reached an average size of ∼1,700 mm3 (Fig. 3B). Although these tumors were almost 7 times larger at the time of treatment, they did shrink in size and were eradicated by the virus. At the end of the virotherapy, histology analysis found that the tumor remnants consisted only of dead tissues (data not shown). We also found that treatment of these larger tumors, compared with smaller tumors, resulted in a 1-week delay in the onset of tumor regression and required a longer overall period of treatment to affect tumor shrinkage.
We also investigated potential virus-caused toxicity by monitoring net body weight (gross body weight minus estimated tumor weight) changes during virotherapy (Fig. 3B). After virus injection, mice gained weight initially followed by transient weight loss at ∼3 to 3.5 weeks. Soon afterwards, net body weight began to increase sharply. We found that the net body weight increases in the treated mice were within the range of normal growth curve provided by the animal vendor. In control mice, net body weight did not increase beyond the weight measured at the time of virus injection. Therefore, the GLV-1h68 virus has shown a promising safety profile even in immunocompromised nude mice with large tumor burdens.
Efficacy of GLV-1h68-Mediated Oncolytic Viral Therapy Was Enhanced by Cisplatin and Gemcitabine
To test whether a chemotherapy drug could enhance oncolytic virus-mediated tumor therapy, we combined the drug cisplatin with GLV-1h68 virus treatment (Fig. 4A). Cisplatin alone was only able to transiently arrest tumor growth before tumor size increased again. The virus alone, on the other hand, was able to permanently reverse the course of tumor growth. When virus treatment was combined with consecutive cisplatin treatments, significantly faster decreases in tumor size and an increased eradication rate were observed. At the end of observation period, 7 of 8 mice showed complete tumor regression without any tissues remaining. At the same time point, only 1 of 8 mice treated with virus alone had a complete tumor remission. Further analysis showed that the differences in tumor volumes between the virus treatment alone group and the combination therapy group at days 75, 84, 90, and 96 are statistically significant (P < 0.02). We also tested combination therapy of tumors using GLV-1h68 and gemcitabine (50 mg/kg twice a week for 3 consecutive weeks, starting 14 days after virus injection; Fig. 4B). Again, gemcitabine alone was only able to transiently arrest tumor growth. The virus alone was able to permanently reverse the course of tumor growth. When the virus treatment was combined with gemcitabine treatments, the initiation of tumor shrinkage was accelerated and tumor growth was significantly inhibited compared with mice treated with either virus or gemcitabine alone. Further analysis showed that the differences in tumor volumes between the virus treatment alone group and the combination therapy group at days 50 and 56 are statistically significant (P < 0.05). Therefore, the combination treatment seemed to enhance and accelerate the tumor therapy more than using the virus alone. However, as shown in the virus and gemcitabine combination therapy experiment, the therapeutic outcome using the virus alone was able to catch up with the combination therapy when given sufficient time.
Figure 4.
