

Abstract
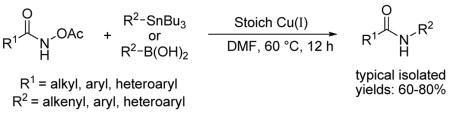
A general non-oxidative N-amidation of organostannanes and boronic acids has been developed. Under non-basic conditions a wide variety of aryl, alkenyl, and heteroaryl organostannanes and boronic acids couple efficiently with O-acetyl hydroxamic acids in the presence of Cu(I) sources.
Mild methods for the formation of carbon-heteroatom bonds are of considerable interest to the synthetic organic community. A great diversity of carbon—nitrogen bonds can now be easily generated by transition metal-catalyzed protocols, 1 most prominantly using the base-promoted conditions developed by Buchwald and Hartwig2 and the oxidative couplings of boronic acids and their derivatives with amines and amides described by Lam, Chan, Evans3 and Collman and Batey.4 Complementary protocols that are differentiated from these C-N bond forming reactions by providing non-basic and non-oxidizing reaction conditions are always of value in complex synthetic settings. The chemistry depicted in Scheme 1 suggests that boronic acids could couple with derivatives of hydroxylamine and deliver new chemoselective methods for C—N bond formation through selective metal-catalyzed cleavage of the N—O bond followed by transmetalation and reductive elimination. Hydroxylamine derivatives as coupling reactants provide a nitrogen moiety “R1NH” for C—N bond formation and an internal “oxygenate” partner “OR’” to pair with the “B(OH)2” fragment. In principle, this would allow C—N bond formation to take place under neutral reaction conditions and avoid the addition of an oxygenate base that is commonly used in boronic acid-based cross-couplings. The development of suitably mild reaction conditions would allow the selective targeting of reactions at an N-O bonded functional group in the presence of other reactive functionalities and facilitate the development of small molecule therapeutics or provide a tactic for the chemical modification and/or tagging of more complex biomolecules.
Scheme 1.

Hydroxylamine-Derived Non-basic, Non-oxidative C—N Bond Formation
Using traditional nucleophilic reagents, Narasaka (organomagnesium) 5 and Johnson (organozinc) 6 have shown that hydroxylamine derivatives can be cleaved by organometallic reagents in the presence of metal catalysts and provide a general process for C-N bond formation. However, until recently boronic acids were not known to participate in similar N-O cleavage-based cross-coupling with hydroxylamine derivatives. 7 Our laboratory disclosed a mild copper-catalyzed N-imination of boronic acids and organostannanes using oxime O-carboxylates as iminating agents 8 and extended that chemistry to establish a simple modular synthesis of substituted pyridines. 9 In an extension of this strategy, we now disclose a new non-basic method for the preparation of N-substituted amides by the Cu-mediated cross-coupling of boronic acids and organostannanes with O-acetyl hydroxamic acids (Scheme 2).
Scheme 2.

Cu(I)-Mediated Amidation of Boronic Acid/Organostannane with O-Substituted Hydroxamic Acid
This new reaction provides a means of generalizing the Lam “oxidative” amidation of boronic acids3a,d by substituting an oxidized form of the amide coupling partner under neutral reaction conditions in place of an external oxidant like stoichiometric Cu(II) or air. In addition to allowing transformations on base-sensitive molecules, the use of N—O moiety-based couplings could also provide useful reaction chemoselectivities in the presence of Lam-condition-reactive “NH” moieties.
