

Abstract
The synthesis and exploration of novel butyrophenones have led to the identification of a diazepane analog of haloperidol, 4-[4-(4-Chlorophenyl)-1,4-diazepan-1-yl]-1-(4-fluorophenyl)butan-1-one (Compound 13) with an interesting multireceptor binding profile. Compound 13 was evaluated for its binding affinities at DA subtype receptors, 5HT subtype receptors, H-1, M-1 receptors and at NET, DAT and SERT transporters. At each of these receptors, compound 13 was equipotent or better than several of the standards currently in use. In in vivo mouse and rat models to evaluate its efficacy and propensity to elicit catalepsy and hence EPS in humans, compound 13 showed similar efficacy as clozapine and did not produce catalepsy at five times its ED50 value.
Keywords: Haloperidol, antipsychotics, butyrophenone, dopamine receptor ligands, serotonin receptor ligands, D2-like receptor ligands, diazepane, atypical antipsychotics
The introduction of clozapine1 (Clozaril, 1) as a treatment option in the 1990s has opened the door to the use of atypical antipsychotics and the replacement of the prototypic agent, haloperidol (2) as a drug of choice for the treatment of schizophrenia. The superior therapeutic profile of clozapine and other atypical antipsychotic drugs and the long-term debilitating side-effects2 associated with haloperidol treatment have conspired to diminish its use in the clinic. Among several hypotheses put forth to explain the long-term extrapyramidal side effects of haloperidol including tardive dyskinesia, is the fact that haloperidol undergoes biotransformations that lead to the formation of toxic metabolites.3,4 Specifically, haloperidol has been shown to undergo CYP450-mediated oxidation to form pyridinium metabolites5 which can potentially damage dopamine receptors in the nigrostriatum of the brain (Chart 1). Based on this hypothesis, we 6-9 proposed a drug design strategy that replaces the piperidine ring in haloperidol with bioisosteric equivalents which could not undergo in vivo biotransformation to pyridinium species known to be associated with neuronal toxicity. A similar strategy has more recently appeared in the literature.10 The outcome of such a strategy was to enhance the pharmacological and side-effect profiles of the resulting agents.
Chart 1.

Our laboratory has previously shown that modification in the piperidine ring of haloperidol results in significant changes in pharmacological activity.6-7 We also identified new bioisosteric groups of the piperidine ring with binding profiles similar or better than those of haloperidol at the D2 dopamine receptor subtype.7-9 Several of these compounds are shown in Chart 2, with new binding affinity data at selected receptors reported in Table 1.
Chart 2.

Table 1.
Binding Affinity Constants of Compounds to Selected 5-HT and H-1 Receptors.
| Compds | Binding Data of Compounds, Ki ± SEM (nM) | Ki D2/Ki D4 Ratio | |||||
|---|---|---|---|---|---|---|---|
| DAD2 | DAD4 | 5HT1A | 5HT2A | 5HT2C | H-1 | ||
| Cloz (1)a | 130 | 54 | 140 | 8.9 | 17 | 1.8 | 2.4 |
| Hal (2)a | 0.89 | 10 | 3600 | 120 | 4700 | 440 | 0.09 |
| 4b | 253.5±38.9 | 17.5±2.0 | 90.9±21.0 | 109.6±16.0 | 3552±943 | 157.6±36.0 | 2.8 |
| 5b | 1.6±0.14 | 5.3±1.0 | 27.7±8.0 | 30.9±6.0 | 872.1±178.0 | 8780±1625 | 0.30 |
| 6b | 2.3±0.28 | 19.2±2.3 | 37.6±6.0 | 12.3±3.0 | >10,000 | 635.4±96.0 | 0.12 |
| 7b | 124.4±16.5 | 176.8±36.0 | 414.4±101.0 | 106.0±21.0 | 1065±244 | 973.5±207.0 | 0.70 |
| (+)-8b | 51.1±6.0 | 3.6±0.48 | 772.3±90.0 | 75.8±12.0 | 3598±1162 | 1467±473 | 14.2 |
| (-)-8b | 489.4±119.4 | 245.5±29.2 | 831.5±126.0 | 241.5±39.0 | 1252±461 | 259.6±34.0 | 2.0 |
Binding data obtained from reference 12b.
Although compounds 5 and 6 have high affinity for 5HT1A and 5HT2A receptors, the demonstration that compound 5 elicits a higher catalepsy than haloperidol dampened our desire to pursue the tropane moiety as a piperidine bioisostere.9 It is important to note that the alcoholic function in compounds 5 and 6 contributes significantly to affinity not only at the D2-like receptors but also at the 5HT1A and 5HT2A receptors (cf compounds 5, 6 and 7). Interestingly, the OH group does not appear to play a significant role in the binding of the tropane analogs to the 5HT2C receptor.
The reaction mechanism by which haloperidol is metabolized in vivo to a pyridinium species or BCPP+ has been suggested to begin with dehydration of the tertiary alcoholic function.5 A drug design strategy to prevent such a biotransformation is to eliminate the alcoholic function. Compound 3, a haloperidol analog without its alcoholic function, was previously synthesized and evaluated at D2-like receptors.7 The results supported the notion that the alcoholic function contributes to affinity but is not an absolute requirement for binding to dopamine receptors. Compound 4 in which the C-OH group was replaced with a nitrogen atom showed a lower binding affinity than haloperidol at the dopamine receptors.8 Compounds 5 and 6 were designed on the premise that the tropane moiety could not form a pyridinium ring due to the fact that a double bond could not form at the ring junctions adjacent to the N atom.9 In addition, bridging the ring restricts conformational mobility and holds the alcoholic function in a preferred conformation.8 Binding evaluation showed that both compounds were as potent or more potent than haloperidol at D2-like receptors. It was further demonstrated that the OH group is in the axial position and this led to the suggestion that haloperidol most likely binds to the dopamine receptors with its OH in the axial position. Comparing 5 and 6 suggests that the chloro group contributes to binding but is also not an absolute requirement. Removal of the OH group from the tropane ring (Compound 7) again decreased affinity but retained sufficient dopamine binding affinity as observed for compound 3. Contraction of the piperidine ring to a pyrrolidine (8) resulted in a decrease in affinity but expansion to a 7-membered homopiperidine ring (9) retained affinity as in haloperidol, thus suggesting that homopiperidine ring is bioisosteric to piperidine ring.7 Separation of the enantiomers of compound 8 and their subsequent evaluation at D2-like receptors revealed that (+)-8 was the eutomer.6
To further explore these changes in the piperidine moiety of haloperidol in our search for new antipsychotic drugs with atypical pharmacological profiles, we have embarked on SAR studies with modifications primarily in the piperidine ring that could lead to the identification of new agents with clozapine-like binding profiles but without the associated weight gain and new onset type II diabetes.11 Thus, it was desirable not only to evaluate the binding of the compounds at dopamine receptors but also at serotonergic receptors (5HT1A and 5HT2A) since binding to these receptors is often associated with therapeutic advantages observed in atypical antipsychotic drugs.12 In addition, evaluation of the compounds at 5HT2C and histamine H-1 receptors will be valuable indicators of whether these compounds have the potential to develop the weight gain side effect associated with atypical antipsychotic drugs.13 In line with the multiple receptor targeting strategy in the development of new antipsychotic agents, we have focused our design efforts on obtaining agents that have the following receptor binding profiles:
Binding to DA D2 receptor with moderate affinity (30 < Ki < 150 nM)
Binding to DA D4 receptor with high affinity (Ki < 10 nM)
Binding to 5HT1A and 5HT2A receptors with high affinity (Ki < 50 nM)
Binding to 5HT2C and H-1 receptors with very low affinity (Ki > 500 nM)
The above criteria (a – d) will also guide the selection of compounds for evaluation in in vivo animal models for efficacy and the propensity of the compounds to produce catalepsy, a condition associated with extrapyramidal side-effects in humans.14 To achieve the drug design objectives, we have utilized observations from previous SAR studies to design compounds 10 - 14 (Chart 3) as probes for the dopamine, serotonin and histamine H-1 receptor subtypes in an attempt to obtain new prototype drugs.
Chart 3.

