

Abstract
Spinal cord GluR2-lacking AMPA receptors (AMPARs) contribute to nociceptive hypersensitivity in persistent pain, but the molecular mechanisms underlying this event are not completely understood. We report that complete Freund's adjuvant (CFA)-induced peripheral inflammation induces synaptic GluR2 internalization in dorsal horn neurons during the maintenance of CFA-evoked nociceptive hypersensitivity. This internalization is initiated by GluR2 phosphorylation at Ser880 and subsequent disruption of GluR2 binding to its synaptic anchoring protein (GRIP), resulting in a switch of GluR2-containing AMPARs to GluR2-lacking AMPARs and an increase of AMPAR Ca2+ permeability at the synapses in dorsal horn neurons. Spinal cord NMDA receptor-mediated triggering of protein kinase C (PKC) activation is required for the induction and maintenance of CFA-induced dorsal horn GluR2 internalization. Moreover, preventing CFA-induced spinal GluR2 internalization through targeted mutation of the GluR2 PKC phosphorylation site impairs CFA-evoked nociceptive hypersensitivity during the maintenance period. These results suggest that dorsal horn GluR2 internalization might participate in the maintenance of NMDA receptor/PKC-dependent nociceptive hypersensitivity in persistent inflammatory pain.
Introduction
AMPA receptors (AMPARs) are responsible for the majority of excitatory synaptic transmission and play a critical role in synaptic plasticity in the mammalian CNS. They are heterotetrameric cation channels composed of a combinational assembly of four subunits, GluR1–GluR4 (Burnashev et al., 1992). In contrast to GluR1, GluR3, and GluR4 mRNA, GluR2 mRNA undergoes efficient editing at a critical residue in the pore-forming M2 segment (Burnashev et al., 1992). Incorporation of these edited GluR2 subunits into AMPARs (GluR2-containing AMPARs) reduces AMPAR conductance, hinders Ca2+ permeation through AMPARs, and confers a linear current–voltage (I–V) relationship. Conversely, AMPARs assembled without the edited GluR2 subunits (GluR2-lacking AMPARs) have higher conductance, are readily Ca2+ permeable, and exhibit inwardly rectifying I–V relationships. Therefore, the presence or absence of synaptic GluR2 in the AMPAR complex greatly influences AMPAR properties and synaptic functions.
In adult mammalian CNS, most AMPARs have low Ca2+ permeability because GluR2 is fully edited throughout development and widely expressed (Burnashev et al., 1992; Greger and Esteban, 2007). However, AMPAR Ca2+ permeability in adult CNS is not static. It can be modified dynamically by neuronal activity, sensory experience, and neuronal insults via changes in trafficking, expression, and RNA editing of the GluR2 subunit (Isaac et al., 2007; Liu and Zukin, 2007). These changes might underlie the central mechanisms during physiological and pathological processes, such as synaptic plasticity and stroke.
Although all four AMPAR subunits are found within the spinal dorsal horn, GluR1 and GluR2 are the most abundant and are highly concentrated on the postsynaptic neuronal membranes of the superficial dorsal horn (Kerr et al., 1998; Lu et al., 2002). Targeted disruption of GluR2 gene produces a deficiency of dorsal horn GluR2-containing AMPARs, facilitates dorsal horn nociceptive plasticity, enhances long-term potentiation of primary afferent neurotransmission, and increases inflammatory hyperalgesia (Hartmann et al., 2004; Youn et al., 2008). In contrast, GluR1 and GluR3 knock-out mice display intact responses to persistent inflammatory insult, although GluR1 deletion decreases expression of dorsal horn GluR2-lacking AMPARs (Hartmann et al., 2004). These genetic studies indicate that GluR2, as a key subunit in the AMPAR complex, determines activity-induced nociceptive hypersensitivity and suggest that removal (or internalization) of synaptic GluR2 in dorsal horn neurons might contribute to spinal central sensitization, a specific form of synaptic plasticity, in persistent inflammatory pain. However, whether and how GluR2 are trafficked away from the synaptic membrane in dorsal horn neurons under inflammatory pain conditions is unknown.
Here we show that complete Freund's adjuvant (CFA)-induced peripheral persistent inflammation causes a time-dependent increase in GluR2 phosphorylation at Ser880 (GluR2-p Ser880) in dorsal horn. This phosphorylation is accompanied by reduced affinity of GluR2 to its synaptic anchoring protein (GRIP), internalization of synaptic GluR2, and a switch to synaptic GluR2-lacking AMPARs in dorsal horn neurons in the maintenance of CFA-induced inflammatory pain. GluR2-p Ser880 and its internalization are dependent on activation of the NMDA receptor (NMDAR)/protein kinase C (PKC) signaling cascade. Interfering with CFA-induced dorsal horn GluR2 internalization by preventing GluR2 PKC phosphorylation at Ser880 blunted CFA-evoked nociceptive hypersensitivity during the maintenance period. Together, our studies imply an involvement of dorsal horn GluR2 internalization in the maintenance of persistent inflammation-induced nociceptive hypersensitivity.
Materials and Methods
Animal preparation.
Male Sprague Dawley rats (250–300 g) were purchased from Hilltop Lab Animals. For chronic intrathecal drug delivery, a polyethylene (PE-10) tube was inserted into the subarachnoid space at the rostral level of the spinal cord lumbar enlargement segment through an incision at the atlanto-occipital membrane according to the method described previously (Tao et al., 2000; Zhang et al., 2003). For persistent sciatic nerve conduction blockade, an indwelling perisciatic catheter system was developed based on previous studies (Chacur et al., 2001; Wei et al., 2007). Perisciatic catheters were constructed from sterile gelfoam (Upjohn) aseptically cut into 15 mm (L) × 5 mm (W) × 9 mm (H) strips. One end was bisected (3.5 mm W) to a depth of 1 cm to allow a 4 cm sterile PE-10 tube to be sutured inside. The assembly was implanted around the sciatic nerve at midthigh level. The animals were allowed to recover for 5–10 d before being used experimentally. Rats showing any neurologic deficits postoperatively were discarded from the study. The position of the PE-10 catheter was confirmed in each animal after behavioral testing.
GluR2K882A knock-in (KI) mice were created as described previously (Chung et al., 2003; Steinberg et al., 2006). Adult GluR2K882A KI mice and control wild-type (WT) littermates (10–12 weeks) were obtained by interbreeding GluR2K882A heterozygous mice. Genotypes were determined by PCR analysis on digested mouse tail samples (Chung et al., 2003; Steinberg et al., 2006). Male and female GluR2K882A KI mice are viable and fertile without any observable developmental defects (Steinberg et al., 2006). They also display normal locomotor functions (supplemental Table S1, available at www.jneurosci.org as supplemental material).
To produce inflammation and nociceptive hypersensitivity, CFA (Mycobacterium tuberculosis; Sigma; 0.1 ml for rats and 0.02 ml for mice) suspended in an oil–saline (1:1) emulsion was injected subcutaneously into the plantar side of one hindpaw. Naive animals and saline (0.9%; 0.1 ml for rats and 0.02 ml for mice) injections were used as controls. The animals were used in accordance with protocols that were approved by the Animal Care and Use Committee at Johns Hopkins University.
Behavioral testing.
Animals were acclimatized to the experimental settings several times before the testing. The experimenters were blinded to the treatment groups and the mouse genotypes. Paw withdrawal responses to thermal and mechanical stimuli were measured in rats and mice as described previously (Tao et al., 2000, 2003; Zhang et al., 2003).
To measure paw withdrawal response to noxious heat stimuli, each animal was placed in a Plexiglas chamber on a glass plate located above a light box. Radiant heat from a Model 336 Analgesic Meter (IITC/Life Science Instruments) was applied by aiming a beam of light through a hole in the light box through the glass plate to the middle of the plantar surface of each hindpaw. When the animal lifted its foot, the light beam was turned off. The length of time between the start of the light beam and the foot lift was defined as the paw withdrawal latency. Each trial was repeated five times at 5 min intervals for each paw. A cutoff time of 20 s was used to prevent paw tissue damage.