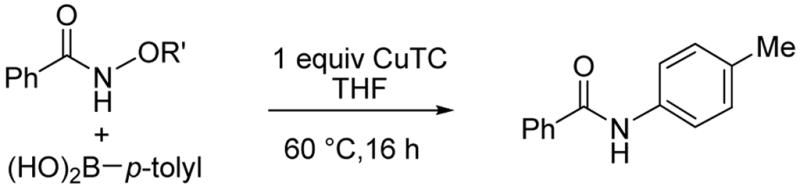
This investigation was initiated by exploring the cross-coupling of O-acetyl benzohydroxamic acid with 1.1 equiv of phenyl boronic acid. A screening of different Cu(I) sources, solvents, additives and reaction temperatures revealed the optimum use of 1 equiv of Cu(I) thiophene-2-carboxylate (CuTC) in THF at 60 °C. Product yields were compromised using less than 1 equiv of Cu(I), but loadings in excess of 1 equiv did not lead to improved yields. CuOAc and CuTC gave similar results, while non-oxygenate Cu(I) sources such as CuCl, CuI and CuCN were ineffective. Solvents such as DMF, DMA and toluene were not as effective as THF. The addition of Lewis acids (BF3·Et2O, Sc(OTf)3, TiCl4, CuPF6(CH3CN)4) or base (Et3N, Cs2CO3) dramatically reduced the product yield. The influence of the hydroxamic acid O-substituent on the reaction efficiency was probed by reacting the O-substituted benzohydroxamic acids with 1.1 equiv p-tolylboronic acid and a stoichiometric amount of CuTC in THF at 60 °C (Table 1).
Table 1.
Reaction Survey: Amidation of p-Tolylboronic Acid with O-Substituted Benzohydroxamic Acids
 | ||
|---|---|---|
| entry | R′ | amide (%)a |
| 1 | -COMe | 69 |
| 2 | -CO(t-Bu) | 61 |
| 3 | -CO(p-tolyl) | 40 |
| 4 | -CO(2-thienyl) | 41 |
| 5 | -CO(2-pyridyl) | 30 |
| 6 | -COC6F5 | 36 |
| 7 | -Me | 62 |
| 8 | -Ph | 60 |
Isolated yield
O-Acetyl benzohydroxamic acid still gave the highest product yield (69%, entry 1). O-(p-methylbenzoyl), O-(thiophene-2-carbonyl) and O-(2-pyridinylcarbonyl) substitutions resulted in moderate yields of amide (entries 3–5). Surprisingly, O-methyl and O-phenyl benzohydroxamic acid also generated the N-substituted amide in about 60% yield (entries 6–7).
To probe generality of this reaction various O-acetyl hydroxamates and boronic acids were reacted with 1 equiv of CuTC in THF at 60 °C for 16 h producing the corresponding amides in moderate to good yields in most cases (Table 2). A 1:1 mixture of hexane and THF was used as the reaction solvent in some cases to keep the effective Cu concentrations in solution low and diminish undesired homocoupling and protodeborylation reactions of the arylboronic acid. Arylboronic acids with electron-donating substitutions generated amide products as desired (entries 3 and 4), while a low yield of the amide product was obtained when using an electronic deficient arylboronic acid (entry 5). Only a trace of the enamide product was observed by GC/MS analysis of the reaction of benzohydroxamic acid with E-β-styrylboronic acid (entry 6).
Table 2.
CuTC-Mediated Amidation of Arylboronic Acids with O-Acetyl Hydroxamic Acids10
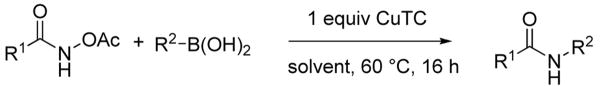 | |||
|---|---|---|---|
| entry | R1 | R2 | amide (%)a |
| 1b | phenyl | phenyl | 74 |
| 2b | phenyl | p-tolyl | 77 |
| 3b | phenyl |
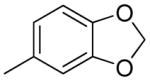
|
63 |
| 4b | phenyl | 4-phenoxyphenyl | 68 |
| 5b | phenyl | 3-formylphenyl | ~10d |
| 6b | phenyl | E–β-styryl | traced |
| 7c | 3-methoxyphenyl | p-tolyl | 58 |
| 8c | (4-CF3)Ph | p-tolyl | 81 |
| 9c | benzyl | p-tolyl | 76 |
| 10c | 1-naphthyl | p-tolyl | 65 |
Isolated yield.
Hexanes/THF (1:1) as solvent.
THF as solvent.
GC/MS analysis using nonadecane as an internal standard.
