

Abstract
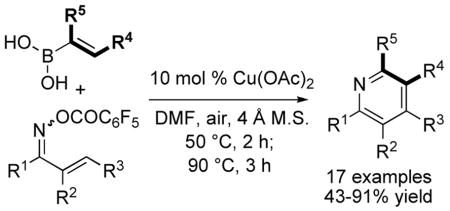
A simple, modular method to prepare highly substituted pyridines is disclosed. The method employs a cascade reaction comprising (1) a novel N-iminative, Cu-catalyzed cross-coupling of alkenylboronic acids at the N—O bond of α, β-unsaturated ketoxime O-pentafluorobenzoates, (2) electrocyclization of the resulting 3-azatriene, and (3) air oxidation affording highly substituted pyridines in moderate to excellent isolated yields (43–91%). Starting materials are readily available and functional group tolerance is very good.
The careful tuning of metal catalysts and mediators to specific bond types can lead to new synthetic transformations possessing significant chemoselectivity in functionally complex systems. Molecules bearing N—O bonds are fruitful candidates for beneficial tuning to metal catalysts and mediators, and such systems have the potential to deliver new, chemoselective methods for C—N bond formation under non-basic and non-oxidative conditions.1 To exemplify this concept, we report herein a versatile synthesis of highly substituted pyridines that takes place through a reaction cascade comprised of (a) a novel and mild C—N cross-coupling of alkenyl boronic acids at the N—O bond of an oxime O-carboxylate generating 3-azatrienes, (b) electrocyclization, and (c) aerobic oxidation (Scheme 1).
Scheme 1.

Proposed C—N Coupling Route to Substituted Pyridines
Substituted pyridines are important components of biologically active molecules2 making the development of mild, efficient, and modular methods for the synthesis of substituted pyridines highly desirable. Most syntheses of pyridine rings are based upon condensation reactions of carbonyl compounds or are designed around thermal or metal catalyzed cycloaddition reactions.3 Existing methods of synthesis allow the construction of specific substitution patterns about the pyridine core, but are often incompatible with various functional groups or general substitution patterns.3l, 4
Construction of a substituted pyridine ring by the thermal electrocyclization of 1-, 2-, and 3-azatrienes has been previously demonstrated.5 The recently disclosed N-imination of boronic acids and organostannanes under neutral reaction conditions via a copper-catalyzed C-N cross-coupling at the N-O bond of oxime carboxylates1a could provide a mild, modular, and novel route to variously substituted 3-azatrienes, if practiced on α,β-unsaturated oxime O-carboxylates and alkenylboronic acids. After in situ thermolysis and oxidation, this new chemistry would provide a simple, straightforward, and potentially powerful method for the modular construction of substituted pyridines with flexible control over the substitution pattern (Scheme 1). The neutral pH reaction conditions could allow broad functional group tolerance and therefore provide access to diverse substitution patterns about the pyridine core.
α,β-Unsaturated ketoxime O-pentafluorobenzoates are easily prepared by the condensation of an α,β-unsaturated ketone with hydroxylamine followed by acylation with pentafluorobenzoyl chloride. As a probe experiment, a mixture of 1,3-diphenyl-2-propen-1-one oxime O-pentafluorobenzoate (1.4:1, E/Z unassigned) and commercially available trans-1-hexen-1-ylboronic acid in DMF was exposed to 10 mol % Cu(OAc)2 at 50°C for 1 h open to air. Both starting materials disappeared and a new spot was observed on TLC.6 The reaction mixture was then heated at 85 °C for 3 h transforming the uncharacterized intermediate into 5-n-butyl-2,4-diphenylpyridine in 82% yield (Scheme 2). Since 1,3-diphenyl-2-propen-1-one oxime O-pentafluorobenzoate exists as a 1.4:1 ratio of geometric isomers, facile E/Z equilibration about the imine bond of the intermediate 3-azatriene under the reaction conditions must allow both isomers of the α,β-unsaturated ketoxime O-pentafluorobenzoate to participate in the reaction.
Scheme 2.