Chemistry
Construction of the ethylene-bridged piperazine moiety was initiated using (2S, 5R)-diethyl 2,5-dibromohexanedioate (16) as the starting material (Scheme 1) as previously reported.15,16 Double alkylation of 4-chlorophenyl aniline with 16 yielded N-chlorophenyl pyrrolidine (17). Amidation of 17 with benzylamine produced the amide, 18 which underwent a ring closure with the remaining ester function to form the bridged piperazine, 19. An attempt to reduce the lactam functions of 19 with LiAlH4 also resulted in the removal of the 4-chloro group on the phenyl ring to form the di-substituted 3,8-diaza-bicyclo[3.2.1]octane (20). Debenzylation of 20 to form 22 was accomplished by reaction with ethyl chloroformate and the subsequent hydrolytic cleavage of the resulting carbamate, 21. Compound 10 was obtained by alkylation of the secondary amine function of 22 with 4-chloro-4′-fluorobutyrophenone. Target compounds 11 and 12 were obtained by alkylating the corresponding starting materials, 23 and 24 respectively (commercially available from Sigma-Aldrich) with 4-chloro-4′-fluorobutyrophenone (Schemes 2 and 3). Scheme 4 describes the synthesis of target compound 13 in five steps. Initial starting material 4-chloroboronic acid (26) was synthesized from 4-chlorobromobenzene by lithiation with n-butyl lithium and treatment with trimethylborate. The chloroboronic acid was coupled to N-Boc protected 1,4-diazepane (28) under palladium-catalyzed amination condition to form 29. Deprotection of 29 in trifluoroacetic acid delivered amine 30 which was alkylated in the usual way with 4-chloro-4′-fluorobutyrophenone to form 13. An alternative synthesis of compound 13 was also explored in which the 4-chlorophenyl homopiperazine (30) was obtained in one step without the protection and deprotection steps. The reaction involved the coupling of 4-chloroiodobenzene directly with unprotected homopiperazine under CuI catalysis to form compound 30 in excellent yield. The same alkylation step delivered the desired compound 13 in 61% overall yield (Scheme 5).
Scheme 1a.

a Reagents: (i) 4-Chloroaniline, DME, KI; (ii) Benzylamine, o-Xylene; (iii) (a) NaOH, EtOH; (b) Ac2O, NaOAc; (iv) LiAlH4; (v) ClCO2Et, Toluene; (vi) KOH, EtOH, NH2NH2; (vii) 4-Chloro-4′-fluorobutyrophenone, KI, K2CO3, DME.
Scheme 2a.

a Reagents: (i) 4-Chloro-4′-fluorobutyrophenone, KI, K2CO3, DME.
Scheme 3a.

a Reagents: (i) 4-Chloro-4′-fluorobutyrophenone, KI, K2CO3, DME.
Scheme 4a.

a Reagents: (i) (a) n-BuLi, -78 °C, THF; (b) B(OMe)3; (ii) t-BuOH, NaOH, (t-BuOCO)2O; (iii) Cu(OAc)2, NEt3, Mol sieves (4Å), rt; (iv) TFA, CH2Cl2; (v) 4-Chloro-4′-fluorobutyrophenone, KI, K2CO3, DME.
Scheme 5a.

Alternative Synthetic Scheme for Compound 13
a Reagents and conditions: (i) CuI, ethylene glycol, K2CO3, iPrOH, 80 °C, reflux, 12 h; (ii): KI, K2CO3, DME, 4-chloro-4′-fluorobutyrophenone, reflux, 12 h.
8-Methyl-8-aza-bicyclo[3.2.1]octa-3-one (31) served as the starting material for the synthesis of target compound 14 (Scheme 5). Compound 31 was converted to the cyclic lactam (32) by insertion of a nitrogen atom followed by reduction using lithium aluminum hydride to form 33 in very good yield (80%). The free secondary amine in 33 was BOC protected prior to its treatment with ethyl chloroformate to form the carbamate protected amine, 35. Compound 35 was BOC deprotected and then coupled with 4-chloroboronic acid in the presence of copper acetate to yield 37. Decarbamylation of 37 was achieved using KOH in ethylene glycol and the resulting secondary amine was alkylated with 4-chloro-4′-fluorobutyrophenone under the usual alkylating condition to produce the desired compound 14.
Results and Discussion
Our previous observation that bridging the piperidine ring to form the tropane analogs of haloperidol resulted in 7- and 3-fold increases in binding affinity at the D2 and D4 receptors respectively informed the design of target compounds 10 and 14. The rationale was to investigate bridged analogs to probe the effect of conformational restriction on piperazine and homopiperazine equivalents of haloperidol. Compound 10 can also be viewed as an analog of 7 with the carbon at position 4 of the ring replaced by a nitrogen atom. The ethylene bridge in compound 10 holds the piperazine ring in the chair conformation while in compound 11 the ring is held in the boat conformation by a methylene group. Binding affinity data for compounds 10 -14 are reported in Table 2.
Table 2.
Binding Affinity Constants of Compounds to Dopamine, 5HT and Histamine Receptors.
| Compds | Binding (Ki±SEM) Data of Synthetic Compounds at Selected Receptors (nM) | ||||||
|---|---|---|---|---|---|---|---|
| D2 | D3 | D4 | 5HT1A | 5HT2A | 5HT2C | H1 | |
| Cloz(1)a | 130 | 240 | 54 | 140 | 8.9 | 17.0 | 1.8 |
| Hal (2) a | 1.4 | 2.5 | 3.3 | 3600 | 120 | 4700 | 440 |
| 10b | 155 | 600 | 2100 | ND | ND | ND | ND |
| 11b | 170 | 220 | 520 | ND | ND | ND | ND |
| 12 | 98.0±15.3 | 244.1±106.0 | 6.53±0.76 | 30.5±5.0 | 22.0±4.0 | 4132±1081 | 911.8±152.0 |
| 13 | 43.3±13.3 | 158.8±35.1 | 6.6±0.6 | 117.4±32.6 | 23.6±2.7 | 1425±207 | 188.6±16.0 |
| 14 | 178.4±29.2 | 548.1±246.0 | 41.8±9.0 | 2332±470 | 194.8±53.0 | 3513±912 | 1014±206 |
ND = Not Determined.; Hal = Haloperidol; Cloz = Clozapine
Data for compounds 1 & 2 were obtained from ref. 12b.
Data from Pfizer Global Research and Development Laboratories by A. W. Schmidt.
Both compounds 10 and 11 have only moderate to weak binding affinity at the D2-like receptors and do not meet the proposed criteria. Interestingly, compound 12, previously reported by Yevich, J. P. et al.17 with the 4-chlorophenyl ring in compound 4 replaced by 1,3-diazine moiety, met the four criteria set of interest (Table 3). The authors pharmacologically evaluated compound 12 and its ketone-reduced analog for their binding affinity for DAD2, sigma and cortical α1 adrenergic receptors but not at the receptors of interest in this study. Thus, its synthesis provides an opportunity to compare the contributions of 4-chlorophenyl moiety in compound 4 with the 1,3-diazine ring in 12 to the binding affinities at the various receptors.
Table 3.
Comparative Binding Affinity Data for Compound 13 and Agents currently in use at Human Receptors and Rat Transporters.
| a Receptor | Binding (Ki±SEM) Data of Compd 13 and standard agents at Selected Receptors (nM)b | ||||||
|---|---|---|---|---|---|---|---|
| Comp 13 | bClozapine | bRisperidone | bZiprasidone | bOlanzapine | bQuetiapine | bHaldol | |
| DA D1 | 162.7±33.1 | 290 | 580 | 130 | 52 | 1300 | 120 |
| DA D2 | 43.3±13.3 | 130 | 2.2 | 3.1 | 20 | 180 | 1.4 |
| DAD3 | 158.8±35.1 | 240 | 9.6 | 7.2 | 45 | 320 | 2.5 |
| DAD4 | 6.6±0.6 | 54 | 8.5 | 32 | 50 | 2200 | 3.3 |
| 5HT1A | 117.4±32.6 | 140 | 210 | 2.5 | 2100 | 230 | 3600 |
| 5HT2A | 23.6±2.7 | 8.9 | 0.29 | 0.39 | 3.3 | 220 | 120 |
| 5HT2B | 495.2±94.0 | NR | NR | NR | NR | NR | NR |
| 5HT2C | 1425.0±207 | 17 | 10 | 0.72 | 10 | 1400 | 4700 |
| 5HT6 | 295.9±48.6 | 11 | 2000 | 76 | 10 | 1400 | 6000 |
| Hist H1 | 188.6±16.0 | 1.8 | 19 | 47 | 2.8 | 8.7 | 440 |
| M1 | 871.6±75.6 | 1.8 | 2800 | 5100 | 4.7 | 100 | 1600 |
| DAT | 1150±133 | NR | NR | NR | NR | NR | NR |
| NET | 850.7±175.0 | 390 | 28000 | 48 | 2000 | 680 | 5500 |
| SERT | 56.6±6.0 | 3900 | 1400 | 53 | >15,000 | >18,000 | 1800 |
Human cloned receptors were used in all cases except for the NT transporters which are from rats.
Data for standard drugs were obtained from Schmidt et al., reference 12b.
NR = Not Reported.