Organostannanes proved to be excellent reaction partners in Cu(I)-mediated couplings with O-acetyl hydroxamic acids. In earlier studies of Cu-mediated cross-couplings involving organostannanes, the beneficial pairing of Cu(I) diphenylphosphinate (CuDPP) with organostannanes was noted and ascribed to the precipitation of (n-Bu3SnO(O)PPh2) from the reaction mixture. 11 In the present system involving organostannane reaction partners, stoichiometric CuDPP proved equally useful (Table 3). Optimal yields were achieved with >1 equiv of Cu(I) – 2.0 equiv of CuDPP was routinely used for convenience. In contrast to the boronic acid system which appeared limited to electron-rich aryl substituents, the organostannane system showed significant generality. Thus, various O-acetyl hydroxamates and organostannanes were heated with 2 equiv of CuDPP in DMF at 60 °C for 12 h, generating amides in moderate to good yields. As shown in Table 3, O-acetyl benzohydroxamic acid coupled efficiently with aryl, heteroaryl, and alkenyl organostannanes providing the desired amides (entries 1–7), while slight lower yields of amides were obtained when using deactivated (electron-deficient) organostannane reagents (entries 3 and 7). Entries 8–14 show that significant variation in the nature of the acyl moiety is accommodated by the reaction system. Heteroaryl O-acetyl hydroxamic acids react well (entries 11–12) and synthetically useful N-alkyl substituted amides may also be obtained efficiently by this method (entries 13–18). Special attention is drawn to entry 14 in which a 4-iodophenyl moiety is tolerated under the reaction conditions.
Table 3.
CuDPP-Mediated Amidation of Organostannanes with O-Acetyl Hydroxamic Acids12
 | |||
|---|---|---|---|
| entry | R1 | R2 | amide (%)* |
| 1 | phenyl | p-tolyl | 80 |
| 2 | phenyl | 4-methoxyphenyl- | 76 |
| 3 | phenyl | 4-chlorophenyl- | 56 |
| 4 | phenyl |
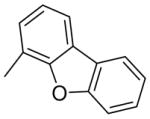
|
73 |
| 5 | phenyl | (Z)-1-propenyl | 66 |
| 6 | phenyl | 2-furyl | 73 |
| 7 | phenyl | 2-pyrazinyl | 42 |
| 8 | 4-nitrophenyl | p-tolyl | 74 |
| 9 | 3-methoxyphenyl | p-tolyl | 71 |
| 10 | 1-naphthyl | p-tolyl | 83 |
| 11 | 2-thienyl | 4-methoxyphenyl | 90 |
| 12 | 2-thienyl | 2-thienyl | 60 |
| 13 | benzyl | p-tolyl | 74 |
| 14 | benzyl | 4-iodophenyl | 60 |
| 15 | benzyl | 2-methyl-1-propenyl | 64 |
| 16 | methyl | p-tolyl | 85 |
| 17 | undecyl | p-tolyl | 66 |
| 18 | undecyl | (E)-β-styryl | 52 |
Isolated yield.
Oxidative addition of the O-acetylhydroxamic acid to Cu(I) 13 is a likely first step in the mechanism of the previously described Cu(I)-catalyzed8,9 as well as in the current Cu(I)–mediated cross-coupling of hydroxyl amine derivatives with boronic acids and organostannanes. We note that heating O-acetyl benzohydroxamic acid with 1 equiv of CuTC in THF resulted in complete conversion to PhCONH2 after aqueous workup.14 Why at least a full equivalent of Cu(I) is required during cross-coupling reactions of O-acetylhydroxamic acids, but is required in only catalytic quantities for cross-couplings with O-acyloximes,8,9 must reside in the nature of the two different amido Cu intermediates A and B shown in Scheme 3.
Scheme 3.

Mechanistic Speculations
Although the detailed mechanism of these Cu-mediated couplings of hydroxylamine derivatives with boronic acids and organostannanes is not known, we speculate that the imido intermediates R2C=NCuX2 are sufficiently electrophilic to engage in a fast transmetalation with boronic acids and organostannanes, while the amido RCONHCuX2 species are likely to form relatively stable and less electrophilic amido bridging dimeric structures that would be more sluggish at transmetalation. This would rapidly consume a full equivalent of Cu(I) and could give the appearance of a non-catalytic process. In fact N-substituted O-acetyl hydroxamic acids do not participate in the Cu(I)-mediated cross-coupling, generating instead (after aqueous workup) the product of N-O bond reduction, suggesting that transmetalation is prevented by steric encumbrance in these cases.
