

Abstract
Modulation of interactions between activated GPCRs (G-protein coupled receptors) and the intracellular (IC) signal transducers, heterotrimeric G-proteins, is an attractive, yet essentially unexplored, paradigm for treatment of certain diseases. Regulating downstream signaling for treatment of congenital diseases due to constitutively active GPCRs, as well as tumors where GPCRs are often overexpressed, requires the development of new methodologies. MEDSET (Modeling, Experimental data, Docking, Scoring and Experimental Testing) was developed to discover inhibitors that target the IC loops of activated GPCRs. As proof-of-concept, MEDSET developed and utilized a model of the interface between photoactivated rhodopsin (R*) and transducin (Gt), its G-protein. A National Cancer Institute (NCI) compound library was screened to identify compounds that bound at the interface between R* and its G-protein. High-scoring compounds from this virtual screen were obtained and tested experimentally for their ability to stabilize R* and prevent Gt from binding to R*. Several compounds that modulate signal transduction have been identified.
GPCRs (G-coupled protein receptors) are seven-helix transmembrane (TM) proteins that transduce extracellular (EC) signals to intracellular (IC) effectors for broad ranges of physiological signal processes, including chemical (hormonal peptides and proteins, as well as neurotransmitters), smell, taste and vision. In GPCRs, a conformational change from an inactive to an activated state upon agonist binding to the EC loops regulates G-protein binding to the IC loops and subsequent activation of signal transduction (1).
GPCRs have historically been targets for drug development. About 50% of recently launched drugs target GPCRs, yielding annual sales greater than $30 billion (2). With data available from the human genome project, the number of drugs that target GPCRs is expected to grow, as several hundred orphan GPCRs, the most common protein family in the human genome, were revealed by sequence comparisons. Only about 30 GPCRs are currently targeted by drugs on the market (2).
Despite the abundance of GPCRs targeted by the pharmaceutical industry, very little 3D structural data exists for the family of GPCRs, making structure-based agonist and antagonist design problematic. In 2000, rhodopsin, in its inactive state (R), was the first GPCR for which a X-ray crystal structure was solved (3). A X-ray crystal structure spectrally similar to the MII-photoactivated state (R*) was solved (4), but the TM structure observed in that crystal conflicted with a large amount of biophysical data that suggests significant movement of helices associated with the conformational change from R→R* (5, 6). The X-ray crystal structure of the human β2-adrenergic receptor, a Type A GPCR similar to rhodosopin, has also been solved (7, 8). Recently, ab initio models (9) and homology models (10-15) of GPCRs based on R have been shown to be adequate for structure-based virtual screening for EC agonist and antagonists. In these cases, the homology models and virtual screening have focused on the TM domain closest to the EC side, usually attempting to find a molecule that will bind and block agonist binding. An alternative target for inhibition of signal transduction is the interface between the IC side of the activated GPCR and its G-protein. Various peptides based on the C-terminal end of the G-protein α-subunit are known to bind to GPCRs and block signaling (16), and only recently, a small molecule, BIM-46174 (2-Amino-1-(8-cyclohexylmethyl-2-phenyl-5,6-dihydro-8H-imidazo[1,2-a]pyrazin-7-yl)-3-mercapto-propan-1-one), was reported that inhibited the binding of a GPCR to G-protein, as well as growth in a number of human cancer cell lines (17). These results demonstrate the feasibility of targeting the interface between an activated GPCR and its G-protein to block signal transduction.
In this study, a combination of experiments and computational techniques were used to find inhibitors that modulate GPCR/G-protein signal transduction by binding to GPCR activated IC loops. The method, MEDSET (integration of Modeling, Experimental data, Docking, Scoring, and Experimental Testing), provides a systematic approach for modeling activated GPCR/G-protein complex interfaces, then virtually screens compound libraries to find lead compounds for treatment of congenital diseases caused by continuous activation of GPCRs. The visual GPCR of the eye, rhodopsin, and its G-protein, transducin (Gt), were used as a model system for proof-of-principle of MEDSET. A working model of photoactivated rhodopsin (R*) bound to Gt was deduced by combining molecular modeling and docking with mutational data and experimental TrNOE structures of the recognition motif of the C-terminal segment of the α-subunit of Gt. To validate this model of the interaction of the IC loops of R* with Gt, a small molecule library was virtually screened for potential leads that could inhibit R*/Gt interactions. This is the first time a model of the IC loops of an activated GPCR bound to a G-protein has been used to identify compounds that inhibit signal transduction by binding at the GPCR/G-protein interface. MEDSET, integrating experimental data with modeling, is generally applicable to modulating transduction mediated by all classes of G-protein/GPCRs of pharmaceutical interest, particularly in oncology where multiple GPCRs are overexpressed on many tumor cells.
