

Abstract
An approach was developed for the quantification of subtle gains and losses of genomic DNA. The approach relies on a process called molecular combing. Molecular combing consists of the extension and alignment of purified molecules of genomic DNA on a glass coverslip. It has the advantage that a large number of genomes can be combed per coverslip, which allows for a statistically adequate number of measurements to be made on the combed DNA. Consequently, a high-resolution approach to mapping and quantifying genomic alterations is possible. The approach consists of applying fluorescence hybridization to the combed DNA by using probes to identify the amplified region. Measurements then are made on the linear hybridization signals to ascertain the region's exact size. The reliability of the approach first was tested for low copy number amplifications by determining the copy number of chromosome 21 in a normal and trisomy 21 cell line. It then was tested for high copy number amplifications by quantifying the copy number of an oncogene amplified in the tumor cell line GTL-16. These results demonstrate that a wide range of amplifications can be accurately and reliably quantified. The sensitivity and resolution of the approach likewise was assessed by determining the copy number of a single allele (160 kb) alteration.
Aneuploidies associated with the development of a variety of malignancies and genetic diseases involve the amplification or deletion of specific DNA sequences (1, 2). Karyotyping, which uses fluorescence in situ hybridization (FISH) on metaphase spreads, allows for a direct enumeration of individual chromosomes (3). Comparative genomic hybridization (CGH), on the other hand, allows for the genome-wide detection of loss or gain of chromosomal sequences greater than several Mb in size (4–6). Other methods for visualizing genomic instabilities recently have been developed, such as M-FISH or spectral karyotyping (7, 8). These methods allow for the genome-wide characterization of aneupoloidies with a resolution of about 1 Mb (9, 10). Recent studies suggest that CGH eventually could be used to detect copy number alterations of single genes by using arrays of cloned DNA on a diagnostic chip, but the ability to reliably quantify subtle alterations on the kilobase scale has proven to be technically difficult (11–13). Fiber-FISH techniques have advanced the resolution of chromosomal analysis to the kilobase range. These techniques rely on methods for aligning the DNA sample that often result in a nonuniform and uncontrolled extension of the molecules (14–19). Consequently, a statistically adequate number of extended molecules can be difficult to obtain with these methods.
In the following, we present an approach for the quantification of subtle gains and losses of DNA sequences in a sample of genomic DNA. Genomic amplifications ranging from subtle duplications as small as 50 kb in size to gross amplifications involving oncogenes or whole chromosomes can be precisely mapped and quantified with this approach. The approach is based on a process called molecular combing, which involves the uniform alignment and extension of DNA molecules on a glass surface by the hydrodynamic force exerted by a receding air/water interface, or meniscus (20, 21). This method of aligning and extending DNA has the advantage that each molecule is extended exactly 1.5 times its crystallographic length, providing a direct correlation between the measured length of the molecule and its actual size in kb:
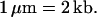 |
1 |
It previously has been shown that up to several hundred haploid genomes can be stretched on a glass surface by using this approach and that fluorescence in situ hybridization analysis is feasible on combed DNA (22, 23).
Materials and Methods
Preparation of Genomic DNA.
Escherichia coli genomic DNA was extracted by standard protocols. Human genomic DNA was prepared and combed as described (20–22, 24).
Probes.
Lambda DNA was labeled with biotin-14-dUTP. A total of 50–100 ng of probe pooled with 10 μg of herring sperm DNA was ethanol-precipitated and dried. The probe then was resuspended in 10 μl of hybridization buffer (50% formamide/10% dextran sulfate/2× SSC/1% Tween-20). Cosmid and bacterial artificial chromosome (BAC) DNA was prepared by alkaline lysis. A total of 0.7–1 μg of each cosmid probe (five probes for each chromosome) was obtained either by nick-translation or by random priming with biotin-14-dUTP or digoxygenin-11-dUTP. In the trisomy 21 experiment labeled probes specific to chromosome 21 were pooled with excess COT-1 DNA (5 μg) and ethanol-precipitated. Labeled probes specific to chromosome 9 were similarly prepared. In the met dosage experiment, 700–800 ng of each cosmid probe and 1.4 μg of the BAC probe were precipitated with 5× COT-1 DNA and 10 μg of herring sperm DNA. The dried probes were resuspended in 10 μl of hybridization buffer.
Hybridization.