Compound 13, which can be viewed as a homopiperazine analog of haloperidol and compound 9, meets all the criteria except for its binding affinity at the 5HT1A receptor. Compound 14, a bridged analog of compound 13, also showed low binding affinity for the receptors of interest (Table 2) and failed to meet the other stated criteria. Thus, it would appear that bridging does not necessarily result in favorable attributes for these compounds. Based on these observations, compound 13 was selected for further pharmacological evaluation.
Table 3 shows a head to head comparison of the binding affinities of compound 13 and several atypical antipsychotic drugs currently in use at several receptors known to have implications for the therapeutic value of atypical antipsychotic drugs. Compound 13 has moderate affinity for the DA D2 receptor and high affinity at the D4 receptor. The effect of compound 13 on apormophine-treated mice (see later) suggests D2 receptor antagonism and thus, would be expected to demonstrate antipsychotic properties in humans.18 5HT1A receptor activation has been suggested to contribute to the improved activity of certain atypical antipsychotic drugs.12b,19
Compound 13 has moderate affinity for 5HT1A (Ki = 117 nM). This binding affinity is higher than that for risperidone and quetiapine but similar to that of clozapine (Ki = 140 nM). Compound 13 also binds with high affinity at the 5HT2A receptor (Ki = 23.6 nM), a receptor implicated in the therapeutic efficacy of atypical antipsychotic drugs in treating the negative symptoms of schizophrenia and preventing motor disturbances associated with antipsychotic treatment.20 While this affinity is 10-fold better than that of quetiapine, other atypical antipsychotics have higher affinity for this receptor. 5HT6 receptors are implicated in the amelioration of the cognitive disturbances in schizophrenia and both clozapine and olanzapine have high affinity for this receptor.21 While 13 has weak affinity for the 5HT6 receptor (Ki = 295.8 nM), it is over 13-fold more potent at this receptor than quetiapine and over 6-fold better than risperidone.
Treatment of schizophrenia with atypical antipsychotic drugs has been associated with weight gain and the onset of type II diabetes mellitus. Two receptors, histamine H-1 and 5HT2C, have been suggested to be involved in this adverse event. Several papers22 have demonstrated that there is significant correlation between affinity for H-1 receptor and weight gain and hence the observation that compound 13 has much lower affinity (Ki = 186 nM) for H1 receptor than risperidone (19 nM), quetiapine (8.7 nM), ziprasidone (47 nM), olanzapine (2.8 nM) and clozapine (1.8 nM) is an indication that compound 13 may have a lower propensity to induce weight gain. In addition, the low affinity of 13 for the 5HT2C receptor (Ki = 1425 nM) compared to ziprasidone (Ki = 0.72 nM), risperidone (Ki = 10 nM) and olanzapine (Ki = 10 nM) suggests a possible decrease in the propensity of 13 for treatment-emergent weight gain. The observation that 5-HT2B receptor agonists are associated with increased risk for valvular heart disease23 will require that antipsychotics that interact with 5HT receptors be evaluated for binding to the 5HT2B receptor. Thus, it is encouraging to note that compound 13 does not bind appreciably to 5HT2B receptor (Ki = 495). Also encouraging is the observation that compound 13 lacks affinity for the cholinergic muscarinic M3 receptor (Ki = 3000 nM), a receptor reported to have a contributing role in new-onset diabetes in schizophrenic patients treated with atypical antipsychotics.12a
Compound 13 was also evaluated at neurotransmitter uptake transporters. Compound 13 has very weak affinity for both the dopamine (DAT) and norepinephrine (NET) transporters but has similar affinity for the serotonin transporter (SERT) (Ki = 56.6 nM) compared to aripiprazole (Ki = 98 nM).24 This binding profile at the transporters suggests that while it has potential for antidepressant properties, it has a much lower potential to induce cocaine-like adverse properties since it has a very weak binding affinity for DAT.
Animal behavioral studies were initiated to provide preliminary evidence on efficacy and to evaluate the propensity of compound 13 to induce catalepsy. The ability to block apomorphine induced stereotypy is an indication of a drug's in vivo capacity to antagonize dopamine D2 receptors while induction of catalepsy is considered to be an indication of a drug's propensity to produce extrapyramidal symptoms in humans. The ED50 values of compound 13, clozapine and haloperidol are estimated from the dose-response curves in Figs 1–3. The ED50 value of compound 13 in inhibiting apomorphine-induced stereotypy was estimated to be 35.8 μmole/kg compared to clozapine's 42.4 μmole/kg suggesting that compound 13 is slightly more potent at blocking the D2 receptors in vivo than clozapine on a molar basis. This is also consistent with their estimated Ki values at the D2 receptor. The ED50 values comparing compound 13, clozapine and haloperidol is reported in Table 4. Compound 13 and clozapine have ED50s (μmole/kg) that are about 500 times that of haloperidol. The results of these behavioral studies support the radioligand binding data that compound 13 does not bind to D2 receptors as well as haloperidol, but does bind to D2 receptors with about the same affinity as clozapine.
Fig 1.

Fig 3.

Table 4.
Comparison of ED50 values for Apomorphine Test
| Test Compound | FW | ED50, mg/kg | 95% Confidence Interval, mg/kg | ED50, μmoles/kg |
|---|---|---|---|---|
| Compound 13 | 411.3 | 14.7 | 13.4 to 16.1 | 35.8 |
| Clozapine | 326.8 | 13.8 | 7.57 to 25.3 | 42.4 |
| Haloperidol | 375.9 | 0.0293 | 0.0112 to 0.0768 | 0.0779 |
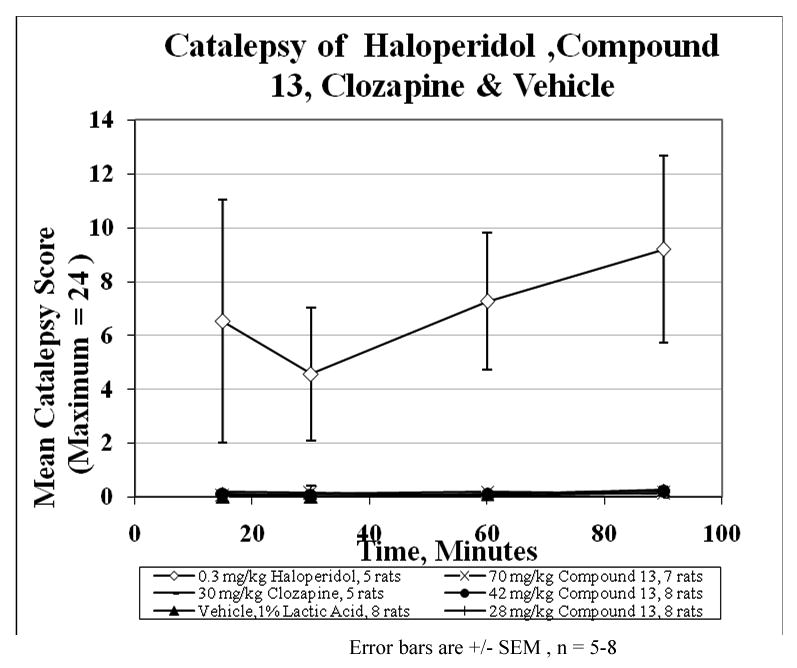
Fig 4 depicts plots of mean catalepsy scores for compound 13 with clozapine and haloperidol as controls. The vehicle served as a negative control in the study. The ED50 value for each compound was used as the basis for the dose used for the first set of comparative evaluations. It is clear that haloperidol showed the highest propensity to induce catalepsy while clozapine and compound 13 had no significant difference between them. The extent of the absence of catalepsy was evaluated by increasing the dose of compound 13 up to 5 times the ED50 value at which point it was impossible to retain the drug in solution for intraperitoneal delivery. At 5× the ED50 value, there was no appreciable cataleptogenic activity observed. Taken together, the combined receptor binding profile and the absence of catalepsy suggest that compound 13, a butyrophenone analogue which does not induce catalepsy up to 5× its ED50 value may serve as an atypical antipsychotic agent.
Fig 4.
Experimental
Melting points were determined on a Gallenkamp (UK) apparatus and are uncorrected. NMR spectra were obtained on a Varian 300 MHz Mercury Spectrometer. Elemental analyses were carried out by Atlantic Microlab, Inc., Norcross, GA and are within 0.4% of theory unless otherwise noted. Flash chromatography was performed with Davisil grade 634 silica gel. N,N-Dimethylformamide was distilled from CaSO4 and stored over 4 Ǻ molecular sieves. 4-Chloro-4′-fluorobutyrophenone was obtained from Sigma-Aldrich but was purified by distillation under reduced pressure to a colorless liquid prior to use. Other starting materials were used without further purification.
Synthesis of compound 10
The synthesis of compound 10 followed the methods previously reported by Cignarella et al.15 and Thompson et. al.16 as depicted in Scheme 1.