Results
Virtual Screening Results
A working model of Gtα(340-350) bound to the R* IC loops was generated in a previous study (18). Out of 1,990 compounds of the NCI Diversity Set docked to the “inferred” model of R* IC loops, 55,680 poses were obtained (Fig. 1, Panel A & B). X-Score, Autodock, and CSCORE were run on all of these poses (Fig. 1, Panel C) and were ranked independently. The top 10% of poses (5,568) from each scoring function were placed in separate sets. CSCORE had many compounds with the same consensus score. The CSCORE Scores ranged from 1 to 4, with 4 being the best score. Compounds that scored a 4 were all in the top 10% of poses, but the total number of compounds that scored either a 3 or a 4 exceeded the top 10% of poses. The compounds with a CSCORE of 3 were also ranked with ChemScore when taking the top 10% of solutions. For X-Score, compounds that had a LogP value calculated by X-Score greater than 5 were eliminated from the list, yielding 48,688 poses, then the top 5,568 poses were taken and placed into a set. The intersection of poses from Autodock, X-Score, and CSCORE sets were calculated, yielding 682 total poses of 133 unique compounds. The intersection of the Autodock and X-Score sets yielded 1,927 poses, the Autodock and CSCORE sets yielded 1,832 poses, and CSCORE and X-Score sets yielded 1,591 poses. Only 177 of the 682 poses passed through the distance filter (Fig. 1, Panel D), yielding 51 unique compounds. A subset of these unique compounds (Fig. 1, Panel E) were chosen for experimental testing by ranking the poses by either the X-Score or the Autodock score, and the top 10 compounds from each ranking were investigated further (1 (NSC1614), 2 (NSC88135), 3 (NSC88915), 4 (NSC93241), 5 (NSC95090), 6 (NSC128437), 7 (NSC159628), 8 (NSC521777), 9 (NSC601364)). The NCI Diversity Set provides information about the purity of a compound and also verifies the theoretical molecular weight experimentally. Only compounds with experimental molecular weights validated by the NCI were ordered from the NCI for testing (Fig. 2). Compounds sometimes were ranked highly by both Autodock and X-Score, so the two sets overlapped (Supporting Information, Fig. 1). The binding pose of Gtα(340-350) found previously (18) was also scored using Autodock, X-Score, and CSCORE for comparison with the most promising docked small molecules. Gtα(340-350) scored higher because the peptide is much larger than compounds in the NCI Diversity Set with more binding interactions from its larger surface interface (Supporting Information, Fig. 1).
Fig. 1.

Flow chart of virtual screening methodology. The final docked binding energy, which consists of the final intermolecular energy of the complex plus the final internal energy of the ligand, was obtained from Autodock3. The average X-Score is an average of three similar scoring functions that calculate hydrophobic contribution differently. The following scoring functions were evaluated in SYBYL (G Score, PMF Score, D Score, and ChemScore), then CSCORE was used to obtain a consensus score.
Fig. 2.

Compounds experimentally tested. Compounds 1-9 are from the NCI Diversity Set. These compounds scored highest with either the X-Score or Autodock scoring functions. Compound 10 is from the Open NCI Database and is similar to 3, and Compounds 11-16 are from the Open NCI Data base and are similar to 2.
Experimental Testing
Compounds ordered from the NCI were assayed by monitoring extra MII stabilization (19). The compounds all had limited solubility with the assay buffer typically used. Instead of dissolving the compounds in assay buffer, the compounds were first dissolved in DMSO, diluted into assay buffer and filtered. Experiments showed that the rhodopsin MII-assay tolerated up to 50% DMSO; however, the equilibrium between MI and MII gradually shifted toward MII with higher concentrations of DMSO, increasing the background level of MII in the assay. In the initial scan for compound activity, the final concentration of DMSO in the assay was 5%. Compound activity was assessed using UV/Visible difference spectroscopy, based on the ability of the compound to stabilize the MII state when compared to a negative control without compound and to a positive control with the Gtα(340-350) peptide. Compounds 2 and 3 were both considered “hits” after the initial scanning round because they showed increased MII stabilization compared to the negative control. Given that NCI compounds vary in purity, a pure sample of 2 was obtained to ensure that 2, rather than some unknown contaminant, was responsible for activation and to accurately determine its EC50 value. The other hit, compound 3 was not available commercially, but the structure was verified using 1-D NMR; EC50 values were measured using 3 obtained from the NCI Diversity Set. The EC50 value of 2 and 3 were 120 μM and 45 μM, respectively (Fig. 3a). Further, the dose-dependent inhibition of R*-Gt showed that both 2 and 3 inhibited Gt with an IC50 value of 180 μM and 15 μM, respectively (Fig. 3b). The dose-response MII stabilization curves for 2 and 3 show a decline in MII stabilization around 20 to 40 μM for 3 and 200 to 250 μM for 2, similar to the decline in MII stabilization for Gtα(340-350) between 1 mM and 2 mM seen previously (20). The decline in Meta II stabilization appears to be due to the stabilization of Meta I at high compound concentrations (21) and partial non-specific effect on the Schiff base in R* (22, 23). High concentrations of compound do not affect R. For 2 and 3, the EC50 value was derived from the initial phase of the curve. The UV/Visible difference spectra are shown in the Supporting Information, Fig. 2. Acid-trapping experiments showed that the compounds stabilized MII, as there was a shift in Amax from 380 nm to 440 nm upon protonation of the Schiff base formed between rhodopsin and retinal.
Fig. 3.