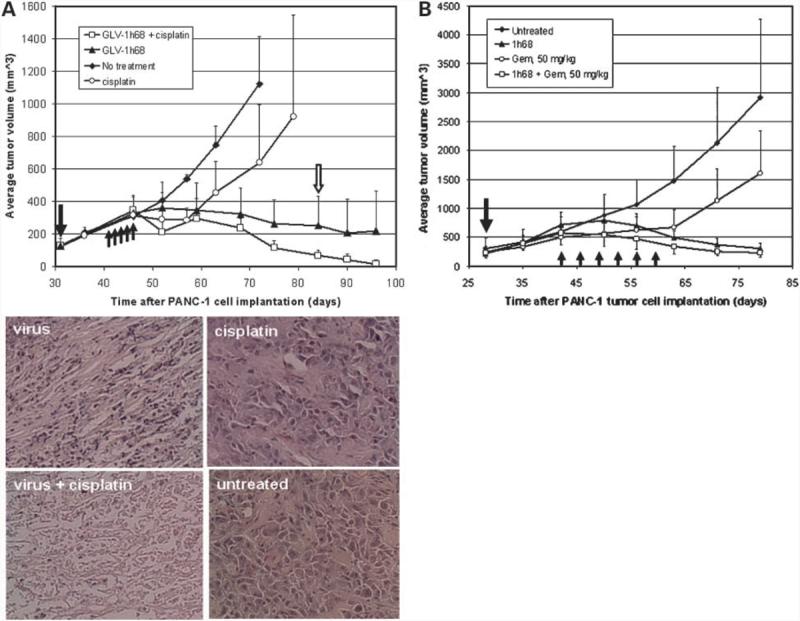
Combination therapy of s.c. PANC-1 tumors with GLV-1h68 virus and cisplatin or gemcitabine. A, combination of virus and cisplatin. Tumorous mice (n = 8 per group) were treated with the virus alone (1 × 106 pfu), with cisplatin alone (6 mg/kg/d for 5 consecutive days), with the virus first (1 × 106 pfu) followed by cisplatin treatment (6 mg/kg/d for 5 consecutive days) 10 d later, or with no treatment. Single solid arrow, time of virus injection; up-pointing arrows, time points of cisplatin treatments. For histology analysis, mice were killed 55 d postvirus injection. Tumors were excised, sectioned, and stained with H&E. Open arrow, time point of histologic analyses (n = 8 per group). Microscopic analyses of virus (top left), cisplatin (top right), and combination therapy (bottom left) treated and untreated (bottom right) tumor sections (5 μm) stained with H&E (bottom). Magnification, ×200. Also shown are examples of two mice after their s.c. PANC-1 tumors were eradicated by the combination therapy. B, combination of virus and gemcitabine. Single solid arrow, time of virus injection; up-pointing arrows, time points of gemcitabine treatments (n = 8 per group).
To further examine the tumor tissues after virus, cisplatin, and combination therapies, we analyzed the tissue sections through histology (Fig. 4A). Without treatment, almost all tumor cells were found to be alive. We did see a few small pockets of dead tissues in the untreated tumors. The majority of cells in tumors treated with cisplatin alone were also alive and dividing. With virus treatment, extensive cell death was seen throughout the tumor. Occasionally, we observed small islands of living cells in the tumor. In contrast, when we examined the tumors treated with the virus and cisplatin combination, we did not notice any islands of live tumor cells. Moreover, less tumor cell debris and stroma remnants were seen in the tumors after combination therapy than in the tumors treated with virus alone. To summarize, the histology results were in accordance with the above tumor size change data.
Proinflammatory Immune Response from the Host Is Activated in Virus-Infected Tumor Tissues
We examined the expression levels of immune-related protein antigens in tumors at certain time points after virus infection. We found that the concentrations of several proinflammatory cytokines and chemokines were significantly elevated in tumor tissues (Table 2A). Many of these cytokines and chemokines, such as eotaxin, IP-10, IL-6, MCP-1, MCP-3, MCP-5, M-CSF, IL-18, etc., are known to activate macrophages, monocytes, neutrophils, eosinophils, etc., and trigger proinflammatory responses in target tissues. Interestingly, IFN-γ was not found to be elevated in the infected tumor tissues. RANTES and MIP-1β, the Th1-associated chemokines of the CC subfamily, were found to be down-regulated on virus infection (Table 2B). Due to the lack of T- and B-cell functions in nude mice, cytokines (IL-2, IL-3, IL-4, IL-7, IL-10, etc.) that activate T and B cells were found to not be significantly affected by vaccinia virus infection. Production of GM-CSF, TNF-α, EGF, VCAM-1, and MDC (CCL22) in tumors was also not found to be significantly altered after virus infection.
Table 2.