Genomic DNA previously combed on glass surfaces and stored at −20°C were dehydrated for 5 min at room temperature in a series of ethanol baths (70%, 90%, and 100%). The DNA then was denatured for 2 min at 70°C in a formamide bath (70% formamide, 2× SSC, pH 7), and immediately dehydrated in a second series of ethanol baths on ice. The hybridization mixture was heated at 80°C for 2 min followed by incubation at 37°C for 5 min. Fifteen microliters of the probe solution was hybridized to the combed genomic DNA in a humid chamber overnight at 37°C.
Detection by Fluorescent Antibodies.
The hybridized probes were simultaneously detected by fluorescent antibodies by using FITC or Texas red. Successive layers of fluorescence-conjugated antibody were used to obtain easily detectable signals (22).
Measurements.
Fluorescent signals were screened and recorded by using an inverted epifluorescence microscope connected to a video camera (SIT 68, Dage–MTI, Michigan City, IN; spherical deformations caused by the intensified camera were corrected). In the experiment with E. coli DNA, the measurements involved two steps: (i) a prehybridization step in which the average length of the combed E. coli DNA per field of view (FOV) was calculated from a representative number (10–100) of randomly chosen FOVs; and (ii) a posthybridization step that involved measuring the length of hybridized lambda probe more than 100 FOVs to obtain an average number of hybridized lambda signals per FOV. In the experiment with human DNA, successive sets of 100 FOVs were first screened by using the FITC set of filters and subsequently rescreened by using the Texas red set of filters. In both cases, measurements were performed on the recorded images.
Statistical Analysis.
E. coli genome experiment.
The assessment of the number of combed E. coli genomes was performed by dividing the total measured length of the combed E. coli DNA by the known length of the genome (4.64 Mb) from a recorded set of randomly selected FOVs (10–100) (24). After hybridization, a larger number of FOVs was scanned (100 FOVs). As shown in Fig. 1B, the average number of E. coli genomes per FOV (averaged NG/FOV) rapidly converges to a constant value, even if the NG/FOV fluctuates from FOV to FOV with a SD varying from 10% to 30% of the averaged NG/FOV. The average value obtained from 10 randomly chosen FOVs appears to yield a close approximation, demonstrating the uniformity of combing over a single surface. However, the average number of genomes per FOV depends on the DNA solution used and varies from surface to surface (Table 1). Similarly, the average number of (lambda) targets per FOV (average NT/FOV) was calculated after hybridization by measuring the length of the detected signals from each randomly chosen FOV and then dividing by the known length of the lambda genome (48.5 kb). The SD is higher (90–160%) because some FOVs do not contain any signals. However, convergence to a stable average NT/FOV is observable after fewer than 100 FOVs, as shown in Fig. 1B. Both measurements (before and after hybridization) converge to approximately the same number of genomes per FOV, as shown in Fig. 1C. Summary of the final measured ratio NT/NG is presented in Table 1.
Figure 1.
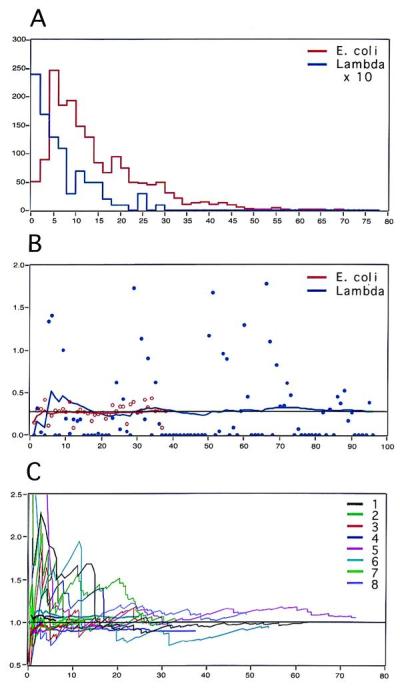
Lambda dosage in E. coli. (A) Histogram of the sizes of genomic E. coli DNA fragments (red) measured on 38 randomly chosen FOVs and a histogram of the sizes of hybridized lambda probes from the same slide measured on 96 random FOVs (blue, vertical scale has been expanded 10 times). Horizontal axis: size of fragments in μm; vertical axis: number of fragments. The size distribution is typical of sheared DNA. (B) Number of genomes per FOV. Horizontal axis: number of FOVs; vertical axis: number of genomes. Before hybridization, the number of E. coli genomes per FOV was obtained from the total length of combed DNA per FOV divided by 2,320 μm (red circles). After hybridization, the number of lambda genomes was obtained from the total length of the hybridized signals divided by 24.3 μm (blue circles). These numbers fluctuate widely, but the average numbers calculated over the scanned FOVs (red and blue curves) converge rapidly to a common value of 0.3 genome per FOV. (C) Summary of measurements performed on eight different slides. The same experiments as in B were performed for eight different slides. For each slide, we represent the average number of genomes per FOV as a function of the effective number of measured genomes. Thick curves correspond to measurements before hybridization (measurements of counterstained E. coli genomic DNA), thin curves correspond to measurements after hybridization (measurements of the hybridized lambda probe signals). All sets of curves (before/after hybridization) are normalized to 1 to facilitate comparison of the convergence, which occurs between 10 and 20 genomes.