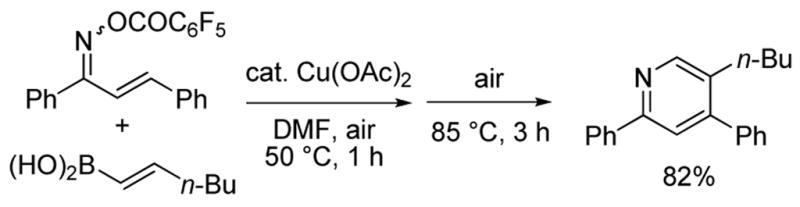
Initial Probe Experiment
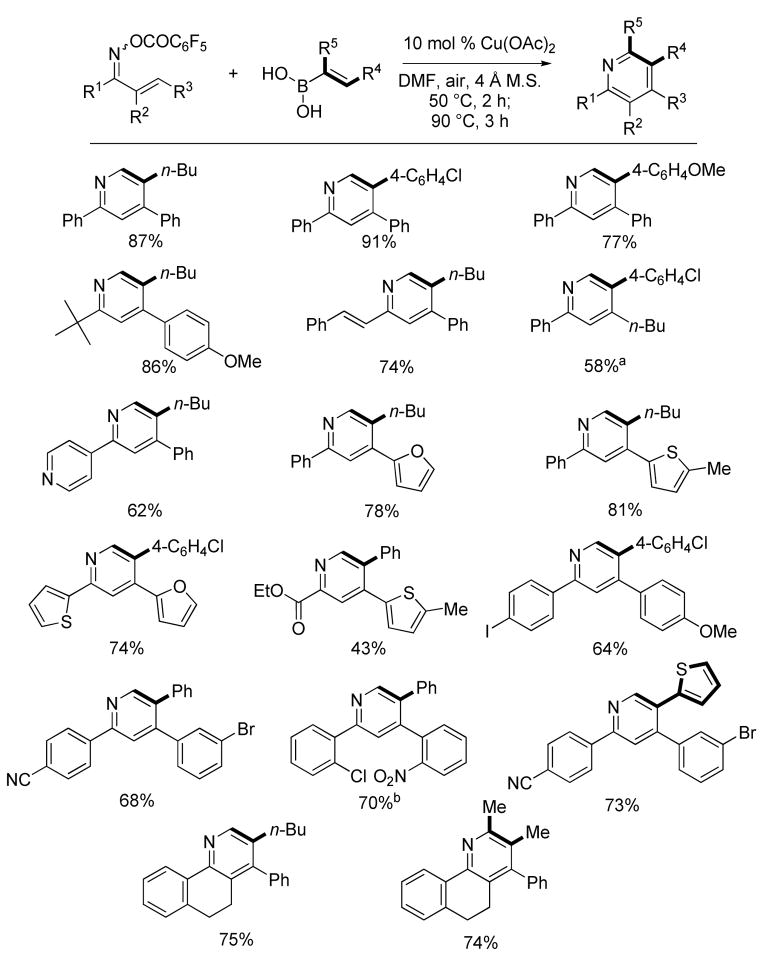
A brief study revealed optimized reaction conditions suitable for most substrates: DMF open to air, 4Å molecular sieves, 50 °C for 2 h for the cross-coupling followed by 90 °C for 3–5 h for the electrocyclization-oxidation. The addition of 4Å molecular sieves protected the transient 3-azatrienes from competitive hydrolysis by water (generated in situ from the boronic acid – boroxine equilibrium7) under the reaction conditions. The scope of this new methodology proved quite general as shown in Table 1.
Table 1.
Modular Synthesis of Substituted Pyridines
|
Final thermolysis carried out for 5 h at 90 °C.
Final thermolysis carried out for 3 h at 150 °C.