To measure paw withdrawal response to repeated mechanical stimuli, each animal was placed in a Plexiglas chamber on an elevated mesh screen. For rats, a single trial of mechanical stimuli consisted of eight applications of a calibrated von Frey filament (8.01 mN; Stoelting) within a 2–3 s period. Each trial was repeated 10 times at 5 min intervals on each hindpaw. For mice, two calibrated von Frey filaments (0.24 and 1.24 mN) were used. Each filament was applied to the hindpaw for ∼1 s and repeated 10 times on each hindpaw. The occurrence of paw withdrawal in each of these 10 repeats was expressed as a percentage response frequency.
Locomotor function testing.
Three reflexes (placing, grasping, and righting) were tested as described previously (Tao et al., 2000; Zhang et al., 2003). (1) Placing reflex: the experimenter held the mouse or rat with hindlimbs slightly lower than the forelimbs and brought the dorsal surfaces of the hindpaws into contact with the edge of a table. The experimenter recorded whether the hindpaws were placed on the table surface reflexively. (2) Grasping reflex: the experimenter placed the animal on a wire grid and recorded whether the hindpaws grasped the wire on contact. (3) Righting reflex: the experimenter placed the animal's back on a flat surface and noted whether it immediately assumed the normal upright position. Scores for placing, grasping, and righting reflexes were based on counts of each normal reflex exhibited in five trials. In addition, the animal's general behaviors, including spontaneous activity (e.g., walking and running), were observed.
Antibodies.
The following antibodies were used: rabbit anti-GluR2 (Millipore Bioscience Research Reagents), mouse anti-GluR2 (Millipore Bioscience Research Reagents), rabbit anti-GluR2-p Ser880 (Chung et al., 2003; Steinberg et al., 2006), rabbit anti-PICK1 (Steinberg et al., 2006), rabbit anti-GRIP1 (Millipore), mouse anti-PSD-95 (Millipore), rabbit anti-PSD-93 (Alomone Labs), rabbit anti-NR2B (Millipore Bioscience Research Reagents), rabbit anti-NR1 (Millipore Bioscience Research Reagents), rabbit anti-NR2A/2B (Millipore Bioscience Research Reagents), rabbit anti-PKCα (Santa Cruz Biotechnology), rabbit anti-PKC β (Santa Cruz Biotechnology), rabbit anti-PKCγ (Santa Cruz Biotechnology), rabbit anti-N-cadherin (BD Transduction Laboratories), rabbit anti-stargazin (Millipore), mouse anti-α-adaptin (Sigma), and mouse anti-β-actin (Sigma).
Surface expression assay.
Surface biotinylation expression experiments were performed in dorsal horn neuronal culture and spinal cord slices as previously described (Tao et al., 2003). High-density cultured spinal dorsal horn neurons (1.5 × 106 cells/ml) and live transverse dorsal horn slices (400 μm) were prepared. Briefly, the cultured neurons or slices were incubated for 45 min on ice with artificial CSF (ACSF) containing 1.5 mg/ml sulfo-NHS-LC-biotin (Pierce) and then rinsed in ACSF to quench the biotin reaction. The cultured neurons were lysed and the slices homogenized in modified RIPA buffer (1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, 50 mm NaPO4, 150 mm NaCl, 2 mm NaF, 1 mm PMSF, 1 mg/ml leupeptin, and 10 mm sodium pyrophosphate). The homogenates were centrifuged at 10,000 × g for 15 min at 4°C, and the supernatant was harvested. After the measurement of protein concentration, 20% of the supernatant was removed to detect total expression of GluR2, NR1, and NR2B. The remaining supernatant was incubated with 50% NeutrAvidin agarose (Pierce) for 4 h at 4°C, and washed three times with RIPA buffer. Total and biotinylated surface proteins were detected using quantitative Western blots as described below. The surface/total ratio was calculated.
Subcellular fractionation.
Subcellular fractionation was performed as described previously (Matsuda et al., 2000) with minor modification. Briefly, the tissues were homogenized in homogenization buffer [250 mm sucrose, 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm PMSF, 1 mm benzamidine]. The homogenate was centrifuged at 1000 × g for 10 min at 4°C, and then the supernatant (S1, total soluble fraction) was collected. After centrifugation at 20,000 × g for 20 min at 4°C, the supernatant (S2, crude cytosolic fraction) and pellet (P1, crude plasma membrane fraction) were collected. The S2 was subsequently centrifuged at 150,000 × g for 1 h at 4°C, and the pellet was used as the 150,000 × g (150 k-g) fraction.
Immunoprecipitation and immunoblotting.
The sample proteins from different fractions were prepared as described above. For immunoprecipitation, the affinity-purified antiserum, with or without preincubation with excess fusion protein, was incubated with 100 μl of a 1:1 slurry of protein A-Sepharose for 1 h, and the protein–antibody complex was spun down at 2000 rpm for 4 min. The solubilized fractions were then added to the Sepharose beads and the mixture incubated for 2–3 h at 4°C. The mixture was washed with the modified RIPA buffer. The proteins were separated on 4% stacking/7.5% polyacrylamide gels by SDS-PAGE and then electrophoretically transferred onto nitrocellulose membrane. The membrane was blocked with 3% nonfat dry milk and subsequently incubated with the primary antibody (see antibodies listed above) overnight. The proteins were detected with horseradish peroxidase-conjugated secondary antibody and visualized by chemiluminescence reagents provided with the ECL kit (GE Healthcare) and exposure to film. The intensity of blots was quantified with densitometry.
Postembedding immunogold labeling.
Postembedding immunogold labeling was performed as described previously (Petralia et al., 1997; Tao et al., 2003; Ho et al., 2007). Briefly, rats were perfused transcardially with 4% paraformaldehyde + 0.5% glutaraldehyde 1 d after CFA or saline. Cryoprotected sections from the ipsilateral L5 superficial dorsal horns were frozen in Leica CPC and freeze-substituted into Lowicryl HM-20 in a Leica AFS freeze substitution instrument. Thin sections were labeled with monoclonal mouse (Ho et al., 2007) or polyclonal rabbit (Petralia et al., 1994) anti-GluR2 serum (Millipore Bioscience Research Reagents). Controls for specificity and selectivity of these antibodies were performed as described previously (Petralia et al., 1994; Ho et al., 2007). Areas for study were selected at random from the superficial dorsal horn at low magnification (i.e., synapses not visible) and then micrographs of synapses were taken at high magnification. Immunogold counts at synapses included all gold particles found in the synaptic cleft and postsynaptic density (Ho et al., 2007).
Electrophysiology.