Cis-2,5-dicarboethoxy-1-[4-chlorophenyl]pyrrolidine, 17
To a solution of (2S, 5R)-diethyl 2, 5-dibromo-hexanedioate (16) (10g, 27.8 mmol) in anhydrous DME (80 mL), was added 4-chloroaniline (14.2g, 111.1 mmol) and KI (1g, 6.02 mmol). The resulting mixture was refluxed under N2 with stirring for 16 h. and allowed to cool to room temperature. Excess DME was removed under vacuum and the residue was triturated in Et2O (100 mL) to yield a solid. The solid obtained was filtered, washed with Et2O and the organic phase was treated with 100 mL of ice cold 5 N HCl and H2O, dried over MgSO4 and solvent removed in vacuo. The crude compound was purified by chromatography on silica gel (8:2 hexane :EtOAc) to give compound 17 (8.5g, 93.5%) as a mixture of trans-2,5-dicarboethoxy-1-[4-chlorophenyl]pyrrolidine and cis-2,5-dicarboethoxy-1-[4-chlorophenyl]pyrrolidine.
2-Benzylcarbamyl-5-carboethoxy-1-[4-chlorophenyl]pyrrolidine, 18
A mixture of cis- and trans-2,5-dicarboethoxy-1-[4-chlorophenyl]pyrolidine, 17 (8.5g, 26 mmol) was dissolved in 10 mL of o-xylene. Benzylamine (3.61 g, 33.74 mmol) was added to the reaction mixture and refluxed for 18 h. under N2, cooled and solvent evaporated. The residue was subjected to column chromatography (silica gel) eluting with (1:1) hexane: EtOAc to give compound 18 (4.4 g, 39.8 %). 1H NMR (300 MHz, CDCl3) δ 1.19 (t, J = 7.13 Hz, 3H), 2.01-2.45 (m, 4H), 4.11-4.37 (m, 4H), 4.40-4.46 (dd, J = 6.0, 3.2 Hz, 2H), 6.36-6.44 (m, 2H), 7.09-7.24 (m, 7H), 8.52 (m, 1H).
3-Benzyl-8-[4-chlorophenyl]-3,8-diazabicyclo[3.2.1]octane-2,4-dione, 19
The solution of compound 18 (4.0 g, 10.35mmol) in 30 mL of 1N NaOH (75% aq. ethanol) was stirred at room temperature for 2 h. The reaction was quenched with HCl gas to the end point of phenolphthalein and the solid was filtered. The solvent was removed in vacuo and the solid material was re-dissolved in absolute alcohol. The solution was partially removed in vacuo and the residue was treated with Ac2O (5 mL) and NaOAc (1 g). After refluxing overnight under N2, the excess Ac2O was removed and the reaction mixture was quenched with aq. Na2CO3, extracted with CH2Cl2 (3 × 100 mL) and dried over anhydrous MgSO4. The solvent was removed in vacuo and separated on a silica gel column (7:3 hexane : EtOAc) to give compound 19 (2.56 g, 56.7%, white solid, mp 163-164 °C). 1H NMR (270 MHz, CDCl3) δ 2.04-2.12 (m, 2H), 2.46-2.55 (m, 2H), 4.63-4.66 (dd, J = 5.1, 2.7 Hz, 2H), 4.70 (s, 2H), 6.70-6.82 (m, 4H), 7.03-7.12 (m, 5H).
3-Benzyl-8-phenyl-3, 8-diazabicyclo[3.2.1]octane, 20
To a solution of compound 19 (0.58 g, 1.70 mmol) in anhydrous THF (3 mL), was added slowly, 1 M LiAlH4 (10.24 mL, 10.24 mmol) and refluxed for 2 days under N2. The reaction was quenched in ice bath with 10% NaOH (20 mL) and extracted with CH2Cl2. The organic phase was separated, dried over MgSO4 and removed in vacuo. The residue was subjected to column chromatography (silica gel, 9:1 hexane:ethyl acetate) to give compound 20 (0.4 g, 84.6%, white crystals, mp 115-117 °C). 1H NMR (270 MHz, CDCl3) δ 1.87-2.08 (m, 4H), 2.51-2.52 (d, J = 2.0 Hz, 4H), 3.41 (s, 2H), 4.13-4.15 (m, 2H), 6.65-6.78 (m, 3H), 7.15-7.30 (m, 7H).
3-Carboethoxy-8-phenyl-3, 8-diazabicyclo[3.2.1]octane, 21
To a solution of compound 20 (0.77 g, 2.8 mmol) in anhydrous toluene (15 mL), was added ethyl chloroformate (1.6 mL, 16.6 mmol) and refluxed for 16 h under N2. The resulting solution was allowed to cool to RT and excess solvent was removed under vacuum. The resulting residue was subjected to column chromatography (silica gel, 8:2 hexane:ethyl acetate) to give compound 21 (0.65 g; 90.5%) as white crystals, mp 124.4 °C. 1H NMR (270 MHz, CDCl3) δ 1.22 (t, J = 8.1 Hz, 3H), 1.79-1.84 (m, 2H), 1.98-2.04 (m, 2H), 3.29 (t, J = 13.5 Hz, 2H), 3.60-3.76 (dd, J = 13.5 Hz, 2H), 4.07-4.18 (m, 4H), 6.70-6.80 (m, 3H), 7.18-7.26 (m, 2H).
8-Phenyl-3,8-diazabicyclo[3.2.1]octane, 22
To a solution of compound 21 (0.73 g, 2.48 mmol) in EtOH (5 mL), was added KOH [3 g in H2O (3 mL)], EtOH (25 mL) followed by NH2NH2 (3 mL). The resulting mixture was refluxed for 16 h. Excess alcohol was removed and the residue was extracted with EtOAc (4 × 50 mL) and H2O (20 mL) to give compound 22 (0.4 g 86%). 1H NMR (270 MHz, CDCl3) δ 1.88-2.07 (m, 4H), 2.51-2.57 (dd, J = 13.5, 2.7 Hz, 2H), 3.19-3.24 (d, J = 13.5 Hz, 2H), 4.09 (br s, 2H), 6.67-6.78 (m, 3H), 7.17-7.24 (m, 2H).
8-Phenyl-3,8-diazabicyclo[3.2.1]octan-1-yl-1-(4-fluorophenyl)butan-1-one, 10
To a mixture of compound 22 (0.56 g, 2.96 mmol), K2CO3 (1.4g, 10 mmol) and KI (0.3 g) in DME (10 mL), was added 4-chloro-4-fluorobutyrophenone (2.0 g, 10.01 mmol) and stirred. The mixture was refluxed for 16 h under N2 atmosphere and the reaction mixture was allowed to cool to room temperature. Excess DME was removed in vacuo and the residue was extracted with CH2Cl2 (3 × 50 mL) followed by brine (50 mL). The organic phase was collected and dried over anhydrous MgSO4 and the solvent evaporated. The resulting crude product was subjected to column chromatography (silica gel, 8:2 EtOAc:MeOH) to give an oily residue (1.21 g, 68.2 %) and subsequently converted to an oxalate salt as a white solid, mp 194.7-195.5 °C. 1H-NMR (270 MHz, CDCl3) δ 1.89-1.99 (m, 6H), 2.39 (t, J = 6.8, 2H), 2.47-2.65 (m, 4H), 3.06 (t, J = 7.3, 2H), 4.21 (m, 2H), 6.73-6.85 (m, 3H), 7.17-7.33 (m, 4H), 8.05-8.10 (m, 2H). Anal. (C24 H26CFN2O5:H2O) calcd. C 64.62, H 6.19, N 6.28. Found C 64.66, H 6.11, N 6.14.