(A and B) mouse (host) and(C and D) human (from xenograft tumor) immune-related protein antigen profiling
| (A) Protein expression level up-regulated | |||
|---|---|---|---|
| Antigen names | GLV-1h68/untreated ratio |
Classification | |
| Day 21 | Day 42 | ||
| MCP-5 (CCL12) | 25.59 | 39.05 | Proinflammatory cytokine |
| IL-18 | 14.1 | 33.98 | Proinflammatory cytokine |
| MCP-1 (CCL2) | 14.02 | 21.8 | Proinflammatory chemokine |
| MCP-3 (CCL7) | 10.59 | 12.23 | Proinflammatory chemokine |
| IP-10 (CXCL10) | 10.48 | 11.24 | Proinflammatory chemokine |
| Eotaxin (CCL11) | 7.44 | 5.56 | Proinflammatory chemokine |
| GCP-2 (CXCL6) | 5.49 | 3.69 | Proinflammatory cytokine |
| Gro-β (MIP-2) | 4.82 | 3.59 | Proinflammatory chemokine |
| IL-6 | 4.08 | 7.82 | Proinflammatory cytokine |
| C-reactive protein | 3.98 | 3.29 | Proinflammatory protein |
| M-CSF (KC/GROα) | 2.20 | 4.06 | Proinflammatory chemokine |
| IL-11 | 6.02 | 4.06 | Pleiotropic cytokine |
| Thrombopoietin | 2.22 | 2.07 | Lineage-specific cytokine |
| Apolipoprotein A1 | 6.87 | 6.77 | Anti-inflammatory protein |
| MPO (myeloperoxidase) | 5.62 | 2.65 | Peroxidase enzyme |
| Matrix metalloproteinase-9 | 10.7 | 6.38 | Enzyme |
| TIMP-1 | 14.26 | 13.8 | Protein inhibitor |
| (B) Protein expression level down-regulated | |||
| Antigen names | Untreated/GLV-1h68 ratio |
Classification | |
| Day 21 | Day 42 | ||
| MIP-1γ (CCL9) | 9.70 | 13.11 | Cytokine |
| RANTES (CCL5) | 4.41 | 7.45 | Proinflammatory chemokine |
| MIP-1β | 2.89 | 2.17 | β-Chemokine |
| (C) Protein expression level up-regulated | |||
| GLV-1h68/untreated | |||
| Ferritin | 5.1 | ||
| Plasminogen activator inhibitor-1 | 4.6 | ||
| Tissue factor | 3.4 | ||
| von Willebrand factor | 2.8 | ||
| MCP-1 (CCL2) | 2.7 | ||
| TIMP-1 | 2.6 | ||
| Brain-derived neurotrophic factor | 2.3 | ||
| PAPP-A | 2.3 | ||
| Basic fibroblast growth factor | 2.1 | ||
| IL-1α | 2.1 | ||
| (D) Protein expression level down-regulated | |||
| Untreated/GLV-1h68 | |||
| Epidermal growth factor | 10.7 | ||
| Insulin-like growth factor-I | 8.2 | ||
| Fatty acid binding protein | 5.7 | ||
| TNF-β | 3.9 | ||
| IL-12p40 | 3.2 | ||
| MIP-1β | 3.2 | ||
| IL-2 | 3.0 | ||
| Factor VII | 2.3 | ||
| IL-1ra | 2.2 | ||
| β2-Microglobulin | 2.1 | ||
| Creatine kinase-MB | 2.1 | ||
| RANTES | 2.1 | ||
| Prostatic acid phosphatase | 2.0 | ||
| Note: | |||
| Fold of enhancement | Fold of suppression | ||
| 2-3 | 2-3 | ||
| 3-5 | 3-5 | ||
| >5 | >5 | ||
NOTE: S.c. PANC-1 tumors in nude mice with or without GLV-1h68 treatment. Time points were taken 21 d after virus i.v. injection. Folds of enhancement or suppression of protein expression after virus injection are shown (n = 2).
We also examined the profiles of immune proteins of human origin in xenograft tumors with or without GLV-1h68 treatment. In contrast to the large panel of activated host immune response proteins, very few immune-related proteins were activated in the tumor tissues in response to virus infection (Table 2C and D). There was down-regulation of growth factors. However, in general, the levels of most immune-related proteins of human origin (including IL-1β, IL-6, IL-7, IL-8, IL-10, IL-16, IL-18, MIP-1α, ICAM-1, eotaxin, GM-CSF, IFN-γ, and TNF-α) remained steady.