Interesting chemoselectivity of this new N—O-based cross-coupling is seen in the two reactions depicted in Scheme 4 (where DMF was used as solvent to ensure solubility of both Cu sources, rather than THF/hexanes as described in Table 2). Upon exposure of a mixture of O-acetyl benzohydroxamic acid and p-tolylboronic acid to Cu(II), Lam-like N-arylation reactivity is predominantly observed. In contrast, the use of Cu(I) allows divergence of this chemistry from Lam-like reactivity to exclusive formation of the product of cross-coupling at the N—O bond.
Scheme 4.

Examples of Chemoselectivity
In summary, a general copper-mediated cross-coupling of O-acetyl hydroxamic acids with boronic acids and organostannanes under non-basic and non-oxidizing conditions has been developed. This methodology allows a variety of amides to be easily prepared and is a useful complement to existing metal-catalyzed methods for C-N bond formation. Given the mild reaction conditions and its inherent chemoselectivity this new method might be useful as a novel bioorthogonal peptide ligation protocol of use in the construction of peptides and peptidomimetics. Additional studies are ongoing.
Supplementary Material
Acknowledgments
The National Institutes of General Medical Sciences, DHHS supported this investigation through grant No. GM066153. Dr. Gary Allred of Synthonix provided the boronic acids and organostannanes used in our studies.
Footnotes
Supporting Information Available Complete description of experimental details and product characterization (20 pages) and photocopies of spectra (54 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For some recent reviews see: Beletskaya IP. Pure Appl Chem. 2005;77:2021–2027.Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337–2364.Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594.
- 2.Leading reviews: Buchwald SL, Mauger C, Mignani G, Scholzc U. Adv Synth Catal. 2006;348:23–39.Hartwig JF. Synlett. 2006:1283–1294.Muci AR, Buchwald SL. Topics in Current Chemistry. 2002;219:131–209.Hartwig JF. Comprehensive Coordination Chemistry II. 2004;9:369–398.Schlummer B, Scholz U. Adv Synth Catal. 2004;346:1599–1626.
- 3.For some examples see: Lam PYS, Vincent G, Bonne D, Clark CG. Tetrahedron Lett. 2003;44:4927–4931.Chan DMT, Monaco KL, Li R, Bonne D, Clark CG, Lam PYS. Tetrahedron Lett. 2003;44:3863–3865.Lam PYS, Bonne D, Vincent G, Clark CG, Combs AP. Tetrahedron Lett. 2003;44:1691–1694.Lam PYS, Deudon S, Hauptman E, Clark CG. Tetrahedron Lett. 2001;42:2427–2429.Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937–2940.
- 4.(a) Collman JP, Zhong M. Org Lett. 2000;2:1233–1236. doi: 10.1021/ol000033j. [DOI] [PubMed] [Google Scholar]; (b) Collman JP, Zhong M, Zeng L, Costanzo S. J Org Chem. 2001;66:1528–1531. doi: 10.1021/jo0016780. [DOI] [PubMed] [Google Scholar]; (c) Collman JP, Zhong M, Zhang C, Costanzo S. J Org Chem. 2001;66:7892–7897. doi: 10.1021/jo010615u. [DOI] [PubMed] [Google Scholar]; (d) Antilla JC, Buchwald SL. Org Lett. 2001;3:2077–2079. doi: 10.1021/ol0160396. [DOI] [PubMed] [Google Scholar]; (e) Quach TD, Batey RA. Org Lett. 2003;5:4397–4400. doi: 10.1021/ol035681s. [DOI] [PubMed] [Google Scholar]; (f) Bolshan Y, Batey RAA. ngew Chem Int Ed. 2008;47:2109–2112. doi: 10.1002/anie.200704711. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kitamura M, Suga T, Chiba S, Narasaka K. Org Lett. 2004;6:4619–4621. doi: 10.1021/ol0479951. [DOI] [PubMed] [Google Scholar]; (b) Narasaka K. Pure Appl Chem. 2002;74:143–149. [Google Scholar]; (c) Tsutsui H, Hayashi Y, Narasaka K. Chem Lett. 1997:317–318. [Google Scholar]
- 6.(a) Campbell MJ, Johnson JS. Org Lett. 2007;9:1521–1524. doi: 10.1021/ol0702829. [DOI] [PubMed] [Google Scholar]; (b) Berman AM, Johnson JS. J Org Chem. 2006;71:219–224. doi: 10.1021/jo051999h. [DOI] [PubMed] [Google Scholar]; (c) Berman AM, Johnson JS. J Am Chem Soc. 2004;126:5680–5681. doi: 10.1021/ja049474e. [DOI] [PubMed] [Google Scholar]
- 7.Hydrazines and hydroxyamines are known to undergo Lam-like oxidative N- and O-arylation with boronic acids without cleavage of the heteroatom-heteroatom bond: Petrassi HM, Sharpless KB, Kelly JW. Org Lett. 2001;3:139–142. doi: 10.1021/ol0003533.Suzuki H, Yamamoto A. J Chem Res-S. 1992;8:280–281.