Table 1.
Lambda to E. coli ratio
| Experiment | # E. coli/FOV | # FOV | # Lambda/FOV | # FOV | # Lambda/# E. coli |
|---|---|---|---|---|---|
| 1 | 0.60 | 10 | 0.61 | 107 | 1.02 |
| 2 | 0.31 | 50 | 0.30 | 102 | 0.97 |
| 3 | 0.28 | 38 | 0.29 | 96 | 1.04 |
| 4 | 0.78 | 10 | 0.72 | 46 | 0.92 |
| 5 | 0.58 | 36 | 0.65 | 116 | 1.12 |
| 6 | 0.61 | 40 | 0.56 | 100 | 0.92 |
| 7 | 0.31 | 41 | 0.31 | 110 | 1.00 |
| 8 | 0.38 | 100 | 0.47 | 107 | 1.24 |
Results of measurements of combed genomic DNA of RW597(λa)1 E. coli (20). The total length in μm was divided by 2,320 and by the number of FOVs to get an average number of E. coli genomes per FOV (# E. coli /FOV). The same measurement was performed after hybridization of a lambda probe on a larger number of randomly chosen FOV to obtain an average number of lambda genomes per FOV (# Lambda/FOV). Although this average number varies from slide to slide, the final ratio of lambda genomes to E. coli genomes remains close to the expected 1:1 ratio (1.03 ± 0.11).
Human genome experiment.
The number of haploid genomes Ng (per FOV) can be counted by measuring the total length of the hybridized control sequences (per FOV) according to:
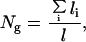 |
2 |
where li stands for a measured (possibly incomplete) signal, and l represents the length of an intact target sequence determined by using Eq. 1 (Fig. 1). It is possible to calculate the ratio between the target and control sequences and to observe the convergence of the ratio as the number of scanned FOV increases, as illustrated in Fig. 3. Theoretically, if intact control and target sequences were randomly distributed on the slide, a simple calculation shows that for an actual ratio R, the observed ratio Robs as a function of Ng, R(Ng), is with 95% confidence in the interval (R−ΔR, R+ΔR), where:
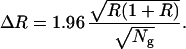 |
3 |
To take into account that broken DNA fibers lead to hybridized signals of various lengths, different size distributions can be used. In the case of a uniform distribution of lengths instead of a bimodal one (0 or 1 intact signals detected per FOV), or any other kind of regular distribution, the 1/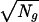 dependence of ΔR is conserved. In the case of a uniform distribution (which is more realistic; see Figs. 1A and 3D), the numerical coefficient in Eq. 3 simply changes to 1.61. However, one has to carefully check the statistical reliability of such a procedure, because, for instance, a cosmid probe (40 kb) represents between 10−3 and 10−4 times the size of a whole human chromosome. In the real experiment, successive regions of finite area are scanned and the total number of probes in these regions is calculated by using Eq. 2. Because there is no chromosome-dependent combing of DNA, the ratio of the number of a given chromosome species present in the scanned area over that of another species will reflect the ratio between these two species in the DNA solution. These numbers are well approximated by the number of detected probes as soon as a sufficient number of probe signals has been measured.
dependence of ΔR is conserved. In the case of a uniform distribution (which is more realistic; see Figs. 1A and 3D), the numerical coefficient in Eq. 3 simply changes to 1.61. However, one has to carefully check the statistical reliability of such a procedure, because, for instance, a cosmid probe (40 kb) represents between 10−3 and 10−4 times the size of a whole human chromosome. In the real experiment, successive regions of finite area are scanned and the total number of probes in these regions is calculated by using Eq. 2. Because there is no chromosome-dependent combing of DNA, the ratio of the number of a given chromosome species present in the scanned area over that of another species will reflect the ratio between these two species in the DNA solution. These numbers are well approximated by the number of detected probes as soon as a sufficient number of probe signals has been measured.