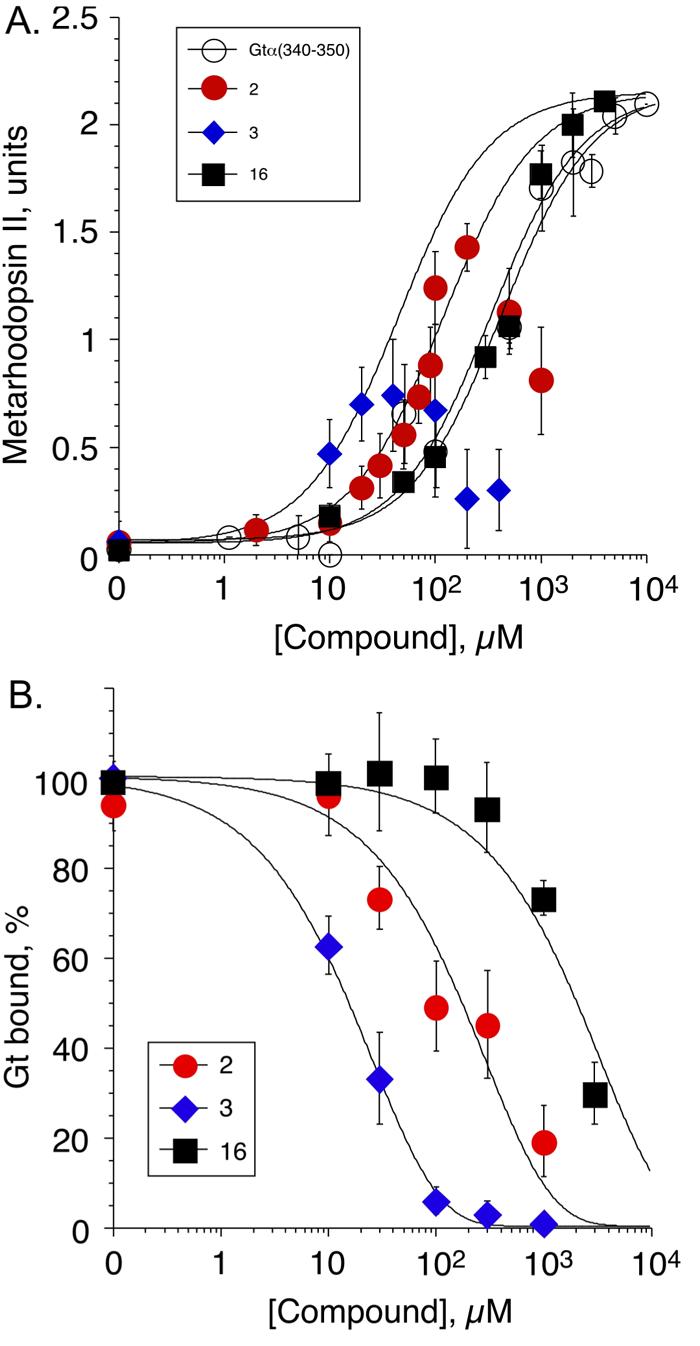
Experimental results of extra MII stabilization and inhibition of R*-Gt. a. Dose dependent stabilization of extra MII in the presence of specified compounds. For compounds 2 and 3, the EC50 values were fit based on the initial phase of the curve. b. Dose-dependent inhibition of R*-Gt interactions by the specified compounds.
The calculated docked conformations for 2 and 3 are shown in Fig. 4a & b. Compounds 2 and 3 dock in generally the same position as the docked conformation of Gtα(340-350) found previously (Fig. 4c) (18). Further, compound 2 makes several important interactions with the IC loop structure: hydrogen bonds with K245, Q312, P71; hydrophobic contacts with L72, A233, V138, K231, E249; and a salt bridge with K141. Compound 3 docks in two equally good poses. The main scaffold remains docked in the same position, but the aromatic group with Br in the para position can occupy either of two positions. Both conformations have the following contacts with the IC loop structure: hydrophobic contacts with K245, A246, A233, K231, K141, V137, V138. Conformation 1 has the following additional contacts: hydrophobic contacts with P71, F148; conformation 2 has the following additional contacts: hydrophobic contacts with T70, Q312.
Fig. 4.
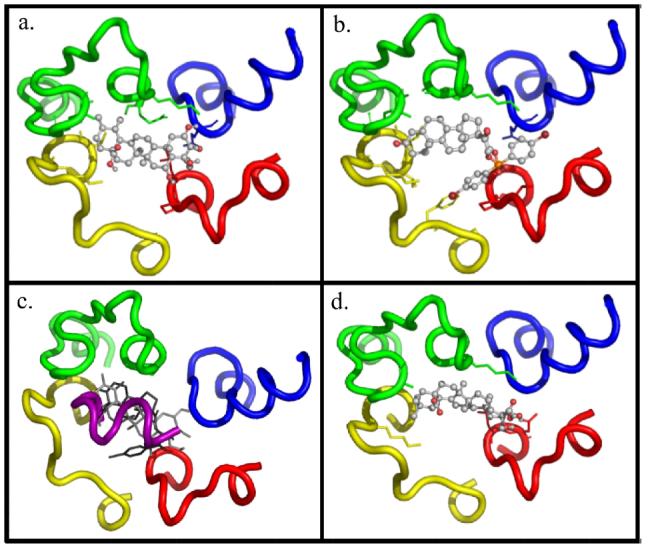
Compounds docked to the R* IC loops. Docked pose of Compound a. 2, b. 3, c. Overlap of Gtα(340-350), compound 2 and 16 on the loop structure, and d. 16. (Rendered using PyMol)
Similarity Search
Searching the entire Open NCI Database of 140,000 compounds (the NCI Diversity Library of 1990 compounds is a subset of this database) for compounds similar to 3, only compound 10 (NSC81395) (Fig. 2) was similar by 90% or greater (based on the Tanimoto index) and obtained from the NCI. The similarity search of the entire Open NCI Database yielded 15 additional compounds that were similar to 2 by 88% or greater based on the Tanimoto index. However, only 6 of these compounds were available through the NCI: 11 (NSC4060), 12 (NSC35347), 13 (NSC35350), 14 (NSC82878), 15 (NSC114945), 16 (NSC382025) (Fig. 2).
These seven similar compounds were assayed using the extra MII stabilization assay in 5% DMSO in the same manner as the initial compounds, except the concentration of compounds were 250 μM. Some compounds still precipitated, so all compounds were filtered. Thus, many of the compounds were at unknown saturating concentrations; therefore, estimated activity was a maximum. Compound 16 (Fig. 2), was significantly active in extra MII assay. Compounds 12 and 13 also showed some activity in the extra MII assay in the initial screen; however, neither showed significant activity when assayed using pure compound. In the docking experiment, compound 16 docks in the same position as 2 and 3 (Fig. 4d), making the following contacts with the IC loop: hydrophobic contacts with L72, K141, A233; hydrogen bonds with N73, K245; and a salt bridge with K141. Compound 16 yielded an EC50 value of 350 μM (Fig. 3a). Further, 16 inhibited the interaction between R* and Gt with an IC50 value of 2 mM in the R*-Gt assay (Fig. 3b).