Transverse slices (300–400 μm thick) of the L4–5 spinal cord with attached long (8–15 mm) dorsal roots were prepared from 17- to 25-d-old male rats at 1 d after CFA or saline as described previously (Vikman et al., 2008). Whole-cell patch-clamp recordings were taken from neurons located in lamina II [substantia gelatinosa (SG)]. Slices were continuously superfused with external solution [(in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 26 NaHCO3, and 10 glucose (dextrose), pH 7.4; osmolarity adjusted to 310–320 mOsm/L with sucrose, equilibrated with 95% O2, 5% CO2]. Patch pipettes (6–9 MΩ) were filled with internal solution [(in mm) 130 Cs-methylsulfonate, 10 NaCl, 10 EGTA, 2 CaCl2, 10 HEPES, 0.1 spermine tetrahydrochloride, 2 Mg-ATP, and 0.1 Na-GTP (pH adjusted to 7.2 with CsOH, osmolarity adjusted to 290 mOsm/L with sucrose)]. QX-314 (5 mm) was added to the internal solution to block voltage-activated sodium currents. Cells were held at −70 mV under voltage-clamp control. EPSCs were recorded at different membrane potentials from −70 mV to +50 mV using 20 mV steps. Dorsal roots were stimulated with a suction electrode. In most cases, stimulation intensities ranged from 70 to 400 μA (constant current output and consisting of 0.1 ms duration at frequency 0.1 Hz). Stimulus intensity was gradually increased until the maximal response was identified. The intensity was then adjusted slightly higher to ensure consistent axon activation under control conditions. Monosynaptic responses were selected based on the absence of synaptic failures and low variability of latency at a stimulation frequency of 10 Hz. At this frequency, polysynaptic responses tended to fail, and their latency markedly changed, but monosynaptic responses kept a constant latency. The latency was defined as the time interval between the stimulus artifact and the response onset. Latency values ranged from 5 to 20 ms. Neurons with high or low reversal potentials (|Erev| > 10 mV) for AMPA evoked EPSCs (eEPSCs) were not included in the analysis.
To study eEPSCs at different voltages, the membrane potential was stepped to a new value 350 ms before the dorsal root was stimulated. Short hyperpolarizing voltage steps to −75 mV were applied every minute to monitor input and access resistance. To isolate the synaptic current, membrane currents recorded in the absence of dorsal root stimulation at each membrane potential were subtracted from the synaptic record. Peak amplitude measurement of synaptic currents was performed on averages of five eEPSCs. The rectification index (RI) of the AMPA eEPSC was determined by dividing the AMPA eEPSC amplitude at +40 mV by the AMPA eEPSC amplitude at −70 mV. In experiments that used the polyamine toxin philanthotoxin-433 (PhTx 433; Sigma), EPSCs were evoked at a stimulation frequency of 0.1 Hz, and PhTx 433 was bath applied after the baseline was recorded. Clampfit 8.0 software (Molecular Devices) was used to analyze evoked synaptic currents.
Statistics.
All data are presented as mean ± SEM. Student's t tests or a one-way or two-way ANOVA followed by the post hoc Tukey tests were used to determine statistically significant differences (p < 0.05). The statistical software package SigmaStat (Systat) was used to perform all statistical analyses.
Results
CFA injection induces a time-dependent increase in dorsal horn GluR2 phosphorylation at Ser880
Evidence from in vitro studies shows that GluR2-p Ser880 is required for GluR2 internalization (Perez et al., 2001; Dev et al., 2004). To characterize GluR2 internalization in dorsal horn neurons in persistent inflammatory pain, we first examined whether CFA-induced peripheral inflammatory insult induced GluR2-p Ser880 in dorsal horn. The ipsilateral and contralateral L4–5 dorsal horn tissues were collected from naive rats and rats at 0.5 h to 14 d after injection of CFA or saline into a hindpaw. Consistent with the previous studies (Guo et al., 2002; Zhang et al., 2003), CFA, but not saline, injection produced mechanical hypersensitivity on the ipsilateral side, which developed over 0.5–2 h, reached a peak level around day 1, lasted for at least 7 d, and disappeared at day 14 (Fig. 1A). A time-dependent change in the level of GluR2-p Ser880 in the ipsilateral dorsal horn was observed when compared with naive rats (0 h) (Fig. 1B) (n = 5/time point). A significant increase in GluR2-p Ser880 began at 2 h (160 ± 20% of naive group; p < 0.05) and was maintained for at least 7 d after CFA injection (Fig. 1B). No significant changes were observed at 0.5 h (120 ± 8%; p > 0.05) or 14 d (95 ± 10%; p > 0.05) after CFA. As expected, the expression level of GluR2-p Ser880 in the contralateral dorsal horn was not markedly altered during the observed period (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Saline injection did not change the basal level of GluR2-p Ser880 in either side of dorsal horn (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). These results indicate that the increase in dorsal horn GluR2-p Ser880 is associated with CFA-induced peripheral inflammation.
Figure 1.
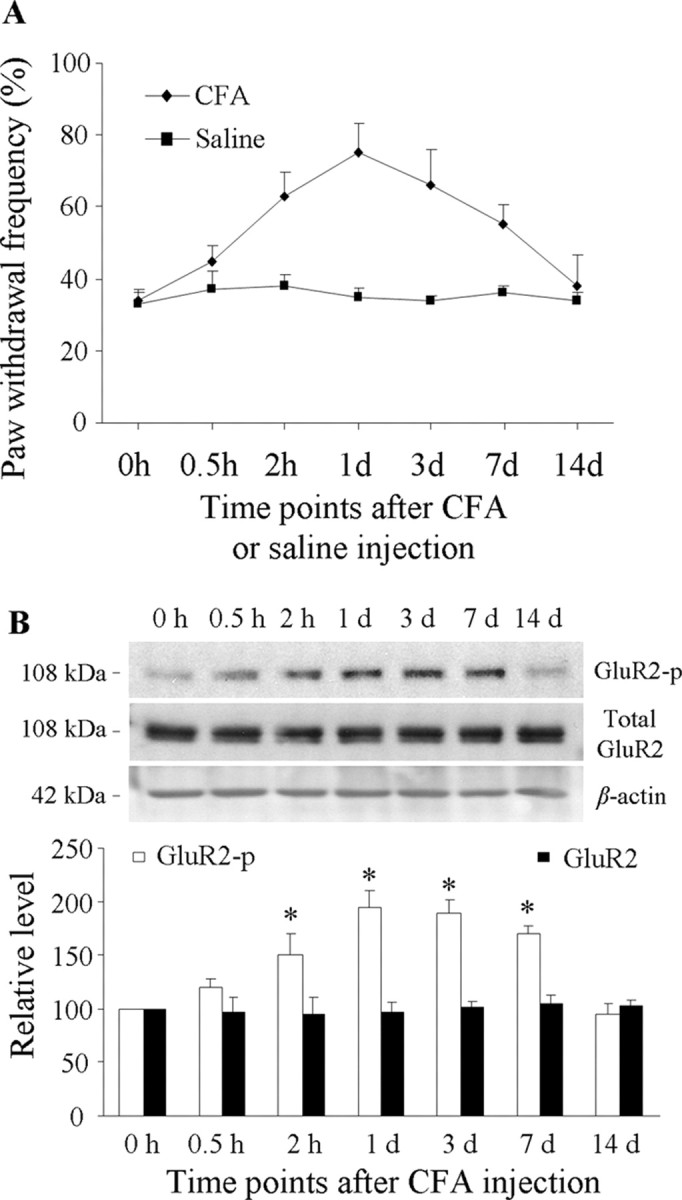
Increased mechanical hypersensitivity and dorsal horn GluR2 phosphorylation at Ser880 (GluR2-p) after CFA injection. A, Paw withdrawal frequency changed over time in response to mechanical stimulation on the ipsilateral side after CFA (n = 10/time point), but not saline (n = 10/time point), injection into a hindpaw. B, Top, Representative Western blots showing time-dependent changes of GluR2 phosphorylation at Ser880, but not of total GluR2, in the ipsilateral dorsal horn after CFA injection; bottom, statistical summary of the densitometric analysis expressed relative to the naive animals (0 h). *p < 0.05 versus the naive group.
We also examined whether persistent inflammatory insult affects total GluR2 protein expression in dorsal horn. Consistent with previous studies (Lee and Ro, 2007; Lu et al., 2008), quantitative Western blot analysis indicated that CFA injection did not lead to a significant change in the level of total GluR2 in either side of dorsal horn within a 14 d period (Fig. 1B; supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), indicating that CFA-induced inflammation alters GluR2 phosphorylation status rather than total GluR2 protein expression in dorsal horn neurons.