A variety of substituents were successfully introduced around the pyridine ring. Examples are shown where (1) R1 is aryl (electron-rich and electron-deficient), heteroaryl (electron-rich and electron-deficient), alkenyl, alkoxycarbonyl, and tert-butyl, (2) R2 is H and alkyl, (3) R3 is aryl (electron-rich and electron-deficient), heteroaryl, and alkyl, (4) R4 is aryl (electron-rich and electron-deficient), heteroaryl, and alkyl, and (5) R5 is H and alkyl. Benefiting from the mild, neutral pH reaction conditions used, the new method tolerates many functional groups, like chloride, bromide, ester, nitrile, and nitro. Iodides are particularly reactive in Suzuki, Stille, and Ullmann reactions,8 but are untouched under these reaction conditions. Hence, functionalization complementary to classic cross-coupling tactics can be achieved in this methodology, which provides substrates for further elaboration. Amides are tolerated since DMF is the solvent. As disclosed in our previous study,1a alcohol and aldehyde functional groups are also compatible with the reaction conditions. It is noteworthy that the aerobic reaction conditions did not lead to over oxidation in the preparations of 5,6-dihydro-4-phenyl-3-butyl-benzo[h]quinoline and 5,6-dihydro-4-phenyl-2,3-methyl-benzo[h]quinoline.
A simple, straightforward mechanism for this transformation is that suggested earlier in Scheme 1. C—N coupling of the E/Z α,β-unsaturated ketoxime O-pentafluorobenzoate and an alkenylboronic acid with catalytic copper produces a 3-azadiene intermediate. The E- and Z- geometric isomers equilibrate under the reaction conditions and the Z-isomer undergoes thermal electrocyclization to afford a transient dihydropyridine. Rapid aerobic oxidation delivers the observed pyridine products.
Although the pyridine synthesis appears quite general in scope, a substrate bearing CH3 as R1 generated product mixtures from which only low yields of the anticipated pyridine were isolated (Scheme 3). It therefore appears that the ketoximine substitutent R1 cannot be attached to the imine carbon via an sp3 C-H. The inefficiency of this reaction when R1 is methyl is presumed to result from a thermally favorable 1,5-hydrogen shift in the 3-azatriene intermediate.9 The unproductive E-3-azatriene isomer would be favored over the more congested Z-3-azatriene geometric isomer at equilibrium further compromising the desired pyridine pathway. α,β-Unsaturated ketoxime O-pentafluorobenzoates in which the double bond was part of an aromatic system (phenyl, furan, indole) were also ineffective substrates, at least under the current reaction conditions.
Scheme 3.

Side Reactions Observed with Alkyl Ketoximines
In conclusion, a simple, modular method to prepare highly substituted pyridines has been disclosed. The method employs a cascade reaction comprising (1) a novel N-iminative, Cu-catalyzed cross-coupling of alkenylboronic acids at the N—O bond of α,β-unsaturated ketoxime O-pentafluorobenzoates, (2) electrocyclization of the resulting 3-azatriene, and (3) air oxidation affording highly substituted pyridines in moderate to excellent isolated yields (43–91%). Starting materials are readily available and functional group tolerance is quite good. The ease of construction of α,β unsaturated ketoximines and the broad availability of alkenylboronic acids implies that an extensive range of substituents can be selectively incorporated around the pyridine ring, but the full potential of this methodology and an exploration of a greater variety of substituents must await additional studies. In addition to its use in pyridine synthesis, this new and simple Cu-catalyzed N-iminoalkenylation invites additional mechanistic and synthetic studies of relatively unexplored 3-azatrienes.
Supplementary Material
Acknowledgments
The National Institutes of General Medical Sciences, DHHS supported this investigation through grant No. GM066153. Dr. Gary Allred of Synthonix provided the boronic acids used in our studies.