Discussion
The current studies showed that vaccinia virus is a promising oncolytic agent. In this study, we showed that the recombinant vaccinia virus GLV-1h68 was able to effectively infect, replicate in, and lyse several pancreatic tumor cell lines in culture. Cell lysis correlated well with viral replication. There is, however, variability in efficiency in lysing different tumor cells. Such differences may be the result of the levels of protection of the cells against virus. Once these defenses are activated, the cell lines may limit the efficient replication or spread of the vaccinia virus. Further experimentation will be required to determine the molecular mechanism that restricts virus replication.
The current study also showed the ability of GLV-1h68 to provide highly effective therapy in vivo. When systemically delivered, the virus caused the regression of established pancreatic tumors in mice. These results are significant for two reasons. First, the targeting of tumors appeared to be highly specific even when the virus was injected systemically. Because the virus carries the marker genes encoding β-gal, GFP, and Renilla luciferase, respectively, three independent readouts were available to verify the appearance of virus in tumors and the absence of virus in healthy tissues. The positive correlation between the observed marker gene expressions and viral replication has been documented previously (13). Secondly, the replication-competent GLV-1h68 not only successfully infected a well-established tumor but also continued to amplify in tumors to achieve an evident regression of tumors. Whether multiple viral injections would result in clinically apparent tumor regression in immunocompetent humans can only be determined by clinical studies. One of the most likely explanations of tumor-specific colonization by GLV-1h68 after systemic delivery may be the differential immune clearance of virus particles in healthy tissues compared with tumor tissues. This concept is currently under investigation. A limitation of the current study is that it was carried out in an immunocompromised nude mouse host. We plan to repeat these therapy studies in immunocompetent transgenic mice with spontaneous tumors. Because the GLV-1h68 therapy is currently designed as a single dose treatment and the vaccination of the general population against smallpox stopped more than three decades ago, we do not expect that preexisting vaccinia neutralizing antibody titer (generally low) will impede GLV-1h68 virotherapy. However, clinical trials may require that all patients vaccinated >25 years ago against smallpox be tested for preexisting titers.
Gemcitabine-based therapy is an acceptable standard for unresectable locally advanced/metastatic pancreatic cancer, but the average median survival is only 6 months. Experiments using combined viral and chemotherapy indicate that the addition of gemcitabine or cisplatin significantly enhanced and/or accelerated the likelihood of complete response to therapy, although the virus monotherapy was able to catch up with the combination therapy when given enough time. Potential mechanisms, which were found to be responsible for the synergistic actions of other oncolytic viruses with chemotherapy, include changes in apoptosis (15), changes in nucleotide pools (16), and changes in DNA repair pathways (17). The i.p. cisplatin dose used in this study is higher than the usual i.v. dose for human patients. Additional dose escalation studies need to be conducted to find the optimal dose range that will achieve desirable combination therapeutic effects without causing additional toxicity. It should be noted that, because many chemotherapy drugs are nucleoside analogues, which target DNA replication, combination of such drugs into a combined therapy scheme may cause inhibition of viral replication.
In the immune-related protein antigen profiling study, we identified several proinflammatory cytokines and chemokines, whose expression by the host were elevated after virus infection. This expression shows that vaccinia virus infection of the tumorous host is accompanied by a robust activation of innate immune response/defense. Although we used an immunocompromised nude mouse model, the resulting cytokine profile was strikingly similar to that in immunocompetent BALB/c mice after vaccinia virus infection (18). Many proteins, such as MCP-1, MCP-3, MCP-5, M-CSF, IL-18, and C-reactive protein are involved in activation of macrophages and neutrophils for phagocytosis of virus-infected cells. IP-10 has been shown to enhance the cytolytic activity of NK cells and contribute to vaccinia virus clearance in vivo (19). IL-18 is also known to activate NK cells to eliminate virus-infected cells (20). Some of the up-regulated proteins have antiangiogenic functions. Matrix metalloproteinase-9 (21) has been shown to increase the generation of antiangiogenic fragments and decreased angiogenesis. In addition, gro-β (22) and IP-10 (23) have been shown to suppress tumor-induced neovascularization. Active remodeling of the tumor environment is also evident based on the elevated levels of matrix metalloproteinase-9 and its inhibitor, TIMP-1. Matrix metalloproteinase-9 has been shown to mediate tumor regression after being expressed in tumors through adenovirus (21). To determine whether the innate immunity alone is sufficient to eliminate tumors, we are repeating the protein antigen profiling experiments in an immunocompetent host using the GLV-1h68 virus strain.