CFA injection disrupts binding of spinal GluR2 to GRIP1 and promotes synaptic GluR2 internalization in dorsal horn neurons
The C terminus of GluR2 contains a PDZ-binding motif that interacts with the PDZ domains of two scaffolding proteins, PICK1 and ABP/GRIP. ABP/GRIP anchors and stabilizes GluR2 at synaptic membranes (Dong et al., 1997, 1999a,b), whereas PICK1 presents PKC to ABP/GRIP–GluR2 complexes, leading to phosphorylation of GluR2 at Ser880 (Staudinger et al., 1997; Perez et al., 2001; Dev et al., 2004). GluR2 phosphorylated at Ser880 loses its ability to interact with ABP/GRIP, but the interaction of GluR2 with PICK1 remains intact (Chung et al., 2000; Perez et al., 2001). This differential effect may arise from the different structure of the ABP/GRIP and PICK1 PDZ domains (Chung et al., 2000). These in vitro results suggest that a CFA-induced increase in GluR2-p Ser880 might affect the binding affinity of GluR2 to ABP/GRIP (but not PICK1) and promote GluR2 internalization in in vivo dorsal horn neurons.
To test this hypothesis, we examined the interaction of spinal GluR2 with GRIP1 and PICK1 under CFA-induced inflammatory conditions. Coimmunoprecipitation was performed in ipsilateral L4–5 dorsal horn tissues at 1 d after CFA (n = 4) or saline (n = 4). In the saline-treated groups, GluR2 was immunoprecipitated with GRIP1 antibody or PICK1 antibody, indicating that GluR2 binds to both proteins in dorsal horn (Fig. 2). Interestingly, CFA injection significantly reduced the amount of GluR2 immunoprecipitated by GRIP1 antibody, although the amount of GRIP1 immunoprecipitated remained unchanged (Fig. 2A). The amount of GluR2 and PICK1 immunoprecipitated by PICK1 antibody in the tissues from CFA-treated rats was similar to that from the saline-treated rats (Fig. 2B). Total expression of GluR2, PICK1, and GRIP1 in dorsal horn from the CFA-treated rats was similar to that from the saline-treated groups (Fig. 2). These findings indicate a reduction in the binding affinity of GluR2 to GRIP, but not to PICK1, in dorsal horn neurons under persistent inflammatory pain conditions.
Figure 2.

Reduced binding affinity of GluR2 to GRIP1, but not PICK1, in dorsal horn 1 d after CFA. A, Left, Coimmunoprecipitation of GluR2 and GRIP1 with anti-GRIP1; middle, total expression of GluR2 and GRIP1; right, statistical summary for the ratios of the densities from the CFA-treated groups to those from the saline-treated groups. B, Left, Coimmunoprecipitation of GluR2 and PICK1 with anti-PICK1; middle, total expression of GluR2 and PICK1; right, statistical summary for the ratios of the densities from the CFA-treated groups to those from the saline-treated groups. IB, Immunoblotting; IP, immunoprecipitation; Sal, saline. *p < 0.05 versus the saline-treated groups. β-Actin was used as a loading control.
Next, we investigated whether this reduced affinity affects synaptic GluR2 membrane expression in dorsal horn neurons in vivo. Because GluR2 is transported away from the synaptic membrane mainly via clathrin-coated pit-dependent endocytosis (Matsuda et al., 2000), we used differential centrifugation to collect a 150 k-g spin fraction that contained abundant endocytosed, clathrin-coated vesicles (Matsuda et al., 2000) from the ipsilateral L4–5 dorsal horn tissues of naive rats (0 h) and rats at 2 h to 3 d after CFA (n = 5/time point) or saline (n = 5/time point). The crude membrane fraction was also fractionated. Consistent with previous studies (Matsuda et al., 2000; Tao et al., 2000), the 150 k-g spin proteins were effectively separated from the plasma membrane proteins (data not shown). We found that the amounts of GluR2 in the 150 k-g fraction were 195 ± 11% and 185 ± 5% higher than those of the naive group on days 1 and 3 after CFA, respectively, and correspondingly 67 ± 6% and 63 ± 8% lower in the membrane fraction at the same time points (Fig. 3A,B). Unexpectedly, the GluR2 level was not significantly changed in either the 150 k-g (96 ± 9% of the naive group; p > 0.05) or membrane (94 ± 4% of the naive group; p > 0.05) fractions at 2 h after CFA (Fig. 3A,B). No significant differences were observed between the saline-treated and naive groups in either the 150 k-g or membrane fractions at 2 h, 1 d, or 3 d after saline (Fig. 3A,B).
Figure 3.

Dorsal horn GluR2 internalization after CFA injection. GluR2 expression in 150 k-g spin fraction (A) and plasma membrane fraction (B) from ipsilateral dorsal horn at 2 h, 1 d, and 3 d after saline or CFA. Top, Representative Western blots; bottom, statistical summaries of the densitometric analysis expressed relative to the naive animals (0 h). *p < 0.05, **p < 0.01 versus the naive group. C, Surface expression of GluR2 and NR1 in dorsal horn neurons at 1 d after CFA or saline. Top, Representative Western blot; bottom, statistical summary of the densitometric analysis. The amount of sample loaded for the total (T) was 10% of that for the biotinylated surface (S). *p < 0.05 versus the saline-treated group. α-Actin, an unbiotinylated intracellular protein, was used as a control. D, Examples of postsynaptic immunogold labeling for GluR2 in superficial dorsal horn synapses illustrate labeling (arrowheads) in both the synapse and adjacent cytoplasmic structures 1 d after saline and only in cytoplasm 1 d after CFA. Pre, Presynaptic terminal; Post, postsynaptic structure. Scale bars, 100 nm.
To further confirm GluR2 loss in the membranes of dorsal horn neurons, we used a surface biotinylation expression assay to compare the surface expression of GluR2 in adult dorsal horn neurons between the saline-treated and CFA-treated groups. Live slices were prepared from the ipsilateral L4–5 dorsal horn 1 d after CFA or saline. The surface receptors were labeled with biotin and then precipitated (Tao et al., 2003; Holman and Henley, 2007). The ratio of surface to total GluR2 was determined by quantitative Western blotting. The biochemical analysis showed that surface GluR2 in the CFA-treated group was 56% lower than that in the saline-treated group (n = 5/group; p < 0.05) (Fig. 3C). CFA injection had no effect on surface expression of NMDAR subunit NR1 in dorsal horn 1 d after CFA (Fig. 3C), ruling out the possibility of nonspecific changes.
To directly display the changes in synaptic expression of GluR2 in the superficial dorsal horn neurons in persistent inflammatory pain, we used a combined approach of postembedding immunogold labeling with electron microscopy. Ultrathin sections were prepared from rat ipsilateral L4–5 superficial dorsal horn 1 d after CFA (n = 2) or saline (n = 2). Consistent with previous reports (Petralia et al., 1997; Morrison et al., 1998; Lu et al., 2002), GluR2 immunogold labeling was distributed mainly in postsynaptic profiles with few gold particles located presynaptically in the saline-treated group (Fig. 3D). CFA injection produced a decrease in the number of GluR2-labeled particles in the postsynaptic membrane and a corresponding increase in the cytoplasm (Fig. 3D). The number of GluR2-labeled particles/synapse in the CFA-treated group (n = 99 synapses) was 39% lower than that of the saline-treated group (n = 109 synapses) (p < 0.05). In addition, the ratio of the number of GluR2-labeled particles in the synapse to the number in the cytoplasm plus extrasynaptic membrane of the postsynaptic structure in the CFA-treated group was 54% lower than that in the saline-treated group (n = 47–64 profiles/group, p < 0.05). These findings further support the results obtained from the biochemical studies described above, indicating GluR2 internalization in dorsal horn neurons during the maintenance period of persistent inflammatory pain.