- 8.Liu S, Yu Y, Liebeskind LS. Org Lett. 2007;9:1947–1950. doi: 10.1021/ol070561w. See also Cu(I)-catalyzed couplings of boronic acids with nitrosoaromatics Yu Y, Srogl J, Liebeskind LS. Org Lett. 2004;6:2631–2634. doi: 10.1021/ol048982q.
- 9.Liu S, Liebeskind LS. J Am Chem Soc. 2008;130:6918–6919. doi: 10.1021/ja8013743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Typical experimental procedure: O-acetyl benzohydroxamic acid (0.10 mmol), phenylboronic acid (0.12 mmol) and CuTC (0.10 mmol) were added to a Schlenk tube. After flushing with argon, THF (3 mL) was added via syringe. The reaction mixture was stirred under argon at 60 °C for 12 h. Ethyl ether (10 mL) was added to the mixture. The organic layer was washed with brine, dried over MgSO4 and evaporated. The residue was then subjected to preparative plate silica chromatography using hexanes/EtOAc as eluent.
- 11.(a) Allred G, Liebeskind LS. J Am Chem Soc. 1996;118:2748–2749. [Google Scholar]; (b) Wittenberg R, Srogl J, Egi M, Liebeskind LS. Org Lett. 2003;5:3033–3035. doi: 10.1021/ol034962x. [DOI] [PubMed] [Google Scholar]
- 12.Typical experimental procedure: O-acetyl benzohydroxamic acid (0.10 mmol), tributyl p-tolyltin (0.12 mmol) and CuDPP (0.20 mmol) were added to a Schlenk tube. After flushing with argon, DMF (3 mL) was added via syringe. The reaction mixture was stirred under argon at 60 °C for 12 h. Ethyl ether (10 mL) was added to the mixture. The organic layer was washed with brine, dried over MgSO4 and evaporated. The residue was then subjected to preparative plate silica chromatography using hexanes/EtOAc as eluent.
- 13.For oxidative addition of N-O bond to various transition metals such as Re, Pd and Cu, see: Kusama H, Yamashita Y, Uchiyama K, Narasaka K. Bull Chem Soc Jpn. 1997;70:965–975.Ferreira CMP, Guedes da Silva MFC, Kukushkin VY, Fraústo da Silva JJR, Pombeiro AJL. J Chem Soc, Dalton Trans. 1998:325–326.Tsutsui H, Narasaka K. Chem Lett. 1999:45–46.Tsutsui H, Narasaka K. Chem Lett. 2001:526–527.Kitamura M, Zaman S, Narasaka K. Synlett. 2001:974–976.Kitamura M, Narasaka K. Chem Rec. 2002;2:268–277. doi: 10.1002/tcr.10030.Chiba S, Kitamura M, Saku O, Narasaka K. Bull Chem Soc Jpn. 2004;77:785–796.Tsutsui H, Hayashi Y, Narasaka K. Chem Lett. 1997:317–318.Narasaka K. Pure Appl Chem. 2002;74:143–149.
- 14.This reduction has been commonly found with various O-substituted hydroxamic acids, even with O-methyl hydroxamic acid: A Ti(III)-mediated reduction of O-methyl hydroxamic acid has been reported: Fisher LE, Caroon JM, Jahangir, Russell Stabler JS, Lundberg S, Muchowski JM. J Org Chem. 1993;58:3643–3647.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.