4-[5-(4-Chloro-phenyl)-2,5-diaza-bicyclo[2.2.1]hept-2-yl]-1-(4-fluoro-phenyl)-butan-1-one, 11
A mixture of compound 23 (0.5g, 2.4 mmol), 4-chloro-4-fluorobutyrophenone (1.9g, 9.6 mmol) KI (50mg) and K2CO3 (138 mg) in DME (10 ml) was refluxed in an atmosphere of N2 for 60 hours. Excess DME was removed under vacuum and residue was partitioned between CH2Cl2 (3 × 75 ml) and brine (50 ml). The organic phase was pooled, dried over anhydrous Na2SO4 and the solvent removed in vacuo to afford a residue as the crude product. The crude product was purified by column chromatography using 0 - 20% MeOH in EtOAc to yield compound 11 (755mg, 84.5%), which was subsequently converted to the oxalic acid salt as a white crystal, mp 116.4 -116.8 °C. 1H-NMR (300 MHz, DMSOd6) δ 1.86-1.93 (m, 4H), 2.09-2.21 (AB system, J=10.7, 2H), 3.00-3.37 (m, 6H), 3.11 (t, J=6.7, 2H), 3,37-3.58 (AB system, J=9.3, 2H). 4.42 (m, 1H), 4.60 (m, 1H), 6.65-6.68 (Part of AAXX, 2H), 7.32-7.38 (Part of AAXX, 2H), 8.00-8.04 (Part of AAXX, 2H). Anal. C21H22ClFN2O.1.5C2H2O4 calcd: C 56.73, H 4.96, N 5.52; Found: C 56.49, H 4.91, N 5.42
4-[4-(2-Pyrimidine)piperazin-1-yl]-1-(4-fluorophenyl)butan-1-one, 12
A mixture of 2-(piperazin-1-yl)pyrimidine dihydrochloride (0.5g, 2.1 mmol), 4-chloro-4′-fluorobutyrophenone (1.3 g, 6.3 mmol), KI (25 mg), K2CO3 (1.5 g, 10.9 mmol), in DME (10 mL) was refluxed with stirring under N2 for 12 h. After cooling to room temperature, the mixture was diluted with EtOAc (200 mL) and washed with H2O. The organic layer was dried (Na2SO4) and filtered. The filtrate was concentrated in vacuo to dryness and the residue was purified by chromatography on silica gel to give compound 12 (563 mg, 78%), mp 109 -110 °C. 1H NMR (CDCl3): 8.29 (2H, d, J = 4.5 Hz), 8.02 (2H, dd, J = 5.1, 8.7 Hz), 7.13 (2H, t, J = 8.7 Hz), 6.47 (1H, t, J = 4.5 Hz), 3.76 (4H, m), 3.01 (2H, t, J = 7.5 Hz), 2.46 (6H, m), 1.99 (2H, m). Calcd for C18H21FN4O: C, 65.84; H, 6.45; N, 17.06. Found: C, 65.91; H, 6.51; N 16.86.
4-Chlorophenylboronic Acid, 26
To a solution of 4-bromochlorobenzene, 25 (4 g, 20.9 mmol) in THF (80 mL) was added n-BuLi (22 mL, 1.6 M) at −78 °C under N2 and stirred for 1h at the same temperature. The reaction mixture was treated with B(OMe)3 (398.8 ml, 78.3 mmol) at −78 °C, allowed to warm up to room temperature and then stirred overnight. The pH of the mixture was then adjusted with a solution of NaHCO3 to pH 6.5 and extracted with CH2Cl2 (3 × 150 mL). The pooled organic phase was dried (MgSO4), the solvent was removed in vacuo and the crude product was column chromatographed (silica gel, 7:3, hexane:EtOAc) to yield compound 26 (4-chlorophenyl-boronic acid) (2 g, 61 %) as a white solid. 1H-NMR (270 MHz, CD3OD) δ 7.30-7.77 (m, 4H).
1-t-Butyl homopiperazine carboxylate, 28
To a solution of homopiperazine, 27 (10g, 0.1 mol) in H2O (104 mL) and t-BuOH (119 mL) containing 2.5 N NaOH (16 mL) was added di-tert-butyl dicarbonate (8.55 g, 0.039 mol) at 0 °C for 40 min. The resulting mixture was stirred for 1 h at room temperature and the solution was concentrated in vacuo. The crude material was extracted with CH2Cl2 (3 × 100 mL), dried over MgSO4 and the solvent evaporated. The crude product was subjected to column chromatography (silica gel, 9:1:1 MeOH : CH2Cl2 : NH4OH to give 1-t-Butyl homopiperazine carboxylate, 28 (5 g, 64 %). 1H-NMR (270 MHz, CD3OD) δ 1.46 (s, 9H), 1.79 (q, J=5.9, 2H), 2.78-2.87 (m, 4H), 3.41-3.49 (m, 4H).
1-t-Butyl-4-(4-chlorophenyl)homopiperazine carboxylate, 29
The mixture of 4-chlorophenyl-boronic Acid, 26 (1.65 g, 10.54 mmol), 28 (1.05g, 5.27 mmol), Cu(OAc)2 (1.43 g, 7.87 mmol), triethylamine (0.726 mL) and molecular sieves (4 g) in CH2Cl2 (50 mL) was stirred at room temperature under air. The reaction was quenched with a 4 ml of NH4OH in 4 mL of MeOH. The mixture was filtered through celite and purified by column chromatography using hexane:EtOAc (7:3) as eluent on silica gel to give compound 29 (1.2 g, 73%) 1H-NMR (300 MHz, CDCl3) δ 1.35 (s, 4H), 1.42 (s, 5H), 1.91-1.98 (m, 2H), 3.18 (t, 1H, J = 6.2 Hz), 3.29 (t, 1H, J = 6), 3.48-3.7 (m, 6H), 6.58 (d, 2H, J = 8.7 Hz), 7.12 (m, 2H, J = 9 Hz)
1-(4-Chlorophenyl)homopiperazine, 30
Trifluoroacetic acid (5 mL) was slowly added in a drop-wise manner to a solution of compound 29 in CH2Cl2 (5 mL) at ambient temperature and the resulting mixture was allowed to stir overnight. The reaction was quenched with aq. NaOH and extracted with EtOAc (3 × 100 mL), washed with NaHCO3 solution (50 mL) and brine (50 mL). The organic phase was collected and dried over MgSO4 and evaporated in vacuo. The crude product was subjected to column chromatography (silica gel, 9:1:1 MeOH: CH2Cl2 :NH4OH to yield 1-(4-chlorophenyl)homopiperazine, 30 (0.720 g, 88%). 1H-NMR (300 MHz, CDCl3) δ 1.92-1.98 (m, 2H), 2.86 (t, 2H, J = 5.7), 3.04 (t, J = 5.4 Hz), 3.53 (q, 4H, J = 6.4 Hz), 5.50 (br s, 1H), 6.58 (d, 2H, J = 9), 7.14 (d, 2H, J = 9.2 Hz).
4-[4-(4-Chlorophenyl)-1,4-diazepan-1-yl]-1-(4-fluorophenyl)butan-1-one, 13
A mixture of compound 30 (709 mg, 3.4 mmol) in dry DME (15 mL) and K2CO3 (1.9 g, 13.9 mmol) was heated for 20 min. after which 4-chloro-4-flourobutyrophenone (2.8 g, 13.9 mmol) was added followed by KI (0.3 g). The reaction mixture was refluxed for 18 h under an atmosphere of N2. Excess DME was removed in vacuo and the residue was extracted with CH2Cl2 (3 × 100 mL) and brine (50 mL). The organic phase was collected and dried over MgSO4 and solvent removed under vacuum. The crude product was subjected to column chromatography (silica gel, 9:1 EtOAc:MeOH) to yield compound 13 (1.19 g, 87%) as an oil. 1H-NMR (300 MHz, CDCl3) δ 1.84-1.95 (m, 4H), 2.53-2.62 (m, 4H), 2.75 (t, 2H, J = 5.1 Hz), 2.93 (t, 2H, J = 7.2 Hz) 3.39-3.48 (m, 4H), 6.58 (d, 2H, J = 9.6 Hz), 7.05-7.13 (m, 4H), 7.96-7.92 (dd, 2H, J = 9, 5.1 Hz). Subsequently, compound 13 was converted to the oxalate salt, crystallized from MeOH/Et2O as a white solid, mp 146.6-146.4 °C. Anal. (C23H26ClFN2O2) calcd. C 59.42, H 5.64, N 6.03 found C 59.27, H 5.70, N 6.05.
Alternative Method for Compound 13
A mixture of homopiperazine (2 g, 20 mmol), CuI (380 mg, 2 mmol), K2CO3 (5.5 g, 40 mmol), ethylene glycol (2.4 g, 40 mmol), 1-chloro-4-iodobenzene (4.76 g, 20 mmol) in iPrOH (50 mL) was refluxed for 12 h under N2. The mixture was diluted with EtOAc (500 mL) and washed with water. The organic phase was dried (Na2SO4), and filtered. The filtrate was concentrated in vacuo to dryness followed by chromatography on silica gel (EtOAc then MeOH/EtOAc =10/90) to afford 1-(4-chlorophenyl)-1,4-diazepane (3.8 g, 90%).