To examine whether CFA-induced inflammatory noxious input was required for dorsal horn GluR2 internalization, we infused vehicle (saline) or the nerve conduction blocker bupivacaine (0.5%; dissolved in saline; AstraZeneca) to the ipsilateral sciatic nerve for 1 d via an osmotic minipump (0.1 ml/h) beginning 3 h before or 2 d after CFA injection. The mechanical behavioral test was performed before infusion and on days 1, 2 or 3 after injection. L4–5 dorsal horns ipsilateral and contralateral to injection were collected on days 1 and 3 after behavioral tests, and the 150 k-g fraction was prepared. Consistent with previous reports (Beloeil et al., 2006; Wen et al., 2007), infusion of bupivacaine induced sensory (responses to pinch and von Frey filaments) and motor (responses to placing and grasping reflexes) blockade within 2.5–3 h. This effect lasted for at least 5–6 h, without signs of nerve damage, because all of these rats recovered to full sensory and motor responsiveness after the block wore off (data not shown). Therefore, none of the rats that were infused with bupivacaine for 1 d responded to ipsilateral mechanical stimulus on days 1 or 3 after CFA (Fig. 4A,C) (n = 5/group). Neither bupivacaine nor vehicle affected basal response to mechanical stimulus on the contralateral side (data not shown). Importantly, GluR2 protein expression in the ipsilateral dorsal horn 150 k-g fraction was significantly greater in the vehicle + CFA group than in the vehicle + saline group on days 1 and 3 after CFA (Fig. 4B,D) (p < 0.01; n = 4/group). CFA injection increased the amount of GluR2 by 1.75-fold on day 1 and by 1.9-fold on day 3 compared with saline injection. These increases were abolished in the bupivacaine + CFA group (94 ± 6.5% on day 1 and 90 ± 8% on day 3) (Fig. 4B,D). Bupivacaine treatment alone did not alter the expression of GluR2 in the 150 k-g fraction (data not shown). These findings indicate that peripheral inflammatory input is required to induce and maintain GluR2 internalization in dorsal horn neurons.
Figure 4.

Requirement of CFA-induced inflammatory input for induction and maintenance of dorsal horn GluR2 internalization. Perisciatic nerve infusion of bupivacaine (Bup) for 1 d beginning 3 h before (A, B) or 2 d after (C, D) CFA injection blocked paw withdrawal responses to mechanical stimulation on the ipsilateral side at times shown (A, C). Bup also blocked CFA-induced increases in the amount of GluR2 in the 150 k-g fraction (B, D). **p < 0.01 versus the vehicle (Veh) + saline group; ##p < 0.01 versus the vehicle + CFA group.
Synaptic GluR2-containing AMPARs in dorsal horn neurons of substantia gelatinosa decrease strikingly after CFA injection
GluR2 determines the properties of AMPARs, including Ca2+ permeability (Burnashev et al., 1992; Swanson et al., 1997). If CFA-induced inflammation drives synaptic GluR2 internalization, dorsal horn neurons would be expected to lose synaptic GluR2-containing AMPARs after CFA-induced inflammation. To test this hypothesis, we recorded EPSCs evoked by primary afferent stimulation from visually identified SG neurons of ipsilateral L4–5 dorsal horn 1 d after CFA or saline. The AMPAR component was isolated by application of 50 μm APV (an NMDAR antagonist), 10 μm bicuculline methiodide (a GABAR antagonist), and 2 μm strychnine hydrochloride (a glycine receptor antagonist). There were no significant differences between the saline-treated and CFA-treated groups in resting membrane potential (−60 ± 0.8 mV, n = 38 vs −58 ± 0.7 mV, n = 31, p > 0.5), neuronal input resistance (511.8 ± 49.9, n = 38 vs 586.1 ± 74.3, n = 31, p > 0.5), or threshold to evoked action potentials (−33.5 ± 0.9 mV, n = 38 vs −34.0 ± 1.4 mV, n = 31, p > 0.5). The eEPSCs were dramatically blocked by 95 ± 2% by an AMPAR antagonist, GYKI52466 (100 μm), but were not affected by a kainate receptor antagonist, SYM 2081 (3 μm), in either the saline-treated (n = 4) or CFA-treated (n = 4) rats (Fig. 5A), indicating that eEPSCs were mediated predominantly by AMPARs. Decay and rise times of eEPSCs revealed no significant differences between the saline-treated and CFA-treated rats (decay time: 1.3 ± 0.1 ms, n = 11 vs 1.2 ± 0.2 ms n = 10, p > 0.5; rise time: 4.7 ± 0.5 ms, n = 11 vs 5.7 ± 0.6 ms n = 9, p > 0.2).
Figure 5.

AMPAR-mediated evoked EPSCs at synapses between primary afferents and the SG neurons of dorsal horn. A, The evoked EPSCs were dramatically blocked by GYKI52466 (n = 4), but not by SYM 2081 (n = 4). Examples of EPSCs under control conditions and blockers. B, Pooled data for EPSC amplitude (normalized to mean amplitude before PhTx-433 application) versus time for experiments in which PhTx-433 (40 μm) was bath applied to spinal cord slices from rats 1 d after CFA (n = 4) or saline (n = 4). C, I–V curves at 1 d after saline or CFA. Inset at bottom, Examples of the eEPSCs at −70 mV and +40 mV holding potentials. **p < 0.01 versus the saline-treated group.
Polyamine toxin PhTx-433 is a selective blocker of Ca2+-permeable (GluR2-lacking) AMPARs (Tóth and McBain, 1998). Bath application of PhTx-433 (40 μm) inhibited AMPAR-mediated eEPSCs to a greater degree in SG neurons from the CFA-treated groups than in those from the saline-treated groups (Fig. 5B). Sixteen minutes after application of PhTx-433, eEPSC amplitude was inhibited by 51 ± 4% from baseline in the CFA-treated groups (n = 4) but only by 23 ± 3% from baseline in the saline-treated group (n = 4; p < 0.01). Given that AMPAR Ca2+ permeability is greatly influenced by the presence or absence of synaptic GluR2, this result indicates a reduction in the proportion of synaptic GluR2-containing AMPARs in SG neurons at 1 d after CFA.
As described above, another prominent feature of GluR2-lacking AMPARs is an inwardly rectifying I–V relationship (curve) of their eEPSCs, which can be quantitatively expressed as a RI. As shown in Figure 5C, I–V curves obtained from SG neurons of the saline-treated group had weak rectification at positive potentials, whereas a well marked inward rectification was observed in the SG neurons of the CFA-treated group. The SG neurons from the saline-treated animals had an average RI value of 0.26 ± 0.01 (range: 0.17–0.37, n = 16), whereas the average RI in SG neurons from the CFA-treated animals was 0.18 ± 0.02 (range: 0.08–0.29, n = 12; p < 0.01). Given that a reduced RI reflects the loss of synaptic GluR2-containing AMPARs (Tong and MacDermott, 2006), this finding further demonstrates CFA-induced synaptic GluR2 internalization in SG neurons of dorsal horn at 1 d after CFA.
Interestingly, the amplitudes of synaptic AMPAR-mediated eEPSCs at −70 mV (−191 ± 27 pA; n = 10) in the SG neurons of the CFA-treated animals were not markedly different from those of the saline-treated animals (−220 ± 45 pA; n = 14; p > 0.6). This suggests that the number of synaptic GluR2-lacking AMPARs in the SG neurons should be increased at 1 d after CFA. This increase may partially compensate for synaptic AMPAR EPSC reduction when synaptic GluR2-containing AMPARs are eliminated by synaptic GluR2 internalization. Indeed, the data from our laboratory and that of others showed that amount of GluR1 was increased in the postsynaptic density fraction from spinal cord 1 d after CFA (Katano et al., 2008; Park et al., 2008). These findings indicate a switch of GluR2-containing AMPARs to GluR2-lacking AMPARs at the synapses of dorsal horn neurons at 1 d after CFA.