It is worth noting that, although MIA PaCa-2 may be less sensitive to viral lysis than PANC-1 in vitro, similar antitumor efficacy was observed in vivo. Unpublished in vitro virus growth curves indicated that GLV-1h68 can replicate well in both PANC-1 and MIA PaCa-2 cells with even slightly higher virus yield in the latter. Whether these different results are related to other factors such as in vitro cell growth rate, in vitro cell confluence at the time of virus infection, angiogenesis, growth factors, or action on tumor stroma cannot be dissected by the current study design. Nevertheless, these data would suggest that in vitro tumor sensitivity cannot be a sole criterion for selecting patients for therapeutic trials.
During our study, we noticed that, at the end of tumor therapy, the residue tumor nodules gradually turned whitish in appearance. When examined histologically, we found that the whitish tissues were the remains of dead tumor cells and stroma. Pancreatic cancer in vivo tends to be high in stromal components, with sparse cellularity. These data indicate that the vaccinia virus must be capable of intratumoral dissemination around stromal areas to infect and kill the cellular portions and produce remnants of fibrous scaffold. The results may also have significant implications for future clinical testing of GLV-1h68. Because the absorbance of these residual dead tissues takes a longer time, tumor size measurements may not be the most precise determination variables for the evaluation of efficacy.
In summary, these data indicate that GLV-1h68 is a promising candidate virus in the treatment of pancreatic cancer. In vitro tumor cell killing is documented for a broad spectrum of tumor cell lines. In vivo efficacy can be documented by systemic injection with limited toxicity, and these effects can be augmented by systemic chemotherapy. These data are therefore encouraging support for trials of this novel agent in humans.
Acknowledgments
We thank Terry Trevino and Melody Jing for excellent technical support, Thu Ha Le for histology analysis, James Chang, and Andrea Feathers for editorial assistance.
Grant support: Genelux, NIH grants RO1 CA 75416 and RO1 CA/DK80982, Susan G. Komen Breast Cancer Foundation grant IMG0402501, and Flight Attendant Medical Research Institute grant 032047 (Y. Fong).
Footnotes
Disclosure of Potential Conflicts of Interest
Y.A. Yu, N. Chen, Q. Zhang, and A.A. Szalay: received grant support from and employees and shareholders of Genelux.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. 2007. [DOI] [PubMed] [Google Scholar]
- 2.Fong Y, Gonen M, Rubin D, Radzyner D, Brennan MF. Long-term survival is superior after resection for cancer in high volume centers. Ann Surg. 2005;242:540–7. doi: 10.1097/01.sla.0000184190.20289.4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 4.Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–10. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 5.Woo Y, Adusumilli PS, Fong Y. Advances in oncolytic viral therapy. Curr Opin Investig Drugs. 2006;7:549–59. [PubMed] [Google Scholar]
- 6.Thorne SH, Hwang TH, Kirn DH. Vaccinia virus and oncolytic virotherapy of cancer. Curr Opin Mol Ther. 2005;7:359–65. [PubMed] [Google Scholar]
- 7.DiStefano AD, Buzdar AU. Viral-induced remission in chronic lymphocytic leukemia? Arch Intern Med. 1979;139:946. [PubMed] [Google Scholar]
- 8.Yettra M. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch Intern Med. 1979;139:603. [PubMed] [Google Scholar]
- 9.Hansen RM, Libnoch JA. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch Intern Med. 1978;138:1137–8. [PubMed] [Google Scholar]