Figure 3.

Localization of probes and probe length measurements. (A) Approximate locations of cosmid probes for chromosomes 9 and 21. All cosmid sizes were determined by length measurements made on the combed molecules, or, in the case of the 9q34 cosmids, by direct measurement of the hybridized contig from previous experiments (see Table 1). (B) One FOV (×100 objective) of combed genomic DNA before hybridization, stained with YOYO-1. The number of genomes combed per coverslip at this density is estimated to be 100 diploid genomes. (C) Shown is a typical 32-kb cosmid probe signal (fragment) detected by FITC fluorescence after hybridization to the combed genomic DNA. Because the copy number ratio depends on the summed lengths of the signals rather than on their relative intensities variability in fluorescence intensities is negligible. (Scale bar = 10 μm.) (D) Histogram of the measured probe lengths for one experiment (T21a). Cosmid probes hybridizing to chromosome 9 were detected in red (dashed curve), and cosmid probes hybridizing to chromosome 21 were detected in green (plain curve). Both histograms look similar and are characterized by a mean value of 13–14 μm. A few intact hybridized cosmids are observed in the scanned region of the surface (right part of the histogram, 20–22 μm).
To increase the resolution of the approach and the reliability of the measurements, more probes can be used. Taking more probes per sequence increases the respective probability of detecting one probe per unit area. One finds that for p (homogeneously distributed over the chromosome) probes for the target chromosome, and q probes for the control chromosome, Eq. 3 has to be modified as:
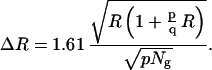 |
4 |
As a result, the gain in convergence rate scales as 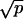 . For five probes for each chromosome, the gain is a factor of 2.2, whereas if five probes are used for the control (q = 5) and one for the target (p = 1), a factor of 1.4 is gained. Practically, the ratio measured in the first case should lead to the same uncertainty found in the second, although with 2.6 fewer genomes scanned.
. For five probes for each chromosome, the gain is a factor of 2.2, whereas if five probes are used for the control (q = 5) and one for the target (p = 1), a factor of 1.4 is gained. Practically, the ratio measured in the first case should lead to the same uncertainty found in the second, although with 2.6 fewer genomes scanned.
Finally, an assessment of the reliability of the method for detecting different ratios (such as an incremental increase in the copy number of the region of interest) is theoretically obtained by using Eq. 4. Detecting an incremental increase in copy number requires being able to unambiguously discriminate between two different ratios R1 > R2. This is feasible when the two curves R1−ΔR and R2+ΔR separate from one another as a function of the number of detected genomes, Ng. Beyond a certain number of genomes, Ng, the experimental convergence curves will correspond unequivocally to either R1 or R2. Equivalently, the precision δR with which an experimental ratio R is determined after the measurement of Ng genomes can be calculated by finding δR such that the curves for (R+δR)−ΔR and (R−δR)+ΔR separate from the curves for R+ΔR and R−ΔR, respectively.
Results
The ability to uniformly stretch hundreds of diploid genomes on a single coverslip suggested the possibility of developing a quantitative assay for assessing the copy numbers of specific DNA sequences in a sample of genomic DNA. The simple principle behind this approach is to determine the effective copy number of an amplified region and to compare that to the effective copy number of a nonamplified region. This is done by measuring the lengths of individual hybridization signals from sequence-specific probes. Therefore, the effective copy number of each region is the sum of the lengths of the measured hybridization signals divided by the known length of the respective regions (Eq. 2).
In a pilot experiment using E. coli strain RW597(λa)1 (25) containing one lambda insert, prestained bacterial DNA was combed and measured. As shown by the histogram in Fig. 1A and the curve in Fig. 1B, a statistically meaningful average number of E. coli genomes per FOV can be obtained from measurements made over a few random FOVs, even if the DNA is significantly sheared (expected length, 2,320 μm; observed mean length, 12 μm). This feature of the method has important implications for samples of DNA that are of poor quality. After hybridization with a lambda probe, only signals from the hybridized probe are visible and the average number of lambda genomes per FOV can similarly be computed from a representative number of FOVs (Fig. 1 A and B). Control experiments performed with E. coli strain RW592 (26) containing no inserted lambda genomes yielded an average of 0.01–0.02 detected inserts per FOV (data not shown). As shown in Fig. 1C and Table 1, this approach is not affected by the density of the combed molecules. This experiment demonstrates that a large region can be reliably quantified by using a relatively small sequence as a probe (here the ratio between the sizes of the lambda and E. coli genomes is 1/100).