Spinal NMDA receptor and PKC activation are involved in inflammation-induced phosphorylation and internalization of GluR2 in dorsal horn neurons
The increase of [Ca2+]i that is required for PKC activation can result from an inflow through NMDAR channels (Basbaum and Woolf, 1999). Because spinal cord NMDARs and PKC are activated under persistent inflammatory pain conditions (Basbaum and Woolf, 1999), we hypothesized that spinal NMDAR-mediated triggering of PKC activation would induce GluR2-p Ser880 and GluR2 internalization in dorsal horn neurons during the maintenance of CFA-induced inflammatory pain.
To study this hypothesis, we challenged an in vitro spinal dorsal horn neuronal culture with NMDAR agonist (NMDA, 40 μm) and antagonist (APV, 50 μm) or with PKC activator (PMA/TPA, 1 μm) and inhibitor (chelerythrine chloride, 10 μm). The drug doses used were based on previous studies of in vitro cultured brain neurons (Chung et al., 2000, 2003; Tigaret et al., 2006). PMA significantly induced phosphorylation of GluR2 at Ser880 and GluR2 internalization in dorsal horn neurons (Fig. 6A,B). Compared with the control groups (GluR2-p Ser880: 100%, n = 4; surface GluR2: 40 ± 5%, n = 4), the level of GluR2-p Ser880 increased by 2.3-fold (n = 4; p < 0.01) and surface GluR2 decreased by 75% (n = 4; p < 0.01). PMA-induced effects were abolished by chelerythrine chloride (GluR2-p Ser880: 110 ± 12.5%, n = 4; surface GluR2: 42 ± 4%, n = 4) (Fig. 6A,B). Chelerythrine chloride treatment alone did not affect basal expression of GluR2-p Ser880 (n = 4) or surface GluR2 (n = 4) (Fig. 6A,B). NMDA stimulation markedly increased the level of GluR2-p Ser880 (170 ± 6%, n = 4; p < 0.01) and reduced surface GluR2 (15 ± 6%, n = 4; p < 0.01) compared with the control groups (GluR2-p Ser880: 100%, n = 4; surface GluR2: 50 ± 2%, n = 4) (Fig. 6C,D). Moreover, NMDA-stimulated effects were attenuated dramatically not only by APV (GluR2-p Ser880: 107 ± 10%, n = 4; surface GluR2: 40 ± 8%, n = 4), but also by chelerythrine chloride (GluR2-p Ser880: 105 ± 7%, n = 4; surface GluR2: 42 ± 2%, n = 4) (Fig. 6C,D). These findings suggest that NMDAR activation induces PKC-mediated GluR2-p Ser880 and GluR2 internalization in dorsal horn neurons. APV treatment alone did not affect basal expression of GluR2-p Ser880 (n = 4) or surface GluR2 (n = 4) (Fig. 6C,D). PMA, chelerythrine chloride, NMDA, APV, and their combined treatment failed to change total GluR2 protein or surface NMDAR subunit NR2B expression (used as a control) (Fig. 6).
Figure 6.

Induction of GluR2 phosphorylation at Ser880 (GluR2-p Ser880) and GluR2 internalization in cultured dorsal horn neurons after NMDAR and PKC activation. PMA stimulation increased expression of GluR2-p Ser880 (but not total GluR2) (A) and reduced surface expression of GluR2 (but not NR2B) (B); these changes were abolished by chelerythrine chloride (Chy). NMDA stimulation increased expression of GluR2-p Ser880 (but not total GluR2) (C) and reduced surface expression of GluR2 (but not NR2B) (D); these changes were abolished by both APV and Chy. **p < 0.01 versus control group.
Activation of AMPARs and group 1 metabotropic glutamate receptors (mGluRs) induced GluR2 internalization in cultured hippocampal neurons (Lin et al., 2000; Snyder et al., 2001). [Ca2+]i may increase as a result of an inflow of extracellular Ca2+ through GluR2-lacking AMPARs and mobilization of intracellular stores through the activation of the IP3 pathway following group 1 mGluR activation. Thus, one might expect GluR2-lacking AMPAR and group 1 mGluR activation in dorsal horn neurons to induce PKC GluR2-p Ser880 and GluR2 internalization. To test this possibility, cultured dorsal horn and cortical neurons were treated with AMPA (100 μm), GYKI52466 (30 μm), DHPG (50 μm; agonist of group 1 mGluRs), and MPEP (100 μm; antagonist of group 1 mGluRs). The dosage of these drugs was based on previous in vitro studies (Lin et al., 2000; Snyder et al., 2001). In cultured cortical neurons, stimulation with AMPA and DHPG reduced surface GluR2 expression and was reversed by GYKI52466 and MPEP, respectively (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). However, in cultured dorsal horn neurons, these agonists did not significantly change basal levels of GluR2-p Ser880 or surface GluR2 expression (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These results suggest that in dorsal horn neurons, activation of AMPARs and group 1 mGluRs is not linked to induction of PKC GluR2-p Ser880 and GluR2 internalization.
Next we examined whether blocking spinal NMDAR and PKC activation affects CFA-induced increases in GluR2 internalization in dorsal horn during the maintenance of inflammatory pain. The rats were infused intrathecally with vehicle (n = 30), APV (25 nmol/μl/h, n = 15), or chelerythrine chloride (3 nmol/μl/h, n = 15) for 1 d via an osmotic minipump beginning 1 h before or 2 d after CFA (or saline) injection. The dosage of these two drugs was based on previous studies (McNally and Westbrook, 1998; Granados-Soto et al., 2000; Sun et al., 2006). The mechanical behavioral test was performed before drug infusion and on days 1, 2, or 3 after injection. L4–5 dorsal horns ipsilateral and contralateral to injection were collected on days 1 and 3 after behavioral tests, and the 150 k-g fraction was prepared. Pretreatment and posttreatment with APV or chelerythrine chloride significantly attenuated CFA-induced mechanical pain hypersensitivity on the ipsilateral side on days 1 and 3 after CFA; neither drug affected basal paw withdrawal responses to mechanical stimulation on either the ipsilateral or contralateral side (Fig. 7A,B,E). As expected, CFA injection markedly increased the amount of GluR2 in the 150 k-g fraction on days 1 and 3 after CFA compared with saline injection; these increases were markedly blocked by pretreatment or posttreatment with chelerythrine chloride (pretreatment: 105 ± 10% of the vehicle + saline group; posttreatment: 90 ± 7% of the vehicle + saline group) or APV (pretreatment: 116 ± 12% of the vehicle + saline group; posttreatment: 105 ± 4% of the vehicle + saline group) (Fig. 7C,D,F). Neither chelerythrine chloride (96 ± 6% of the vehicle + saline group) nor APV (110 ± 4% of the vehicle + saline group) treatment alone altered the expression of GluR2 in the 150 k-g fraction (Fig. 7C,D). These in vivo findings indicate that spinal NMDARs and PKC activation are required for induction and maintenance of CFA-induced GluR2 internalization in dorsal horn neurons.
Figure 7.

NMDAR/PKC-dependent mechanical hypersensitivity and dorsal horn GluR2 internalization on days 1 and 3 after CFA. Intrathecal infusion of Chy (A) or APV (B) for 1 d beginning 1 h before CFA injection attenuated CFA-induced ipsilateral mechanical hypersensitivity. **p < 0.01 versus the saline + vehicle group; #p < 0.05 versus the CFA + vehicle group. Intrathecal infusion of Chy (C) or APV (D) for 1 d beginning 1 h before CFA injection abolished CFA-induced increases in the amount of GluR2 in the 150 k-g fraction. **p < 0.01 versus the saline + vehicle group; ##p < 0.01 versus the CFA + vehicle group. Intrathecal infusion of APV and Chy for 1 d beginning on day 2 after CFA reduced CFA-induced mechanical hypersensitivity (E) and increased the amount of GluR2 in the 150 k-g fraction (F). **p < 0.01 versus the vehicle + saline group; #p < 0.05, ##p < 0.01 versus the vehicle + CFA group.