- 10.Mastrangelo MJ, Maguire HC, Jr., Eisenlohr LC, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–22. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 11.Arakawa S, Jr., Hamami G, Umezu K, Kamidono S, Ishigami J, Arakawa S. Clinical trial of attenuated vaccinia virus AS strain in the treatment of advanced adenocarcinoma. Report on two cases. J Cancer Res Clin Oncol. 1987;113:95–8. doi: 10.1007/BF00389974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Yu YA, Timiryasova T, et al. Eradication of solid human tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007;67:10038–46. doi: 10.1158/0008-5472.CAN-07-0146. [DOI] [PubMed] [Google Scholar]
- 13.Yu YA, Shabahang S, Timiryasova TM, et al. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat Biotechnol. 2004;22:313–20. doi: 10.1038/nbt937. [DOI] [PubMed] [Google Scholar]
- 14.Yu YA, Szalay AA. A Renilla luciferase-Aequorea GFP (ruc-gfp) fusion gene construct permits real-time detection of promoter activation by exogenously administered mifepristone in vivo. Mol Genet Genomics. 2002;268:169–78. doi: 10.1007/s00438-002-0750-x. [DOI] [PubMed] [Google Scholar]
- 15.Stanziale SF, Petrowsky H, Adusumilli PS, Ben Porat L, Gonen M, Fong Y. Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clin Cancer Res. 2004;10:3225–32. doi: 10.1158/1078-0432.ccr-1083-3. [DOI] [PubMed] [Google Scholar]
- 16.Petrowsky H, Roberts GD, Kooby DA, et al. Functional interaction between fluorodeoxyuridine-induced cellular alterations and replication of a ribonucleotide reductase-negative herpes simplex virus. J Virol. 2001;75:7050–8. doi: 10.1128/JVI.75.15.7050-7058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown SM, MacLean AR, McKie EA, Harland J. The herpes simplex virus virulence factor ICP34.5 and the cellular protein myD116 complex with proliferating cell nuclear antigen through the 63-amino-acid domain conserved in ICP34.5, myD116, and GADD34. J Virol. 1997;71:9442–9. doi: 10.1128/jvi.71.12.9442-9449.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knorr CW, Allen SD, Torres AR, Smee DF. Effects of cidofovir treatment on cytokine induction in murine models of cowpox and vaccinia virus infection. Antiviral Res. 2006;72:125–33. doi: 10.1016/j.antiviral.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Mahalingam S, Farber JM, Karupiah G. The interferon-inducible chemokines MuMig and Crg-2 exhibit antiviral activity in vivo. J Virol. 1999;73:1479–91. doi: 10.1128/jvi.73.2.1479-1491.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.French AR, Holroyd EB, Yang L, Kim S, Yokoyama WM. IL-18 acts synergistically with IL-15 in stimulating natural killer cell proliferation. Cytokine. 2006;35:229–34. doi: 10.1016/j.cyto.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Bendrik C, Robertson J, Gauldie J, Dabrosin C. Gene transfer of matrix metalloproteinase-9 induces tumor regression of breast cancer in vivo. Cancer Res. 2008;68:3405–12. doi: 10.1158/0008-5472.CAN-08-0295. [DOI] [PubMed] [Google Scholar]
- 22.Cao Y, Chen C, Weatherbee JA, Tsang M, Folkman J. gro-β, a -C-X-C- chemokine, is an angiogenesis inhibitor that suppresses the growth of Lewis lung carcinoma in mice. J Exp Med. 1995;182:2069–77. doi: 10.1084/jem.182.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neville LF, Mathiak G, Bagasra O. The immunobiology of interferon-γ inducible protein 10 kD (IP-10): a novel, pleiotropic member of the C-X-C chemokine superfamily. Cytokine Growth Factor Rev. 1997;8:207–19. doi: 10.1016/s1359-6101(97)00015-4. [DOI] [PubMed] [Google Scholar]