The experimental scheme used for quantifying an amplification in human genomic DNA is shown in Fig. 2. We decided to test the feasibility and precision of this approach by quantifying a 1.5 ratio between the target and control region of an autosomal trisomy 21. Two sets of five cosmid probes were chosen to be evenly spaced over each chromosome so that each probe represented one-fifth of either the target (chromosome 21) or the control chromosome (chromosome 9) (Fig. 3A). Cosmid probes specific to chromosome 9 (145 Mb) were used to quantify the number of genomes present on the surface, and cosmid probes specific to chromosome 21 (50 Mb) were used to quantify the relative copy number of that chromosome. Before these experiments, each cosmid probe was separately combed and measured to determine its individual size as well as the respective total lengths of the two sets of probes (Table 2). Total human genomic DNA from a trisomy 21 lymphoblastoid cell line was combed (Fig. 3B), and the two differentially labeled sets of probes specific to chromosomes 9 and 21 were simultaneously hybridized to the combed genomic DNA. The target and control sequences are detected by using different fluorophores for each set of probes (Fig. 3C and Materials and Methods). Individual hybridization signals from each set of probes then were measured and summed to obtain their respective total lengths (Table 3). Fig. 3D shows the histogram of length measurements of the chromosome 9 fluorescent signals in red (plain curve) and chromosome 21 fluorescent signals in green (dashed curve).
Figure 2.

Experimental scheme. Dashed boxes (ri) represent labeled probes hybridized to fragments of genomic DNA and detected by Texas red fluorescence (r, red detection; index i, rank of the observed fragment). These probes are used to count the number of genomes present on the coverslip. This number is given by the sum of the lengths of the corresponding hybridization signals divided by the known length of the region (Nr = R/lr; lr = normal length of region). The black boxes (gi) represent linear FITC fluorescent signals specifically hybridized to the region whose copy number is to be determined (g, green detection; index i, rank of the observed fragment). Again, it is the total length of the measured hybridization signals divided by the known size of the region that yields its respective copy number per coverslip (Ng = G/lg). The copy number of the region present in the sample of genomic DNA then is obtained by dividing the number of target regions present on the coverslip by the number of genomes scanned. The boxes are shown to be of varying sizes to emphasize that detected signals often are truncated because of shearing of the genomic DNA during its preparation.
Table 2.
Cosmid lengths
| Chromosome 9 probes | Length ± SD, μm | Chromosome 21 probes | Length ± SD, μm |
|---|---|---|---|
| 108H11 | 16.9 ± 1.6 | F08132 | 20.4 ± 1.8 |
| 212F2 | 20.5 ± 0.6 | H0973 | 15.8 ± 0.9 |
| 269C11 | 16.6 ± 1.4 | A03115 | 14.6 ± 1.1 |
| 20D5 | 13.6 ± 1.3 | F0812 | 21.2 ± 0.8 |
| 149B8 | 21.7 ± 0.4 | F02103 | 16.1 ± 0.9 |
| Sum of probes | 89.3 ± 1.2 | Sum of probes | 88.1 ± 1.2 |
| Contig | 80.7 ± 3.8 |
Cosmid probe lengths measured by molecular combing. Cosmids were combed on individual surfaces from a batch whose stretching parameter was verified by using a test lambda DNA. Approimately 100 molecules of each cosmid were measured and their respective sizes were determined by fitting the main peak of the histogram to a Gaussian distribution. The chromosome 9 cosmid contig length was obtained by direct end-to-end measurements made on the intact contig hybridization signals.
Table 3.
Measured ratio and number of chromosomes
| Experiment | G 19/22 | WS 212 | T 21 a | T 21 b | T 21 c |
|---|---|---|---|---|---|
| Chr 21 | 21.04 | 61.95 | 34.05 | 35.75 | 117.78 |
| Chr 9 | 20.63 | 68.83 | 21.97 | 21.93 | 71.82 |
| Chr 21/Chr 9 | 1.02 | 0.90 | 1.55 | 1.63 | 1.64 |
Cosmids were hybridized and detected as described. Hybridization fragments were recorded and measured, and the respective sums of each set of signal lengths were used to calculate the corresponding number of chromosomes. The ratio is the number of (target) chromosome 21 (Chr 21) equivalents to the number of (control) chromosome 9 (Chr 9) equivalents.