NMDARs and PKCα are physically coupled to AMPARs in dorsal horn
The above findings suggest a physical link between AMPARs and NMDARs or PKC at synapses in spinal cord dorsal horn. Using a coimmunoprecipitation technique, we demonstrated a complex assembled by PICK1 with GluR2 and PKCα or by a PSD-95-stargazin linkage with GluR2 and NMDAR subunit NR2B in rat spinal cord dorsal horn. PICK1 antibody immunoprecipitated PICK1, GluR2, and PKCα, but not PKCβ1 and PKCγ (Fig. 8A). This finding is in agreement with previous in vitro studies demonstrating that, among PKC isoenzymes, only PKCα contains a unique PDZ ligand that binds to PICK1 PDZ domain (Staudinger et al., 1997). In addition, GluR2 antibody immunoprecipitated GluR2 as well as PICK1 and PKCα (Fig. 8B), indicating a triple GluR2-PICK1-PKCα complex in dorsal horn. Immunoprecipitation also revealed that NR2B and stargazin bind to PSD-95 and that PSD-95 and GluR2 bind to stargazin in dorsal horn (Fig. 8C,D), indicating that PSD-95 interaction with stargazin acts as a bridge to couple NMDARs and AMPARs. Our previous studies and those of others showed that NR2A/2B, PSD-95, GluR2, and stargazin are highly expressed in superficial dorsal horn and that GluR2 colocalizes with stargazin and PSD-95 in this region (Tao et al., 2000, 2003, 2006; Polgár et al., 2008). Under electron microscopy, superficial dorsal horn sections exhibited double labeling for GluR2 and NR1 in the synapse (Fig. 8E), indicating colocalization of AMPARs and NMDARs. Together, these findings present a molecular basis for the physical coupling of NMDARs and PKC to AMPARs in dorsal horn.
Figure 8.

NMDARs and PKC couple to AMPAR complex in dorsal horn. A, PICK1 coimmunoprecipitates with GluR2 and PKCα (but not PKCβ or PKCγ). B, GluR2 coimmunoprecipitates with PICK1 and PKCα. C, PSD-95 coimmunoprecipitates with NR2B and stargazin. D, Stargazin coimmunoprecipitates PSD-95 and GluR2. E, NR1 (5 nm gold, arrowheads) and GluR2 (15 nm gold) colocalize at superficial dorsal horn synapses. Pre, Presynaptic terminal; Post, postsynaptic structure. Scale bar, 100 nm.
Targeted mutation of the GluR2 PKC phosphorylation site impairs pain hypersensitivity during the maintenance of CFA-induced inflammatory pain
Finally, we investigated whether inflammation-induced GluR2 internalization in dorsal horn neurons participates in pain hypersensitivity under persistent inflammatory pain conditions. Our findings presented above and previous studies (Chung et al., 2003; Steinberg et al., 2006) indicate that PKC GluR2-p Ser880 is the key initial regulatory step for induction of GluR2 internalization. Direct mutation of GluR2 Ser880 to alanine disrupts binding of GluR2 to GRIP (Dong et al., 1997; Osten et al., 2000), whereas mutation of GluR2 Ser882 to alanine (K882A) disrupts the PKC consensus site (S/T-T-K/R), prevents PKC phosphorylation of GluR2 at Ser880, and interferes with GluR2 internalization (but does not interfere with GluR2 binding to PICK1 and GRIP) in central neurons (Chung et al., 2003; Steinberg et al., 2006). Therefore, we used GluR2K882A KI mice to investigate whether preventing CFA-induced spinal GluR2 internalization affects CFA-induced nociceptive hypersensitivity.
Basal levels of GluR2-p Ser880 and GluR2 in the 150 k-g fraction from either side of the dorsal horn were similar in WT and GluR2K882A KI mice (Fig. 9A; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Consistent with the findings in rats, CFA injection significantly increased the level of GluR2-p Ser880 (170 ± 5%, n = 3, p < 0.05) and the amount of GluR2 in the 150 k-g fraction (180 ± 7%, n = 3, p < 0.05) from the ipsilateral (but not contralateral) dorsal horn of WT mice 1 d after injection (Fig. 9A; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In contrast, those indices were unchanged in GluR2K882A KI mice at the same time point (Fig. 9A; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Saline injection did not alter basal levels of GluR2-p Ser880 or the amount of GluR2 in the 150 k-g fraction in WT or GluR2K882A KI mice (Fig. 9A; supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 9.

Biochemical and behavioral characterization of GluR2 K882A KI mice after CFA injection. A, Level of GluR2 phosphorylation at Ser880 (GluR2-p Ser880; left) and amount of GluR2 in a 150 k-g fraction (right) from ipsilateral dorsal horns of naive (N), wild-type (WT), and GluR2 K882A KI mice 1 d after CFA (C) or saline (S). **p < 0.01 versus naive WT mice. B, Paw withdrawal response to thermal stimulation at times shown. *p < 0.05 versus the corresponding time points on the ipsilateral side in WT mice. C, D, Paw withdrawal responses to 0.24 mN (C) and 1.24 mN (D) mechanical stimuli at times shown. *p < 0.05 versus the corresponding time points on the ipsilateral side in WT mice.
WT mice injected with CFA in one hindpaw developed long-term thermal nociceptive hypersensitivity (indicated by a significant decrease in paw withdrawal latency) and mechanical hypersensitivity (indicated by a marked increase in paw withdrawal frequency) on the ipsilateral side (Fig. 9B–D). The nociceptive hypersensitivity developed at 2 h, reached a peak on day 1, and persisted for at least 7 d after CFA injection (Tao et al., 2003; Chu et al., 2005). The basal paw withdrawal responses to thermal and mechanical stimuli were similar between WT (n = 10/test) and GluR2K882A KI (n = 10/test) mice (Fig. 9B–D). Although GluR2K882A KI mice developed intact thermal and mechanical nociceptive hypersensitivities 2 h after CFA, both types of hypersensitivity were impaired during the remainder of the observation period (Fig. 9B–D). The magnitude of response latencies to thermal stimulation was significantly increased and the magnitude of response frequencies to mechanical stimuli markedly reduced from day 1 to day 7 after CFA in the GluR2K882A KI mice compared with those in the WT mice (p < 0.05). No significant changes in paw withdrawal responses to thermal and mechanical stimuli were seen on the contralateral side in either WT or GluR2K882A KI mice after CFA injection (Fig. 9B–D). These findings indicate that the GluR2K882A KI mice display blunted pain hypersensitivity during the maintenance phase (from day 1 to day 7 after CFA), but not during the development (2 h after CFA) of persistent inflammatory pain.
Discussion
Cumulative evidence suggests that activity-dependent AMPAR trafficking at postsynaptic membranes is critical for synaptic plasticity and structural remodeling (Liu and Zukin, 2007; Santos et al., 2009). However, the role of regulated AMPAR trafficking in spinal central sensitization associated with persistent pain is much less understood. Here, we show that AMPAR subunit GluR2 is internalized in dorsal horn neurons after CFA-induced inflammation. This internalization is dependent on spinal cord NMDAR/PKC activation and is causally linked to CFA-inflammatory pain.