Discussion
By combining both experimental data and computational techniques (MEDSET), compounds that modulate an activated GPCR/G-protein interaction were found. Rhodopsin, the GPCR involved in vision, and its G-protein, Gt, were used for proof-of-principle of MEDSET. Molecular modeling, docking, mutational data, and experimental TrNOE structures were used to create a working model of R* bound to Gt. For the first time, a model of GPCR loops of an activated GPCR was used to virtually screen a compound library for leads that inhibited signal transduction by blocking R*/Gt interactions.
Nine promising compounds were obtained using MEDSET to virtually screen a library of 1990 compounds, and these compounds were tested experimentally for their ability to stabilize R* and block the interaction between R* and Gt. Two of the nine compounds (2 and 3) stabilized R* and inhibited the interaction between R* and Gt. The compounds found in our computational screen had EC50 values similar to those found previously using an experimental high-throughput screen of a different small molecule library containing 10,000 compounds (24). Solubility and the colored nature of some compounds were limiting factors when screening in the present study. Hence, the results presented here are lower estimates for the number and affinities of compounds that stabilize R*. Further, compound 16, which had a 94% similarity score to 2, also stabilized R* and blocked the interaction between R* and Gt. Determining compounds similar to 2 and 3 that bind, or do not bind, experimentally should provide further information regarding the binding mode and the importance of functional groups in the ligand for binding. Modification for greater potency or specificity could be made using a focused combinatorial library based upon these leads, as well as providing indirect information about the binding site on R* to allow refinement of the working model of the complex. The main differences between the two sapogenins 16 and 2 are substitution of a carboxyl group on ring A for a hydroxyl methyl group, relocation of a methyl group from carbon 19 to 20 on ring E and removal of a hydroxyl group from carbon 6 on ring B. This observation suggests that additional affinity or specificity can be introduced by the synthesis of derivatives of 2 and 16.
The binding poses and contacts with the receptor for compounds 2 and 3 were investigated. The compounds dock to the loop structure in much the same way, stretching along the inside of the IC loop structure in a manner similar to our model of Gtα(340-350). The rings of the compounds extend nearly parallel to the helical axis of Gtα(340-350) (Fig. 4c). This is expected, considering the distance cutoffs imposed in the virtual screening process required that the small molecule be in proximity to certain residues throughout the loop structure. Other compounds that were tested experimentally also bound in a pose similar to 2, 3, and Gtα(340-350), so the scoring function played an important role in the screening process. In fact, 2 and 3 had a very high rank with both X-Score and Autodock score when compared to the other seven compounds tested experimentally, whereas the Autodock score for many other compounds had a much better rank than the X-Score or vice versa. Compound 3 has hydrophobic contacts with the receptor, whereas compound 2 makes hydrophobic contacts, some hydrogen bonds, and a salt bridge with the receptor. The difference in interaction may indicate a difference in specificity, as hydrophobic contacts are much less specific, and rationalize the difference in potency between 2 and 3. For 2, greater specificity may be attained through hydrogen bonding and a salt bridge, but this decreases binding affinity when compared to the cumulative hydrophobic contacts 3 makes.
To test how sensitive MEDSET was for the IC loop structure of the model of R*/ Gt and to determine if 2 and 3 were specific for the R* loop structure from Taylor et al. (18), loop models of inactive R were virtually screened as well to determine if 2 and 3 would have passed those filters and been chosen for testing. The R loop model was the “most open” loop structure from Nikiforovich et al (25). Neither 2 nor 3 would have been chosen for experimental testing with this R loop structure. Compound 3 passed through the scoring function and distance cutoffs (only distance cutoffs from experimental data were used); however, it did not rank high enough to be selected for testing experimentally. Compound 3 may have scored well because of the large number of hydrophobic contacts that it makes with both R and R* IC loops.
Compounds 2, 3, and 16 stabilize the MII state with nearly the same EC50 value, but vary much more in potency when tested for their ability to inhibit Gt binding to R*. These results are consistent with previous experimental evidence by Kisselev et al. comparing activities of Gtα(340-350) with Gtγ(60-71) farnesyl (26) and corroborate the hypothesis by Nikiforovich et al. that different binding modes may be necessary to either stabilize the MII or to inhibit Gt binding to the IC loops (27). The results from this study also indicate that it may be necessary to screen for both MII stabilization and G-protein inhibition when testing promising compounds that result from MEDSET.
Two of the three compounds found in this study are natural products, sapogenins. The second most potent compound (2), which had an EC50 value of 120 μM (about 3 times better than the eleven-residue peptide Gtα(340-350)), is a derivative of asiatic acid commonly known as madecassic acid. Found in the plant Centella asiatica in southeast Asia, extracts from this plant have many known medicinal properties, such as antiinflammatory, antineoplastic, ulcer-protective, wound healing, and for treatment of venous hypertension (28). Madecassic acid is orally available as a nutrapharmaceutical in both pill and liquid dosages (28). Compound 16 is also known as medicagenic acid, which is found in Medicago sativa (alfalfa) (28). In China, extract from Medicago sativa has been found to have therapeutic value for use as a diuretic, treating intestinal and kidney disorders, kidney stones, and poor night vision (29), as well as to enhance insulin activity and antibody production (30). It also has antifungal properties (31). Whether these diverse biological activities are related to modulation of GPCR/G-protein signal transduction is an interesting speculation that requires much further experimentation.