The results of these experiments are summarized in Fig. 4, which shows the measured ratio plotted as a function of the number of genomes. These curves show a convergence of the measured ratio to within 10% of the expected ratio (3:2 in the case of a trisomy and 2:2 in the case of an unaffected individual) after 15–20 genomes have been scanned. This finding indicates that a relatively small sample of genomic DNA is sufficient for an accurate assessment of this type of amplification. In one case (WS 212), the observed convergence was slower. A bias in the measurements was observed in this experiment, but it had no significant effect on the final ratio (histogram not shown). Indeed, an arbitrary degree of precision can be achieved simply by screening larger numbers of genomes, which indicates that the copy number of a chromosome can be reliably determined in a mixed sample containing a population of normal and affected cells. For example, in a mixed sample in which 20% of the cells are normal, the corresponding ratio of 1.4 can be distinguished from a ratio of 1.5 if 400 genomes are scanned by using two sets of 10 probes each (see Materials and Methods).
Figure 4.

Results of the gene dosage experiments. (A) The DNA from two different cell lines of normal patients (G19/22 and WS 212) was combed on separate slides. The labeling and revelation scheme was reversed for WS 212. The red curve represents the evolution of the measured ratio as a function of the effective number of detected control chromosomes (one per haploid genome) for patient G19/22. A quick convergence to a final ratio of 1.05 is observed after a transient regime extending over fewer than 10 haploid genomes. For patient WS 212 (green curve), convergence of the ratio occurs after a larger number of genomes (50 genomes) to a limit value of 0.9. This difference might be caused by a bias in the measurements in favor of small signals in the case of Texas red, as observed from the length histogram (data not shown). Both experimental curves are confined within the two 95% confidence level envelopes (CLE, black curves) calculated by using Eq. 4 (see Materials and Methods), for p = q = 5 and r = 1. (B) The same experiment was performed by using the DNA of a trisomy 21 patient. In one experiment (T 21 a), the initial labeling and revelation scheme was used, and the opposite scheme was used in experiment T 21 b. Both experimental curves show a quick convergence to a limit value of 1.6 after 10 genomes. They both are confined within the two 95% CLEs (black continuous curves) calculated for an expected ratio of 1.5 and five probes per chromosome (p = q = 5). Another experiment also was performed on the same combed DNA, using a cosmid contig of 160 kb as a probe for chromosome 9, instead of the five evenly spaced cosmid probes (see dark blue, T 21 c). In this case, it is expected that the convergence of the measured ratio will be slower (95% CLE for p = 5, q = 1, gray curves), because convergence to 1.65 will occur after approximately 35 genomes have been scanned. This indeed was observed to be the case. A simple rescaling of the data to match the two sets of envelopes for p = q = 5 and p = 5, q = 1 transforms the T 21 c curve into the light blue one, which shows an equivalently fast convergence, as expected.
An important advantage of this approach is its ability to detect and quantify single gene alterations without actually measuring the entire length of the amplicon. A gene or gene region can extend over a few tens or hundreds of kb. The duplication of a single allele is detected by measuring 1.5 (1 + 2:2) times as many signals corresponding to the probes hybridizing in the region. Whether the amplification is a local rearrangement or consists of a minichromosome or any other event does not matter because this approach relies solely on counting the effective number of measured probes.
The sensitivity of the approach for single gene dosage was assessed in the same trisomy cell line by using a 160-kb cosmid contig located on chromosome 9 as a probe. Although the expected ratio (2:3) was obtained, the rate of convergence was about 2.6 times slower than in the previous experiments (Table 2, Fig. 4; see Materials and Methods). This experiment demonstrates that micro-deletions or amplifications of a small region of the genome can be detected and quantified by using a single phage artificial chromosome or BAC clone as a probe.