A mixture of 1-(4-chlorophenyl)-1,4-diazepane (30) (540 mg, 2.57 mmol), 4-chloro-4-fluorobutyrophenone (2 g, 10.28 mmol), KI (25 mg), K2CO3 (880 mg, 6.38 mmol) in DME (20 mL) was refluxed under N2 for 12 h. The mixture was diluted with EtOAc (500 mL) and washed with H2O. The organic phase was dried (Na2SO4), and filtered. The filtrate was concentrated in vacuo to dryness followed by column chromatography on silica gel (pure EtOAc) to afford 4-[4-(4-chlorophenyl)-1, 4-diazepan-1-yl]-1-(4-fluorophenyl)butan-1-one, 13. The product was converted to the salt form using ethereal HCl and then crystallized from MeOH/Et2O to give the pure HCl salt (715 mg, 68%) mp 165-166 °C, 1H NMR (DMSO): δ 10.48 (1H, brs), 8.04 (2H, dd, J = 5.7, 9.3 Hz), 7.35 (2H, t, J = 8.7 Hz), 7.19 (2H, d, J = 9.3 Hz), 7.76 (2H, d, J = 9.3 Hz), 3.78 (2H, m), 3.42 (4H, m), 3.15 (6H, m), 2.38 (1H, m), 2.15 (1H, m), 2.03 (2H, m). Calcd for C21H25Cl2FN2O: C, 61.32; H, 6.13; N, 6.81. Found: C, 61.20; H, 5.98; N, 6.78.
9-Methyl-3,9-diazabicyclo[4.2.1]nonan-4-one, 32
Using the method reported by Michaels and Zaugg,25 conc. H2SO4 (37.5 mL) was added drop wise at −20 °C, to a mixture of tropinone, 31 (16.7g, 32 mmol) in anhydrous CHCl3 (120 mL). This was followed by a slow addition of sodium azide (15.6 g) for 30 minutes while maintaining the temperature below −5 °C. After completing the addition, the reaction mixture was stirred at room temperature for 15 min, and then heated at 50 °C overnight under nitrogen. The reaction mixture was then neutralized with NaHCO3, extracted with CH2Cl2, and the organic layer was collected, dried over Na2SO4 and evaporated in vacuo to afford a light yellowish substance as 32 (13.8 g, 78 %); MS: 155 (M+1)+, IR (KBr): 3190 (-NH), 1620 (C=O).
3,9-Diazabicyclo[4.2.1]nonane-3,9-dicarboxylic acid 3-tert-butyl ester 9-ethyl ester, 35
To a solution of compound 32 (15.2g, 30 mmol) in anhydrous THF (100 mL), was added very slowly under nitrogen powdered LAH (14.1 g, 8 eq). The mixture was allowed to stir at room temperature overnight before being quenched with MeOH, followed by H2O, filtered, washed twice with CH2Cl2, extracted, dried over Na2SO4 and evaporated to yield 9-Methyl-3,9-diazabicyclo[4.2.1]nonane, compound 33 (11 g, 80 %). TLC analysis showed a single spot and was used as such without characterization. Compound 33 (12.0 g, 24 mmol) was dissolved in 15 mL of t-BuOH and added a solution of Boc-anhydride (5.8g, 28mmol) in t-BuOH (10 mL) for 5 min. and the reaction mixture was stirred overnight. The reaction was then neutralized with NaHCO3, extracted with CH2Cl2, dried over Na2SO4 and evaporated in vacuo to yield 9-Methyl-3,9-diazabicyclo[4.2.1]nonane-3-carboxylic acid t-butyl ester, 34 (13.8 g, 67 %). Compound 34 (5.8 g, 32 mmol) was dissolved in anhydrous toluene (20 mL) and ethyl chloroformate (3.6 g, 33.3 mmol) was slowly added in a drop-wise manner. The reaction mixture was refluxed overnight and the solvent was evaporated as an azeotrope with alcohol. The resulting product, 3,9-diazabicyclo[4.2.1]nonane-3,9-dicarboxylic acid 3-tert-butyl ester 9-ethyl ester, 35 was dried and used as such without further purification for the next reaction.
3-(4-Chlorophenyl)-3,9-diazabicyclo[4.2.1]nonane-9-dicarboxylic acid ethyl ester, 37
Compound 35 (5.5 g, 18.4 mmol) was dissolved in CF3COOH (3 mL) and stirred for 4h at room temperature. The reaction mixture was then neutralized with NaHCO3, extracted with CH2Cl2, dried over Na2SO4 and evaporated to get a light yellowish oil, 3,9-diazabicyclo[4.2.1]nonane-9-carboxylic acid ethyl ester, 36 (2.85 g, 78 %). Compound 36 (5 g, 25.6 mmol), 4-chlorophenylboronic acid (5.6 g, 35.8 mmol), Cu(OAc)2 (5.7g, 31.4 mmol) and Et3N (2 mL) in CH2Cl2 (50 mL) with molecular sieves (2 g) was stirred in open air for 48 h. The reaction mixture was then quenched with methanolic NH3 solution, filtered over celite, extracted with CH2Cl2 (100 mL), dried and solvent evaporated under vacuum. The crude residue was subjected to column chromatography (silica gel, 4:1 CH2Cl2:MeOH) to give a yellowish compound 37 (4.5 g, 58 %). 1H NMR (300MHz, CDCl3): δ 1.20-1.26 (m, 3H), 1.39-1.6 (m, 2H), 1.75-1.95 (m, 2H), 2.0-2.29 (m, 2H), 3.0-3.30 (m, 2H), 3.54-3.68 (m, 1H), 3.70-3.8 (m, 1H), 4.05-4.20 (m, 2H), 426-4.43 (m, 2H), 6.62-6.66 (dd, 2H, J = 7.8, 3.3 Hz), 7.06-7.12 (m, 2H).
3-(4-Chlorophenyl)-3,9-diazabicyclo[4.2.1]nonane, 38
Compound 37 (2.2 g, 7.12 mmol), 50 % aq. KOH (2 ml) in ethylene glycol (2 mL) was heated at 90 °C overnight. The reaction mixture was then extracted with CH2Cl2 (2 × 80 mL), dried over Na2SO4 and evaporated under vacuum. The crude product was subjected to column chromatography (silica gel, 4:2 CH2Cl2:MeOH) to yield compound 38 (0.99 g, 59 %). 1H NMR (300MHz, CDCl3): δ 1.32-1.76 (m, 4H), 1.90-2.25 (m, 4H), 3.15-3.35 (m, 4H), 6.63 (d, 2H, J = 9 Hz), 7.09 (d, 2H, J = 9 Hz).
4-[3-(4-Chlorophenyl)-3,9-diazabicyclo[4.2.1]non-9-yl]-1-(4-fluorophenyl)butan-1-one, 14
A mixture of compound 38 (0.45g, 1.9 mmol) and K2CO3 (1g, 7.6 mmol) in dry DME (15 mL) was heated at 80 °C for 20 minutes and 4-chloro-4-fluorobutyrophenone (2.2 ml, 7.6 mmol) was added in a drop-wise manner and refluxed overnight under nitrogen. Excess DME was removed in vacuo and the residue was extracted with CH2Cl2 (3 × 60 ml). The pooled organic phase was collected and dried over Na2SO4 and evaporated under vacuum. The crude product was subjected to column chromatography (silica gel, 4:1 CH2Cl2-EtOAc) to yield compound 4 (0.55 g, 68.2 %) as an oil which was converted into the oxalate salt. (White crystalline solid, mp:182-184 °C). 1H NMR (300 MHz, CDCl3): δ 1.20 -1.66 (m, 4H), 1.80-2.00 (m, 4H), 2.50-2.75 (m, 2H), 2.90-3.05 (m, 2H), 3.35-3.60 (m, 6H), 6.58 (m, 2H), 7.05-7.15 (m, 4H), 7.91-7.97 (m, 2H), Anal. Calcd. for C25H26N2O5ClF: 0.25H2O: C, 60.61; H, 5.80; N, 5.65. Found C, 60.53; H, 5.75, N, 5.53.
Biology
Receptor Binding Studies
Binding affinities reported in Tables 1 – 3 were conducted by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP) unless otherwise stated. Details of the methods and radioligands used for the binding assays were previously reported.26
Apomorphine Induced Climbing Stereotypy
A modified climbing test by Needham et al. was used27 and provides the extent a potential antipsychotic agent inhibits the effect of apomorphine induced stereotypy. Swiss Webster male mice (∼30 g) in groups of 5 were administered an intraperitoneal (IP) injection of 0.3 mL of vehicle (filtered 1% lactic acid) or increasing doses of the dopamine antagonists haloperidol (0.001, 0.01, 0.02, 0.05, 0.1, 0.2 mg/kg), compound 13 (12, 15, 17, 20, 25, 50, 80 mg/kg), and clozapine (3, 10, 20, 30, 60 mg/kg). Animals were then challenged at 30 minutes post injection with 1.5- 1.6 mg/kg of the agonist apomorphine in 0.9% NaCl + 0.1% ascorbic acid, placed in cylindrical wire cages (12 cm in diameter, 14 cm in height), and observed for climbing behavior at 10 and 20 minutes post dose. Climbing behavior was assessed as follows: 3-4 paws on the cage floor = 0 score; 2 and 3 paws on the cage = 1 score; 4 paws on the cage = 2 score. Scores were expressed as mean % climbing inhibition, and plotted in figures 1-3. ED50s in Table 4 were calculated using Graph Pad Prism non-linear regression software with sigmoidal dose-response, variable slope curve-fitting.