In conclusion, we have shown that a combination of experimental data and computational techniques allowed us to build a working model for the interacting interface between R* and Gtα(340-350). The model was used to virtually screen for compounds that stabilize R* and inhibit the interaction between R* and Gt. This is the first example of virtual screening on modeled IC loop structures of activated GPCRs. Three compounds both stabilized R* and blocked Gt binding, with one compound having an EC50 nine times better than that of the much larger Gtα(340-350) peptide itself and an IC50 in the low μM range. Using MEDSET, we were able to find compounds with EC50 values similar to those found in an experimental high-throughput screen done with far more compounds (24). The compounds found in this study are powerful leads for treating diseases such as retinitis pigmentosa and congenital night blindness, as the compound will bind and modulate the constitutive downstream activation of the vision cascade that is believed to be the mechanistic basis of the pathology associated with these diseases. Further, MEDSET could be used for fast and economical discovery of small molecule inhibitors of signal transduction for other important GPCRs associated with disease.
Methods
Modeling, Experimental Data, and Docking
The 3D IC loop model used in this study resulted from a previous computational docking study (Supporting Information) (18). Experimental TrNOE structures of Gtα(340-350) and its analogs (32-34) were docked onto the IC loops and revealed a common binding mode that make similar residue-residue interactions necessary for complex formation between R* and Gtα(340-350). The rhodopsin loop structure to which Gtα(340-350) docked in this common binding mode was used for the virtual screening experiments.
Screening
The NCI Diversity Set (35), containing 1,990 compounds, was downloaded in 3D format (36) (Fig. 1, Panel A). Certain compounds in this library were deemed problematic because Autodock3 could not properly process the compounds and were eliminated due to the presence of metal atoms or multiple fragments, yielding 1,856 compounds to use for screening (36, 37). Two additional compounds were found to be problematic after processing, due to the presence of metal atoms (Bis(2,2,6,6-Tetramethyl-4-piperidinone thiosemicarbazon)nickel(II) (NSC: 227147) and Bis(N-(2,4-dimethylphenyl)-3-hydroxy-2-naphthalenecarboxamidato)nickel(II) (NSC: 306698), and were eliminated. The compound structures had non-polar hydrogen atoms merged and Gasteiger charges added for docking with Autodock3. To prepare the loop structures, non-polar hydrogen atoms were merged, and Kollman charges were added to the rhodopsin loop structures using AutoDockTools.
The grid was centered on the receptor and a grid spacing of 0.375 Å with 120×120×120 points in each dimension. Autodock3 was used to sample conformational space around the receptor and within the rotatable bonds of the ligands using the Lamarkian genetic algorithm (37) (Fig. 1, Panel B). The default parameters for Autodock3 were used with the following exceptions: tstep, 0.5; qstep, 5.0; dstep, 5.0; ga_pop_size, 100; ga_num_evals, 5 000 000; ga_num_generations, 35 000; ga_elitism, 5; and ga_run, 30. These parameters were found to be efficient for screening and yield favorable compound enrichment in a MDM2 test case (Zhang and Marshall, unpublished). Any clustering done by Autodock3 was not used, and all poses obtained from the docking runs were considered in subsequent steps.
Scoring Solutions
The final docked binding energy from Autodock3 (37), the average X-Score (38), and CSCORE (39) were used to evaluate the poses generated by Autodock3 (Fig. 1, Panel C). First, the final docked binding energy, which consists of the final intermolecular energy of the complex plus the final internal energy of the ligand, was obtained from Autodock3 (37). Because Autodock merges non-polar hydrogens, hydrogens were added to the docked solutions using OpenBabel 2.0.2 (http://openbabel.sourceforge.net) before scoring with X-score and CSCORE. Secondly, the average X-Score, which is an average of three similar scoring functions that calculate hydrophobic contribution differently, was also used to score the docked poses (38). Third, CSCORE was used to evaluate all the binding poses (39). The following scoring functions were evaluated in SYBYL (G Score, PMF Score, D Score, and ChemScore), then CSCORE was used to obtain a consensus score. The coefficient of determination (R2) between the different scoring functions was found to be 0.23 or less, indicating that inclusion of each of the scoring functions provided additional data that was not present in the other scoring functions. Although this is a novel combination of scoring functions used in a virtual screening application, utilizing combinations of scoring functions has been shown to improve the results of virtual screening studies (40-43).
To choose compounds to test experimentally, poses that scored within the top 10% of poses using each scoring function were noted. In assessing the top 10% of X-Score solutions, a LogP filter was also imposed to try to eliminate large hydrophobic compounds that might be insoluble. Compounds that had a LogP calculated by X-Score greater than 5 were eliminated from consideration, and then the top solutions were noted. The intersection of the top 10% of poses that resulted from each scoring function was determined and was passed to the distance filter.
The distance filters (Fig. 1, Panel D) required that at least one of the atoms of a ligand in a particular pose be within 5 Å of one or more residues in each loop that had been determined to be important for Gt binding to R* via site-directed mutagenesis (Y136, V137, V138, V139, E247, K248, E249, N310, K311, Q312) (44-46). In addition, residues on R* (T70, L72, K141, K231, N244) found to be important for complex formation between R* and Gtα(340-350) during previous docking studies (18) were also included in a distance filter. Atoms of a pose were also required to be within 5 Å of each of these residues. Only polar hydrogens were considered on the compounds during distance measurements; however, all hydrogens were present on the receptor. Poses, meeting all distance cutoffs and filters, were ranked by Autodock score and X-Score, and the top 10 compounds resulting from Autodock and X-Score rankings (Fig. 1, Panel E) were chosen for experimental testing (Fig. 1, Panel F).