Amplifications involving oncogenes often consist of multiple duplications and rearrangements. We next investigated whether the precision of this approach could be extended to the detection of multiple amplifications of a region containing an oncogene. The human gastric carcinoma cell line GTL-16 (27), a type of cancer widespread in the human population, is a solid tumor that contains multiple copies of the met oncogene. This gene, which is located in the region 7q21–31, codes for a protein that is a member of the tyrosine kinase family. Previous studies have shown that multiple copies of c-met are present within a single nonhomogeneous staining region on a marker chromosome that is different from chromosome 7 (27). The copy number had been qualitatively estimated by Southern blotting to be about 10-fold. In these experiments, a 172-kb BAC containing the met oncogene was used as a probe. A set of five cosmid probes specific to chromosome 21 were used to count the number of genomes on the coverslip. Two independent assays revealed that the c-met oncogene is amplified exactly 11.5 times in this cell line, which corresponds to 21 extra copies of the met region per diploid genome (Fig. 5). Currently, we are applying this method to the quantification and characterization of the met amplicon in a variety of different tumor cell lines. Preliminary results confirm that a wide range of amplifications can be accurately quantified and characterized regardless of the size or structure of the amplicon (C.C., unpublished work).
Figure 5.

met oncogene amplification measurements. DNA from the GTL-16 cell lines was combed on several slides. BAC RG253B13 encompassing the met region (FITC detection) and cosmids F08132, H0973, H03115, F08121, and F02103 from chromosome 21 (Texas red detection) were simultaneously hybridized. The blue and purple curves correspond to two different experiments. The curve represents the evolution of the measured ratio (the effective number of measured BAC probes divided by the effective number of chromosome 21) as a function of the effective number of detected control chromosomes (chromosome 21). A convergence to a final ratio of 11.5 is observed after a transient regime extending over fewer than 10 haploid genomes. Experimental curves are confined within the two 95% confidence level envelopes (thin curves) calculated by using Eq. 4 (see Materials and Methods), for p = 1, q = 5, and r = 11.5.
Discussion
In this report we have described an approach for the precise detection and quantification of both subtle and gross genetic imbalances. Because the approach relies on measuring probe length as opposed to relative fluorescence intensities, we have been able to improve the resolution and reliability with which chromosomal analysis can be performed. Gene dosage by fluorescent probe measurement has the advantage that ambiguities and errors caused by nonspecific hybridization and other sources of fluorescence background are effectively eliminated. Another advantage resides in its simplicity because it requires only a single cosmid for quantifying an amplification. We have shown that a large number of measurable fluorescent signals can be detected from a single hybridization assay. This process allows for an arbitrary degree of precision in the assessment of a given region's copy number, because the method's sensitivity depends only on the number of genomes that are scanned. Consequently, a sufficient number of genomes would allow for the precise determination of an amplification in a mosaic of normal and cancerous cells. Likewise, incremental increases in copy number also can be accurately quantified simply by scanning a sufficient number of genomes (see Materials and Methods).
The results presented here indicate that a variety of chromosomal alterations can be detected by using this approach. It therefore has a potentially broad range of applications in a number of different domains. Changes in the DNA copy number of regions associated with tumorigenesis, for example, often include oncogenes or tumor suppressor genes, and chromosomal analysis of these regions can be a useful indicator of tumor type and prognosis. This approach therefore represents another addition to the increasing number of techniques now available for studying the underlying mechanisms of genetic imbalances and oncogenesis.
Acknowledgments
J.H. benefited from a fellowship from the Association Française contre les Myopathies and in part from a fellowship from the Fondation de France. X.M. was partially funded by the Direction des Application de la Recherche, Institut Pasteur.