Footnotes
Supporting Information Available:. Experimental procedures, synthesis and characterization of all new compounds (48 pages) and scanned spectra (97 pages). This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.(a) Liu S, Yu Y, Liebeskind LS. Org Lett. 2007;9:1947–1950. doi: 10.1021/ol070561w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zaman S, Mitsuru K, Ab AD. Org Lett. 2005;7:609–611. doi: 10.1021/ol047628p. [DOI] [PubMed] [Google Scholar]; (c) Narasaka K, Kitamura M. Eur J Org Chem. 2005:4505–4519. [Google Scholar]; (d) Yu Y, Srogl J, Liebeskind LS. Org Lett. 2004;6:2631–2634. doi: 10.1021/ol048982q. [DOI] [PubMed] [Google Scholar]; (e) Narasaka K. Pure Appl Chem. 2002;74:143–149. [Google Scholar]
- 2.(a) O’Hagan D. Nat Prod Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; (b) O’Hagan D. Nat Prod Rep. 1997;14:637–651. [Google Scholar]
- 3.(a) Heller B, Hapke M. Chem Soc Rev. 2007;36:1085–1094. doi: 10.1039/b607877j. [DOI] [PubMed] [Google Scholar]; (b) Movassaghi M, Hill MD, Ahmad OK. J Am Chem Soc. 2007;129:10096–10097. doi: 10.1021/ja073912a. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Gutierrez AC. Org Lett. 2007;9:1473–1476. doi: 10.1021/ol070163t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meketa ML, Weinreb SM. Org Lett. 2007;9:853–855. doi: 10.1021/ol063038a. [DOI] [PubMed] [Google Scholar]; (e) Sydorenko N, Hsung RP, Vera EL. Org Lett. 2006;8:2611–??. doi: 10.1021/ol060932t. [DOI] [PubMed] [Google Scholar]; (f) Movassaghi M, Hill MD. J Am Chem Soc. 2006;128:4592–4593. doi: 10.1021/ja060626a. [DOI] [PubMed] [Google Scholar]; (g) Movassaghi M, Hill MD. J Am Chem Soc. 2006;128:14254–14255. doi: 10.1021/ja066405m. [DOI] [PubMed] [Google Scholar]; (h) Sklenicka HM, Hsung RP, McLaughlin MJ, Wei LL, Gerasyuto AI, Brennessel WW. J Am Chem Soc. 2002;124:10435–10442. doi: 10.1021/ja020698b. [DOI] [PubMed] [Google Scholar]; (i) Ciufolini MA, Hermann CYW, Dong Q, Shimizu T, Swaminathan S, Xi N. Synlett. 1998:105–114. [Google Scholar]; (j) Kelly TR, Liu H. J Am Chem Soc. 1985;107:4998–4999. [Google Scholar]; (k) Boger DL. Chem Rev. 1986;86:781–793. [Google Scholar]; (l) Kröhnke F. Synthesis. 1975:1–24. [Google Scholar]
- 4.Bagley MC, Dale JW, Bower J. Synlett. 2001:1149–1151. [Google Scholar]
- 5.(a) Tanaka K, Mori H, Yamamoto M, Katsumura S. J Org Chem. 2001;66:3099–3110. doi: 10.1021/jo005779+. [DOI] [PubMed] [Google Scholar]; (b) Lee CH, Jiang M, Cowart M, Gfesser G, Perner R, Kim KH, Gu YG, Williams M, Jarvis MF, Kowaluk EA, Stewart AO, Bhagwat SS. J Med Chem. 2001;44:2133–2138. doi: 10.1021/jm000314x. [DOI] [PubMed] [Google Scholar]; (c) Molina P, Pastor A, Vilaplana MJ. J Org Chem. 1996;61:8094–8098. doi: 10.1021/jo960940v. [DOI] [PubMed] [Google Scholar]; (d) Bonini C, D’Auria M, Funicello M, Romaniello G. Tetrahedron. 2002;58:3507–3512. [Google Scholar]; (e) Panunzio M, Bongini A, Tamanini E, Campana E, Martelli G, Vicennati P, Zanardi I. Tetrahedron. 2003;59:9577–9582. [Google Scholar]
- 6.For the current study the initial intermediate from cross-coupling has not been characterized. On thin layer chromatography it has a distinctive yellow color and is UV-active; on that basis it is tentatively assumed to be the 3-azatriene and not the dihydropyridine. Upon further heating the yellow tlc spot disappears and is converted to a new colorless spot that leads to the pyridine product upon workup. Additional studies will clarify the status of the intermediates in this new pyridine synthesis.
- 7.Snyder HR, Konecky MS, Lennarz WJ. J Am Chem Soc. 1958;80:3611–3615. [Google Scholar]
- 8.Yin LX, Liebscher J. Chem Rev. 2007;107:133–173. doi: 10.1021/cr0505674. [DOI] [PubMed] [Google Scholar]
- 9.Saettel NJ, Wiest O. J Org Chem. 2000;65:2331–2336. doi: 10.1021/jo991488t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.