Similarity Search
A similarity search was done using the NCI Enhanced Web Browser (http://129.43.27.140/ncidb2/), which contains 140,000 compounds, to find compounds similar to compounds that stabilized R* in experiments. The browser was used to search the entire Open NCI Database, and compound similarity was assessed using the Tanimoto index (47).
Compounds Tested
The Gtα(340-350) peptide was manually synthesized using standard solid-phase Fmoc methods on Wang resin as described previously (32). The following compounds were ordered from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, NCI for use in the initial screen: 1, 2, 3, 4, 5, 6, 7, 8, 9 (Fig. 2). Subsequently, compound 10 resulted from the similarity search with 3, and 2 yielded the following compounds: 11, 12, 13, 14, 15, and 16. The purity of the compounds supplied by the NCI was used unless otherwise noted. To obtain more accurate EC50 and IC50 values for 2, pure compound was ordered from LKT Laboratories, Inc., St. Paul, MN. The experiments were performed in triplicate to derive error bars.
Rod Outer-Segment Preparation
Dark-adapted frozen bovine retinas were obtained from W. L. Lawson Co., Lincoln, NE. Rod outer segments (ROS) were prepared according to the method of Papermaster and Dreyer (48), and urea-washed ROS membranes as described (49, 50). The purity of rhodopsin was assessed on a silver-stained SDS-PAGE gel. ROS disc membranes were resuspended in buffer containing: 10mM Tris-HCl (pH 7.4), 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 0.1 mM PMSF, then stored at −70 °C.
Stabilization Assay
The absorbance spectra of rhodopsin in urea-washed rod outer-segment membranes (UM) were measured in the presence of the compounds ordered from the NCI; Gtα(340-350) was used as a control. Compounds obtained from the NCI were essentially insoluble in water and were dissolved in DMSO. The final concentration of the peptide in the assay was 3 mM, and the final concentration of the NCI compounds was 6 mM. However, the NCI compounds did not dissolve completely upon adding assay buffer to the DMSO solution, so the mixtures were filtered. Thus, saturating amounts of NCI compounds were used in the initial screen. The final concentration of DMSO in the initial screen was 5%, and the final concentration of rhodopsin in UM in the assay was 5 μM. The final buffer in the assay consisted of 20 mM Tris/HCl, 130 mM NaCl, 1 mM MgCl2, 1 μM EDTA, and 2 mM DTT, pH 8.0. After being dissolved in DMSO initially, compounds 2 and 16 were soluble in buffer with 5% DMSO so accurate dose-response measurements could be made. Compound 3 dissolved in a DMSO/butanol solution and remained soluble after adding buffer so that the final concentration of solvent in the buffer was 3% DMSO/3% butanol. Various concentrations of the compounds tested were added immediately before light activation. The concentration of organic solvents was equalized in all samples and did not exceed 5% DMSO for 2 and 16 and 3% DMSO/3% butanol mixture for 3. During the experiment, the samples were kept on ice and all solutions containing rhodopsin were prepared under a dim red light to prevent the rhodopsin from bleaching prematurely. The absorbance spectra was measured essentially as described (32, 33).
Acid-Trapping MII Intermediate
The MII state was verified by acid trapping (32, 51). Generally, the procedure is the same as the extra MII-stabilization assay with some exceptions. The absorbance spectra of rhodopsin and compound (NCI compound or Gtα(340-350)) was done in the dark, then after light activation. Following the light-activation scan, 1% HCl (v/v) was added and mixed. The sample was allowed to incubate for 5 minutes, and the absorbance spectrum was taken again.
R*-Gt Binding and Release Assay
The amount of Gt bound to R* was measured essentially as before (52). Briefly, the assay was performed in the linear range of dose-dependent Gt binding using the following conditions: 2 μg of Gt was reconstituted with 30 μg of UM in 100 μl of ROSIso buffer (10 mM Tris-HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.1 mM PMSF) under dark-red light on ice, essentially as described. Compounds 2 and 16 were dissolved in DMSO and remained soluble after adding buffer to a final concentration of 5% DMSO. Compound 3 dissolved in a DMSO/butanol solution and remained soluble after adding buffer so that the final concentration of solvent in the buffer was 3% DMSO/3% butanol. Various concentrations of the compounds tested were also added before light activation, and the mixture was incubated at 4 °C for 15 min. The concentration of organic solvents was equalized in all samples, including a positive control with no compound added and did not exceed 5% DMSO for 2 and 16 and 3% DMSO/3% butanol mixture for 3. The reaction was initiated by exposure of the samples to 480 nm light, followed by a 15 min incubation at 4 °C. UM were centrifuged at 109,000 × g, 4 °C for 10 min in a TLA-100.3 rotor on a Beckman TL-100 Ultracentrifuge. The pellet was washed twice with ROS-Iso buffer. UM with Gt bound was resuspended in buffer (10 mM Tris-HCl pH 7.4, 0.5 mM MgCl2, 1 mM DTT, 0.1 mM PMSF, and 250 μM GTPγS), incubated on ice for 30 min, and centrifuged as described above. The supernatant was analyzed for the presence of G-protein subunits by immunoblotting. Quantification was by image analysis of the ECL films. Band intensity calculations were in Image Gauge (FujiFilm). The experiment was performed in triplicate to derive error bars.