Abbreviations
- BAC
bacterial artificial chromosome
- FOV
field of view
References
- 1.Knuutila S, Björkqvist A-M, Autio K, Tarkkanen M, Wolf M, Monni O, Szymanska J, Larramendy M L, Tapper J, Pere H, et al. Am J Pathol. 1998;152:1107–1123. [PMC free article] [PubMed] [Google Scholar]
- 2.Weinberg R A. In: Genetics and Cancer: A Second Look. Ponder B A J, Cavenee W K, Solomon E, Sidebottom E, editors. Vol. 25. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. pp. 3–12. [Google Scholar]
- 3.Hirai M, Suto Y, Kano H. Cytogenet Cell Genet. 1994;66:149–151. doi: 10.1159/000133687. [DOI] [PubMed] [Google Scholar]
- 4.Kallioniemi A, Kallioniemi O-P, Sudar D, Rutovitz D, Gray J W, Waldman F, Pinkel D. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 5.Bentz M, Plesh A, Stilgenbauer S, Döhner H, Lichter P. Genes Chromosomes Cancer. 1998;21:172–175. [PubMed] [Google Scholar]
- 6.Forozan F, Karhu R, Kononen J, Kallioniemi A, Kallioniemi O-P. Trends Genet. 1997;13:405–409. doi: 10.1016/s0168-9525(97)01244-4. [DOI] [PubMed] [Google Scholar]
- 7.Schröck E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith M A, Ning Y, Ledbetter D H, Bar-Am I, Soenksen D, et al. Science. 1996;273:494–497. doi: 10.1126/science.273.5274.494. [DOI] [PubMed] [Google Scholar]
- 8.Speicher M R, Ballard S G, Ward D C. Nat Genet. 1996;12:368–375. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- 9.Ried T, Liyanage M, du Manoir S, Heselmeyer K, Auer G, Macville M, Schröck E. J Mol Med. 1997;75:801–814. doi: 10.1007/s001090050169. [DOI] [PubMed] [Google Scholar]
- 10.Veldman T, Vignon C, Schröck E, Rowley J D, Ried T. Nat Genet. 1997;15:406–410. doi: 10.1038/ng0497-406. [DOI] [PubMed] [Google Scholar]
- 11.Kraus J, Weber R G, Cremer M, Seebacher T, Fisher C, Schurra C, Jauch A, Lichter P, Bensimon A, Cremer T. Hum Genet. 1997;99:374–380. doi: 10.1007/s004390050375. [DOI] [PubMed] [Google Scholar]
- 12.Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Döhner H, Cremer T, Lichter P. Genes Chromosomes Cancer. 1997;20:399–407. [PubMed] [Google Scholar]
- 13.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo W-L, Chen C, Zhai Y, et al. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 14.Heng H H, Squire J, Tsui L C. Proc Natl Acad Sci USA. 1992;89:9509–9513. doi: 10.1073/pnas.89.20.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiegant J, Kalle W, Mullenders L, Brookes S, Hoovers J M, Dauwerse J G, van Ommen G J, Raap A K. Hum Mol Genet. 1992;1:587–591. doi: 10.1093/hmg/1.8.587. [DOI] [PubMed] [Google Scholar]
- 16.Parra I, Windle B. Nat Genet. 1993;5:17–21. doi: 10.1038/ng0993-17. [DOI] [PubMed] [Google Scholar]
- 17.Fidlerova H, Senger G, Kost M, Sanseau P, Sheer D. Cytogenet Cell Genet. 1994;65:203–205. doi: 10.1159/000133632. [DOI] [PubMed] [Google Scholar]
- 18.Houseal T W, Dackowski W R, Landes G M, Klinger K W. Cytometry. 1994;15:193–198. doi: 10.1002/cyto.990150303. [DOI] [PubMed] [Google Scholar]
- 19.Heiskanen M, Hellsten E, Kallioniemi O P, Makela T P, Alitalo K, Peltonen L, Palotie A. Genomics. 1995;30:31–36. doi: 10.1006/geno.1995.0005. [DOI] [PubMed] [Google Scholar]
- 20.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Science. 1994;265:2096–2098. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 21.Bensimon D, Simon A J, Croquette V, Bensimon A. Phys Rev Lett. 1995;74:4754–4757. doi: 10.1103/PhysRevLett.74.4754. [DOI] [PubMed] [Google Scholar]
- 22.Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, Slegtenhorst M v, Slegtenhorst M v, Wolfe J, Povey S, Bensimon A. Science. 1997;277:1518–1523. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]
- 23.Weier H-U G, Wang M, Mullikin J C, Zhu Y, Cheng J-F, Greulich K M, Bensimon A, Gray J W. Hum Mol Genet. 1995;4:1903–1910. doi: 10.1093/hmg/4.10.1903. [DOI] [PubMed] [Google Scholar]
- 24.Allemand J-F, Bensimon D, Jullien L, Bensimon A, Croquette V. Biophys J. 1997;73:2064–2070. doi: 10.1016/S0006-3495(97)78236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blattner F R, Plunkett G, III, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 26.Marchal C, Greenblatt J, Hofnung M. J Bacteriol. 1978;136:1109–1119. doi: 10.1128/jb.136.3.1109-1119.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponzetto C, Giordano S, Peverali F, Della Valle G, Abate M L, Vaula G, Comoglio P M. Oncogene. 1991;6:553–559. [PubMed] [Google Scholar]