Bar Test for Catalepsy
The bar test used by Hoffman and Donovan28 was modified for our use. Male Sprague-Dawley rats (88-207 g) in groups of 5-8 were dosed by IP injection with 1-23 mL/kg of vehicle (filtered 1% lactic acid), haloperidol (0.3 mg/kg), compound 13 (28, 42, 70 mg/kg), or clozapine (30 mg/kg). Catalepsy severity was assessed at time points (15, 30, 45, 60, and 90 min) post injection, by scoring how long the rat maintained both forepaws motionless on a horizontal metal bar (1.1 cm in diameter, 10 cm above the bench top in a box) up to a maximum of 120 seconds. This was repeated 2 more times and the average (n=3 tries) seconds recorded for each rat. The mean of all the average seconds for one dose for one time point (n=5-8 rats) was divided by 5 and a mean catalepsy score assigned and plotted in figure 4 with a maximum possible score of 24.
Fig 2.

Scheme 6a.

a Reagents: (i) H2SO4, NaN3; (ii) LiAlH4, THF; (iii) t-BuOH, (t-BuOCO)2O; (iv) ClCO2Et, Toluene; (v) TFA (vi) 4-Chlorophenylboronic Acid, Cu(OAc)2, Et3N, CH2Cl2, Mol sieves (4Å), RT, 24 – 48 hr; (vii) KOH, Ethylene glycol; (viii) 4-Chloro-4′-fluorobutyrophenone, KI, K2CO3, DME.
Acknowledgments
We gratefully acknowledge the financial support of the National Institute of General Medical Studies (NIGMS) for MBRS grant # GM 08111, NIMH Psychoactive Drug Screening Program, RCMI grant number G12 RR 03020 from NCRR, and a Title III Grant to Florida A&M University. The authors also acknowledge the original binding studies conducted by A. W. Schmidt at Pfizer Global Research. This work was supported in part by the Pharmaceutical Research Center NIH/NCRR 1 C06-RR12512-01 grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crilly J. The history of clozapine and its emergence in the US market: a review and analysis. Hist Psychiatry. 2007;18:39–60. doi: 10.1177/0957154X07070335. [DOI] [PubMed] [Google Scholar]
- 2.a) Peabody CA, Brody D, Warner MD. Tardive dyskinesia after low-dose haloperidol. Biol Psychiatry. 1987;22:111–112. doi: 10.1016/0006-3223(87)90138-7. [DOI] [PubMed] [Google Scholar]; b) Riddle MA, Hardin MT, Towbin KE, Leckman JF, Cohen DJ. Tardive dyskinesia following haloperidol treatment in Tourette's syndrome. Arch Gen Psychiatry. 1987;44:98–99. doi: 10.1001/archpsyc.1987.01800130110023. [DOI] [PubMed] [Google Scholar]; c) 64 Chirita V, Boisteanu P, Pirozynski T. Tardive dyskinesia syndrome in neuroleptic therapy. General considerations and clinical observations on forty cases investigated. Rev Med Chir Soc Med Nat Iasi. 1987;91:53. [PubMed] [Google Scholar]; d) Munyon WH, Salo R, Briones DF. Cytotoxic effects of neuroleptic drugs. Psychopharmacol (Berl) 1987;91:182–188. doi: 10.1007/BF00217059. [DOI] [PubMed] [Google Scholar]; e) Petty LK, Spar CJ. Haloperidol-induced tardive dyskinesia in a 10-year-old girl. Am J Psychiatry. 1980;137:745–746. doi: 10.1176/ajp.137.6.745. [DOI] [PubMed] [Google Scholar]; f) Gerlach J, Simmelsgaard H. Tardive dyskinesia during and following treatment with haloperidol, haloperidol + biperiden, thioridazine, and clozapine. Psychopharmacol (Berl) 1978;59:105–112. doi: 10.1007/BF00427742. [DOI] [PubMed] [Google Scholar]
- 3.Ablordeppey SY, Borne RF. Detection of a quaternary pyridinium metabolite in the liver of a haloperidol-treated rat. Pharmacol Biochem Behavior. 1993;46:739–744. doi: 10.1016/0091-3057(93)90571-a. [DOI] [PubMed] [Google Scholar]
- 4.a) Subramanyam B, Rollema H, Woolf T, Castagnoli N., Jr Identification of a potentially neurotoxic pyridinium metabolite of haloperidol in rats. Biochem Biophys Res Commun. 1990;166:238–244. doi: 10.1016/0006-291x(90)91936-m. [DOI] [PubMed] [Google Scholar]; b) Subramanyam B, Pond SM, Eyles DW, Whiteford HA, Fouda HG, Castagnoli N., Jr Identification of potentially neurotoxic pyridinium metabolite in the urine of schizophrenic patients treated with haloperidol. Biochem Biophys Res Commun. 1991;181:573. doi: 10.1016/0006-291x(91)91228-5. [DOI] [PubMed] [Google Scholar]; c) Subramanyam B, Woolf T, Castagnoli N., Jr Studies on the in-vitro conversion of haloperidol to a potentially neurotoxic pyridinium metabolite. Chem Res Toxicol. 1991;4:123–128. doi: 10.1021/tx00019a017. [DOI] [PubMed] [Google Scholar]
- 5.a) Fang J, Gorrod JW. Dehydration is the first step in the bioactivation of haloperidol to its pyridinium metabolite. Toxicol Lett. 1991;59:117–123. doi: 10.1016/0378-4274(91)90062-b. [DOI] [PubMed] [Google Scholar]; b) Eyles DW, McLennan HR, Jones A, McGrath JJ, Stedman TJ, Pond SM. Quantitative analysis of two pyridinium metabolites of haloperidol in patients with schizophrenia. Clin Pharmacol Ther. 1994;56:512–520. doi: 10.1038/clpt.1994.172. [DOI] [PubMed] [Google Scholar]; c) Eyles DW, Avent KM, Stedman TJ, Pond SM. Two pyridinium metabolites of haloperidol are present in the brain of patients at post-mortem. Life Sci. 1997;60:529–534. doi: 10.1016/s0024-3205(96)00656-x. [DOI] [PubMed] [Google Scholar]; d) Fang J, Zuo D, Yu PH. Comparison of cytotoxicity of a quaternary pyridinium metabolite of haloperidol with neurotoxin N-methyl-4-phenylpyridinium towards cultured dopaminergic neuroblastoma cells. Psychopharmacology. 1995;121:373–378. doi: 10.1007/BF02246077. [DOI] [PubMed] [Google Scholar]; e) Rollema H, Skolnik M, D'Engelbronner J, Igarashi K, Usuki E, Castagnoli N., Jr MPP+-like neurotoxicity of a pyridinium metabolite derived from haloperidol: in vivo microdialysis and in vitro mitochondrial studies. J Pharmacol Exp Ther. 1994;268:380–387. [PubMed] [Google Scholar]
- 6.a) Ablordeppey SY, Lyles-Eggleston M, Bricker B, Zhang W, Zhu XY, Goodman C, Roth BL. Evaluation of the Eutomer of 4-{3-(4-chlorophenyl)-3-hydroxypyrrolidin-1-yl}-1-(4-fluorophenyl) butan-1-one, {(+)-SYA 09}, a Pyrrolidine Analog of haloperidol. Bioorg Med Chem Lett. 2006;16:3219–3223. doi: 10.1016/j.bmcl.2006.03.057. [DOI] [PubMed] [Google Scholar]; b) Ablordeppey SY, Borne RF. Design and Synthesis of Novel analogues of haloperidol incapable of forming MPP+-like metabolites. Med Chem Res. 1993;3:459–467. doi: 10.1021/jm0301033. [DOI] [PubMed] [Google Scholar]
- 7.Lyles-Eggleston M, Altundas R, Xia J, Sikazwe DMN, Fan P, Yang Q, Li S, Zhang W, Zhu X, Schmidt AW, Vanase-Frawley M, Shrihkande A, Villalobos A, Ablordeppey SY. Design, Synthesis, and Evaluation of Metabolism-Based Analogues of haloperidol Incapable of Forming MPP+-like Species. J Med Chem. 2004;47:497–508. doi: 10.1021/jm0301033. [DOI] [PubMed] [Google Scholar]
- 8.Sikazwe DMN, Li S, Mardenborough L, Cody V, Roth BL, Ablordeppey SY. Haloperidol: towards further understanding of the structural contributions of its pharmacophoric elements at D2-like receptors. Bioorg Med Chem Lett. 2004;14:5739–5742. doi: 10.1016/j.bmcl.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 9.Sikazwe DMN, Li S, Lyles-Eggleston M, Ablordeppey SY. The Acute EPS of haloperidol May be Unrelated to Its Metabolic Transformation to BCPP+ Bioorg Med Chem Lett. 2003;13:3779–3782. doi: 10.1016/j.bmcl.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 10.Tacke R, Popp F, Müller B, Theis B, Burschka C, Hamacher A, Kassack MU, Schepmann D, Wünsch B, Jurva U, Wellner E. Sila-Haloperidol, a Silicon Analogue of the Dopamine (D(2)) Receptor Antagonist haloperidol: Synthesis, Pharmacological Properties, and Metabolic Fate. ChemMedChem. 2008;3:152–164. doi: 10.1002/cmdc.200700205. [DOI] [PubMed] [Google Scholar]