Supplementary Material
Acknowledgements
We thank Maureen Downs for technical assistance, and Prof. Jeff Kao for assistance with the NMR work. This work was partially supported by a W.M. Keck Fellowship in Molecular Medicine and NIH NRSA Postdoctoral Fellowship F32GM082200 (C.M.T.), National Institutes of Health grants GM68460, GM53630, and EY1211301 (G.R.M.), and Research to Prevent Blindness and NIH GM63203 grants (O.G.K.). The authors acknowledge the Drug Synthesis and Chemistry Branch of NCI for supplying most of the compounds used in the screening and testing. We would like to thank Prof. Gregory Nikiforovich for his helpful comments and excellent advice, and Nicole Rockweiler for helpful comments.
Abbreviations Footnote
- GPCR
G-protein coupled receptor
- NCI
National Cancer Institute
- R
dark-adapted rhodopsin
- R*
MII-photoactivated rhodopsin
- TrNOE
transferred nuclear Overhauser effect
- IC
intracellular
- EC
extracellular
- TM
transmembrane
- UM
urea-washed rod outer-segment membranes
- ROS
rod outer segment
- RMS
root mean square
- Gt
transducin
- Gtα(340-350)
transducin α-subunit C-terminal region
Footnotes
Supporting Information Available
Supporting Figures and Methods. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21(1):90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 2.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;3(10):928–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 4.Salom D, Lodowski DT, Stenkamp RE, Trong IL, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103(44):16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 6.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274(5288):768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 7.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450(7168):383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 9.Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. G protein-coupled receptors: in silico drug discovery in 3D. Proc Natl Acad Sci U S A. 2004;101(31):11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bissantz C, Schalon C, Guba W, Stahl M. Focused library design in GPCR projects on the example of 5-HT(2c) agonists: comparison of structure-based virtual screening with ligand-based search methods. Proteins. 2005;61(4):938–952. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 11.Bissantz C, Bernard P, Hibert M, Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of G-Protein Coupled Receptors suitable targets? Proteins. 2003;50(1):5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 12.Evers A, Hessler G, Matter H, Klabunde T. Virtual screening of biogenic amine-binding G-protein coupled receptors: comparative evaluation of protein- and ligand-based virtual screening protocols. J Med Chem. 2005;48(17):5448–5465. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- 13.Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the alpha1A adrenergic receptor. J Med Chem. 2005;48(4):1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 14.Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J Med Chem. 2004;47(22):5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 15.Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, Wang S. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J Med Chem. 2003;46(21):4377–4392. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 16.Gilchrist A, Mazzoni MR, Dineen B, Dice A, Linden J, Proctor WR, Lupica CR, Dunwiddie TV, Hamm HE. Antagonists of the receptor-G protein interface block Gi-coupled signal transduction. J Biol Chem. 1998;273(24):14912–14919. doi: 10.1074/jbc.273.24.14912. [DOI] [PubMed] [Google Scholar]
- 17.Prevost GP, Lonchampt MO, Holbeck S, Attoub S, Zaharevitz D, Alley M, Wright J, Brezak MC, Coulomb H, Savola A, Huchet M, Chaumeron S, Nguyen QD, Forgez P, Bruyneel E, Bracke M, Ferrandis E, Roubert P, Demarquay D, Gespach C, Kasprzyk PG. Anticancer activity of BIM-46174, a new inhibitor of the heterotrimeric Galpha/Gbetagamma protein complex. Cancer Res. 2006;66(18):9227–9234. doi: 10.1158/0008-5472.CAN-05-4205. [DOI] [PubMed] [Google Scholar]
- 18.Taylor CM, Nikiforovich GV, Marshall GR. Defining the Interface between the C-terminal Fragment of alpha-Transducin and Photoactivated Rhodopsin. Biophys J. 2007;92(12):4325–4334. doi: 10.1529/biophysj.106.099242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kisselev O, Ermolaeva M, Gautam N. Efficient interaction with a receptor requires a specific type of prenyl group on the G protein gamma subunit. J Biol Chem. 1995;270(43):25356–25358. doi: 10.1074/jbc.270.43.25356. [DOI] [PubMed] [Google Scholar]
- 20.Dratz EA, Furstenau JE, Lambert CG, Thireault DL, Rarick H, Schepers T, Pakhlevaniants S, Hamm HE. NMR structure of a receptor-bound G-protein peptide. Nature. 1993;363(6426):276–281. doi: 10.1038/363276a0. [DOI] [PubMed] [Google Scholar]
- 21.Szundi I, Lewis JW, Kliger DS. Effect of Digitonin on the Rhodopsin Meta I-Meta II Equilibrium. Photochemistry and Photobiology. 2005;81:866–873. doi: 10.1562/2005-02-10-RA-437. [DOI] [PubMed] [Google Scholar]
- 22.Janz JM, Farrens DL. Role of the Retinal Hydrogen Bond Network in Rhodopsin Schiff Base Stability and Hydrolysis. Journal of Biological Chemistry. 2004;279(53):55886–55894. doi: 10.1074/jbc.M408766200. [DOI] [PubMed] [Google Scholar]
- 23.Hubbard R, Brown PK, Bownds D. Methodology of Vitamin A and Visual Pigments. Meth. Enzym. 1971;18:615–653. [Google Scholar]
- 24.Gilchrist A, Hamm HE. US Patent 7208279 Method for identifying inhibitors of G-protein coupled receptor signalling. 2007
- 25.Nikiforovich GV, Marshall GR. Modeling flexible loops in the dark-adapted and activated States of rhodopsin, a prototypical G-protein-coupled receptor. Biophys J. 2005;89(6):3780–3789. doi: 10.1529/biophysj.105.070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kisselev OG, Meyer CK, Heck M, Ernst OP, Hofmann KP. Signal transfer from rhodopsin to the G-protein: evidence for a two-site sequential fit mechanism. Proc Natl Acad Sci U S A. 1999;96(9):4898–4903. doi: 10.1073/pnas.96.9.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikiforovich GV, Taylor CM, Marshall GR. Modeling of the complex between transducin and photoactivated rhodopsin, a prototypical G-protein-coupled receptor. Biochemistry. 2007;46(16):4734–4744. doi: 10.1021/bi700185p. [DOI] [PubMed] [Google Scholar]
- 28.Physician's Desk Reference for Herbal Medicines. 2nd ed. Medical Economics Company; Montvale, NY: 2000. pp. 12–13. 359-360. [Google Scholar]
- 29.Li TSC. Chinese and Related North American Herbs: Phytopharmacology and Therapeutic Values. CRC Press; Washington D.C.: 2002. p. 97. 204. [Google Scholar]
- 30.Lewis WH, Elvin-Lewis MPF. Medical Botany: Plants Affecting Human Health. 2nd ed. John Wiley & Sons, Inc.; Hoboken, NJ: 2003. p. 104. [Google Scholar]
- 31.Houghton P, Patel N, Jurzysta M, Biely Z, Cheung C. Antidermatophyte activity of medicago extracts and contained saponins and their structure-activity relationships. Phytother Res. 2006;20(12):1061–1066. doi: 10.1002/ptr.1995. [DOI] [PubMed] [Google Scholar]
- 32.Kisselev OG, Kao J, Ponder JW, Fann YC, Gautam N, Marshall GR. Light-activated rhodopsin induces structural binding motif in G protein alpha subunit. Proc Natl Acad Sci U S A. 1998;95(8):4270–4275. doi: 10.1073/pnas.95.8.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson MA, Ogbay B, Arimoto R, Sha W, Kisselev OG, Cistola DP, Marshall GR. Relative strength of cation-pi vs salt-bridge interactions: the Gtalpha(340-350) peptide/rhodopsin system. J Am Chem Soc. 2006;128(23):7531–7541. doi: 10.1021/ja058513z. [DOI] [PubMed] [Google Scholar]
- 34.Anderson MA, Ogbay B, Kisselev OG, Cistola DP, Marshall GR. Alternate binding mode of C-terminal phenethylamine analogs of G(t)alpha(340-350) to photoactivated rhodopsin. Chem Biol Drug Des. 2006;68(6):295–307. doi: 10.1111/j.1747-0285.2006.00460.x. [DOI] [PubMed] [Google Scholar]
- 35. http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html.
- 36.Lindstrom WH, Morris GM, Huey RH, Sanner MF, Olson AJ. The NCI Diversity Set for Autodock. 2003 http://www.scripps.edu/pub/olson-web/doc/autodock.
- 37.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian Genetic Algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–1662. [Google Scholar]
- 38.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46(12):2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 39.SYBYL7.3. Tripos International; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 40.Yang JM, Chen YF, Shen TW, Kristal BS, Hsu DF. Consensus scoring criteria for improving enrichment in virtual screening. J Chem Inf Model. 2005;45(4):1134–1146. doi: 10.1021/ci050034w. [DOI] [PubMed] [Google Scholar]
- 41.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson P. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44(3):793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 42.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem. 2000;43(25):4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 43.Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 44.Ernst OP, Meyer CK, Marin EP, Henklein P, Fu WY, Sakmar TP, Hofmann KP. Mutation of the fourth cytoplasmic loop of rhodopsin affects binding of transducin and peptides derived from the carboxyl-terminal sequences of transducin alpha and gamma subunits. J Biol Chem. 2000;275(3):1937–1943. doi: 10.1074/jbc.275.3.1937. [DOI] [PubMed] [Google Scholar]
- 45.Acharya S, Saad Y, Karnik SS. Transducin-alpha C-terminal peptide binding site consists of C-D and E-F loops of rhodopsin. J Biol Chem. 1997;272(10):6519–6524. doi: 10.1074/jbc.272.10.6519. [DOI] [PubMed] [Google Scholar]
- 46.Marin EP, Krishna AG, Zvyaga TA, Isele J, Siebert F, Sakmar TP. The amino terminus of the fourth cytoplasmic loop of rhodopsin modulates rhodopsintransducin interaction. J Biol Chem. 2000;275(3):1930–1936. doi: 10.1074/jbc.275.3.1930. [DOI] [PubMed] [Google Scholar]
- 47.Perez JJ. Managing molecular diversity. Chem. Soc. Rev. 2005;34:143–152. doi: 10.1039/b209064n. [DOI] [PubMed] [Google Scholar]
- 48.Papermaster DS, Dreyer WJ. Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry. 1974;13(11):2438–2444. doi: 10.1021/bi00708a031. [DOI] [PubMed] [Google Scholar]
- 49.Yamazaki A, Bartucca F, Ting A, Bitensky MW. Reciprocal effects of an inhibitory factor on catalytic activity and noncatalytic cGMP binding sites of rod phosphodiesterase. Proc Natl Acad Sci U S A. 1982;79(12):3702–3706. doi: 10.1073/pnas.79.12.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willardson BM, Pou B, Yoshida T, Bitensky MW. Cooperative binding of the retinal rod G-protein, transducin, to light-activated rhodopsin. Journal of Biological Chemistry. 1993;268:6371–6382. [PubMed] [Google Scholar]
- 51.Kito Y, Suzuki T, Azuma M, Sekoguti Y. Absorption Spectrum of Rhodopsin denatured with Acid. Nature. 1968;218:955–956. doi: 10.1038/218955a0. [DOI] [PubMed] [Google Scholar]
- 52.Kisselev OG, Downs MA. Rhodopsin-interacting surface of the transducin gamma subunit. Biochemistry. 2006;45(31):9386–9392. doi: 10.1021/bi060806x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.