- 11.a) McIntyre RS, McCann SM, Kennedy SH. Antipsychotic metabolic effects: weight gain, diabetes mellitus, and lipid abnormalities. Can J Psychiatry. 2001;46:273–81. doi: 10.1177/070674370104600308. [DOI] [PubMed] [Google Scholar]; b) Taylor DM, McAskill R. Atypical antipsychotics and weight gain--a systematic review. Acta Psychiatr Scand. 2000;101:416–32. doi: 10.1034/j.1600-0447.2000.101006416.x. [DOI] [PubMed] [Google Scholar]; c) Wirshing DA, Spellberg BJ, Erhart SM, Marder SR, Wirshing WC. Novel antipsychotics and new onset diabetes. Biol Psychiatry. 1998;44:778–83. doi: 10.1016/s0006-3223(98)00100-0. [DOI] [PubMed] [Google Scholar]
- 12.a) Seeger TF, Seymour PA, Schmidt AW, Zorn SH, Schulz DW, Lebel LA, McLean S, Guanowsky V, Howard HR, Lowe JA, Heym J. Ziprasidone (CP-88,059): a new antipsychotic with combined dopamine and serotonin receptor antagonist activity. J Pharmacol Exp Ther. 1995;275:101–113. [PubMed] [Google Scholar]; b) Schmidt AW, Lebel LA, Howard HR, Jr, Zorn SH. Ziprasidone: a novel antipsychotic agent with a unique human receptor binding profile. Eur J Pharmacol. 2001;425:197–201. doi: 10.1016/s0014-2999(01)01188-8. [DOI] [PubMed] [Google Scholar]; c) McCreary AC, Glennon JC, Ashby CR, Jr, Meltzer HY, Li Z, Reinders JH, Hesselink MB, Long SK, Herremans AH, van Stuivenberg H, Feenstra RW, Kruse CG. SLV313 (1-(2,3-dihydro-benzo[1,4]dioxin-5-yl)-4- [5-(4-fluoro-phenyl)-pyridin-3-ylmethyl]-piperazine monohydro-chloride): a novel dopamine D2 receptor antagonist and 5-HT1A receptor agonist potential antipsychotic drug. Neuropsychopharmacol. 2007;32:78–94. doi: 10.1038/sj.npp.1301098. [DOI] [PubMed] [Google Scholar]
- 13.Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet. 2005;20:368–78. doi: 10.2133/dmpk.20.368. [DOI] [PubMed] [Google Scholar]
- 14.Sanberg PR, Bunsey MD, Giordano M, Norman AB. The catalepsy test: its ups and downs. Behav Neurosci. 1988;102:748–759. doi: 10.1037//0735-7044.102.5.748. [DOI] [PubMed] [Google Scholar]; b) Crocker AD, Hemsley KM. An animal model of extrapyramidal side effects induced by antipsychotic drugs: Relationship with D2 dopamine receptor occupancy. Prog Neuro-Psychophamachol & Biol Psychiat. 2001;25:573–590. doi: 10.1016/s0278-5846(00)00176-7. [DOI] [PubMed] [Google Scholar]; c) Hoffman DC, Donovan H. Catalespy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology. 1995;120:128–133. doi: 10.1007/BF02246184. [DOI] [PubMed] [Google Scholar]
- 15.Cignarella G, Giangiacomo N, Emilio O. Bicyclic homologs of piperazine. II. Synthesis of 3,8-diazabicyclo[3.2.1]octane. New synthesis of 8-methyl-3,8-diazabicyclo[3.2.1]octane. J Org Chem. 1961;26:2747. [Google Scholar]
- 16.Thompson PE, Zeigler JB, McCall JW. Antifilarial Agents. Diazabicyclooctanes and diazabicycloheptanes as bridged analogs of diethylcarbamazine. J Med Chem. 1974;17:481–487. doi: 10.1021/jm00251a002. [DOI] [PubMed] [Google Scholar]
- 17.Yevich JP, New JS, Lobeck WG, Dextraze P, Bernstein E, Taylor DP, Yocca FD, Eison MS, Temple DL., Jr Synthesis and biological characterization of α-(4-fluorophenyl)-4-(5-fluoro-2-pyrimidinyl)-1-piperazinebutanol and analogs as potential atypical antipsychotic agents. J Med Chem. 1992;35:4516–25. doi: 10.1021/jm00102a002. [DOI] [PubMed] [Google Scholar]
- 18.a) Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–9. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]; b) Seeman P, Chau-Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci U S A. 1975;72:4376–80. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Seeman P, Corbett R, Van Tol HHM. Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacol. 1997;16:93–110. doi: 10.1016/S0893-133X(96)00187-X. [DOI] [PubMed] [Google Scholar]
- 19.a) Bardin L, Auclair A, Kleven MS, Prinssen EP, Koek W, Newman-Tancredi A, Depoortère R. Pharmacological profiles in rats of novel antipsychotics with combined dopamine D2/serotonin 5-HT1A activity: comparison with typical and atypical conventional antipsychotics. Behav Pharmacol. 2007;18:103–18. doi: 10.1097/FBP.0b013e3280ae6c96. [DOI] [PubMed] [Google Scholar]; b) Cuisiat S, Bourdiol N, Lacharme V, Newman-Tancredi A, Colpaert F, Vacher B. Towards a new generation of potential antipsychotic agents combining D2 and 5-HT1A receptor activities. J Med Chem. 2007;50:865–76. doi: 10.1021/jm061180b. [DOI] [PubMed] [Google Scholar]
- 20.a) Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin-2 pKi values. J Pharmacol Exp Ther. 1989;251:238–246. [PubMed] [Google Scholar]; b) Rasmussen K, Aghajanian GK. Potency of antipsychotics in reversing the effects of a hallucinogenic drug on locus coeruleus neurons correlates with 5-HT2 binding affinity. Neuropsychopharmacol. 1988;1:101–107. doi: 10.1016/0893-133x(88)90001-2. [DOI] [PubMed] [Google Scholar]
- 21.Meltzer HY, Li Z, Kaneda Y, Ichikawa J. Serotonin receptors : their key role in drugs to treat schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2003;27:1159–1172. doi: 10.1016/j.pnpbp.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 22.a) Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacol. 2003;28:519–526. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]; b) Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet. 2005;20:368–378. doi: 10.2133/dmpk.20.368. [DOI] [PubMed] [Google Scholar]
- 23.a) Setola V, Dukat M, Glennon RA, Roth BL. Molecular determinants for the interaction of the valvulopathic anorexigen norfenfluramine with the 5-HT2B receptor. Mol Pharmacol. 2005;68:20–33. doi: 10.1124/mol.104.009266. [DOI] [PubMed] [Google Scholar]; b) Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation. 2000;102:2836–41. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]; c) http://cardiology.jwatch.org/cgi/content/full/2007/103/2)
- 24.http://www.rxlist.com/cgi/generic/abilify_cp.htm
- 25.Michaels RJ, Zaugg HE. Synthesis of 9-Methyl-3,9-diazabicyclo[4.2.1]-nonane, 25. J Org Chem. 1960:637–638. [Google Scholar]
- 26.Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28:1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]; b) See references in ref 25a
- 27.a) Needham PL, Atkinson J, Skill MJ, Heal DJ. Zotepine: preclinical tests predict antipsychotic efficacy and an atypical profile. Psychopharmacol Bull. 1996;32:123. [PubMed] [Google Scholar]; b) Protais P, Costentin J, Schwartz JC. Climbing Behavior Induced by Apomorphine in Mice: a Simple Test for the Study of Dopamine Receptors in Striatum. Psychopharmacology. 1976;50:1–6. doi: 10.1007/BF00634146. [DOI] [PubMed] [Google Scholar]
- 28.a) Hoffman DC, Donovan H. Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacol (Berl) 1995;120:128–33. doi: 10.1007/BF02246184. [DOI] [PubMed] [Google Scholar]; b) Hoffman DC, Donovan H. D1 and D2 dopamine receptor antagonists reverse prepulse inhibition deficits in an animal model of schizophrenia. Psychopharmacol (Berl) 1994;115:447–53. doi: 10.1007/BF02245567. [DOI] [PubMed] [Google Scholar]