Normally, AMPAR subunits are constitutively recycled between the synaptic membrane and the intracellular compartment via membrane insertion (exocytosis) and clathrin-mediated internalization (endocytosis). The number of subunits expressed on the synaptic membrane is dependent on the balance between these two processes (Adesnik et al., 2005; Greger and Esteban, 2007). Using biochemical and morphological approaches, we found that GluR2 is internalized in dorsal horn neurons during days 1–3 after CFA. These findings indicate that inflammatory insult breaks the balance of GluR2 recycling between membrane insertion and internalization and promotes GluR2 trafficking away from synaptic membrane. Moreover, this inflammatory noxious input may be indispensable for induction and maintenance of dorsal horn GluR2 internalization because this event could not be induced by preinfusion and postinfusion of bupivacaine to the sciatic nerve or by saline injection.
Our results suggest that dorsal horn GluR2 internalization requires PKC phosphorylation of GluR2 at Ser880 in persistent inflammatory pain. Consistent with previous studies on cultured brain neurons (Perez et al., 2001; Hanley and Henley, 2005), PMA increased the level of GluR2-p Ser880 and reduced surface expression of GluR2 in the cultured dorsal horn neurons; these responses were blocked by chelerythrine chloride. Moreover, intrathecal preinfusion and postinfusion of chelerythrine chloride or prevention of GluR2 PKC phosphorylation at Ser880 attenuated the CFA-induced increase in the level of GluR2 in the dorsal horn 150 k-g fraction on days 1 and 3 after CFA. In addition, the binding affinity of GluR2 to GRIP1 in dorsal horn neurons was reduced on day 1 after CFA. These findings suggest that peripheral inflammatory insult activates spinal PKC and induces PKC phosphorylation of GluR2 Ser880 in dorsal horn. This phosphorylation disrupts GluR2 binding to ABP/GRIP and promotes dorsal horn GluR2 internalization under inflammatory pain conditions (Fig. 10).
Figure 10.

Proposed model for the NMDAR/PKC-dependent dorsal horn GluR2 internalization under persistent inflammatory pain conditions. NMDARs couple to AMPARs through PSD-95 (which binds to NR2A/2B) interaction with stargazin (which binds to GluR1, GluR2, and GluR4). Under normal conditions, ABP/GRIP binds to and anchors GluR2 at synapses. Under persistent inflammatory pain conditions, NMDAR activation causes Ca2+ influx and PKCα activation. The latter phosphorylates GluR2 at Ser880 and disrupts GluR2 binding to ABP/GRIP, which leads to GluR2 internalization. GluR2 internalization results in an increase of AMPAR Ca2+ permeability. The increase in [Ca2+]i in dorsal horn neurons should initiate or potentiate a variety of Ca2+-dependent intracellular cascades that are associated with the maintenance of persistent inflammatory pain.
Interestingly, we found no significant change in the amount of GluR2 in the dorsal horn 150 k-g spin fraction 2 h after CFA injection, although the level of GluR2-p Ser880 at 2 h after CFA was significantly increased compared with the naive group. This unexpected result is most likely attributable to the limited sensitivity of Western blot analysis. GluR2 internalization in dorsal horn neurons might be insufficient to be detectable at 2 h after CFA injection. Another possibility is that there may be a long timescale of synaptic AMPAR exchange for intracellular receptors in dorsal horn neurons because synaptic AMPAR trafficking events in vitro and in intact brains have a slow rate time constant of ∼15–18 h (Adesnik et al., 2005; McCormack et al., 2006).
Our findings also suggest that spinal cord NMDAR activation, as an upstream trigger, leads to PKC phosphorylation of GluR2 Ser880 and GluR2 internalization in dorsal horn in persistent inflammatory pain. The AMPARs form multiprotein complexes with postsynaptic density proteins, including receptors, adaptor proteins, and protein kinases, at synapses (Derkach et al., 2007; Santos et al., 2009). These complexes allow for efficient intracellular signaling. The finding that NMDARs and PKCα are coupled with AMPARs via PICK1, PSD-95, and stargazin in the postsynaptic density of dorsal horn neurons provides a molecular basis for an NMDAR-PKC-AMPAR signal cascade (Fig. 10). Moreover, NMDA stimulation increased the level of GluR2-p Ser880 and reduced GluR2 surface expression in cultured dorsal horn neurons; these responses could be significantly attenuated by both APV and chelerythrine chloride. In vivo, intrathecal pretreatment and posttreatment with APV at the dose that significantly attenuated CFA-induced mechanical hypersensitivity markedly suppressed CFA-induced increases in the amount of GluR2 in the dorsal horn 150 k-g fraction at 1 and 3 d after CFA. Given that spinal cord NMDARs are activated after CFA injection (Dubner and Basbaum, 1997; Basbaum and Woolf, 1999), we propose that, after receiving strong primary afferent drive generated from peripheral nociceptors by inflammation, dorsal horn NMDARs are activated, increasing Ca2+ influx. The Ca2+ promptly activates PKC to phosphorylate GluR2 at Ser880 and to promote GluR2 internalization at postsynaptic dorsal horn neurons (Fig. 10). This conclusion is strongly supported by previous in vitro studies (Iwakura et al., 2001; Tigaret et al., 2006) which showed that NMDAR-triggered GluR2 internalization requires Ca2+ influx directly through NMDARs.
GluR2 internalization may result in an increase in synaptic AMPAR Ca2+ permeability in dorsal horn neurons after CFA injection. Under normal conditions, most dorsal horn AMPARs are Ca2+ impermeable. Our patch-clamp recording demonstrated a marked increase in the number of synaptic GluR2-lacking AMPARs in the SG neurons of dorsal horn 1 d after CFA. A previous study also reported an increase of synaptic GluR2-lacking AMPARs in neurokinin 1 receptor-positive lamina I neurons of dorsal horn on day 3 after CFA (Vikman et al., 2008). These results indicate a switch of GluR2-containing AMPARs expressed on many dorsal horn neurons to GluR2-lacking AMPARs (that is, an increase of Ca2+-permeable AMPARs) after CFA injection. Although the cell types were not identified in our study or those of others (Katano et al., 2008; Vikman et al., 2008), it is very likely that this switch occurs on excitatory (but not inhibitory) neurons in the superficial dorsal horn, because most neurons in rat superficial dorsal horn (particularly in SG) are excitatory (Todd and Sullivan, 1990; Santos et al., 2007). It should be noted that our electrophysiological recording showed no changes in the amplitudes of synaptic AMPAR-mediated eEPSCs 1 d after CFA compared with the saline-treated groups. Since single-channel conductance of GluR2-lacking AMPARs is higher than that of GluR2-containing AMPARs (Burnashev et al., 1992; Swanson et al., 1997), it is very likely that after CFA injection, there are fewer GluR2-lacking AMPARs inserted in the synaptic membrane than internalized GluR2-containing AMPARs in dorsal horn neurons. Indeed, we found that the number of internalized GluR2 subunits is greater than the number of GluR1 subunits inserted in the membrane on day 1 after CFA (Park et al., 2008). Thus, GluR2 internalization may be a major player in CFA-induced increase of Ca2+-permeable AMPARs in dorsal horn neurons.
Dorsal horn GluR2 internalization may be involved in the maintenance of CFA-induced inflammatory pain. We found that CFA-induced time-dependent changes in dorsal horn GluR2 internalization correlated with the time course of changes in CFA-induced pain hypersensitivity during the maintenance period. Peripheral inflammatory input was required for induction and maintenance of dorsal horn GluR2 internalization. This finding indicates that dorsal horn GluR2 internalization might be a marker for inflammation-induced pain hypersensitivity during the maintenance period. We also showed that CFA-induced dorsal horn GluR2 internalization during the maintenance period is dependent on spinal cord NMDAR/PKC activation. Given that spinal cord NMDAR/PKC activation plays a critical role in inflammatory pain (Dubner and Basbaum, 1997; Basbaum and Woolf, 1999), dorsal horn GluR2 internalization may contribute to the mechanism by which NMDAR/PKC activation maintains inflammatory pain. As indicated above, GluR2 internalization leads to an increase in AMPAR Ca2+ permeability in dorsal horn neurons. Because the increase in [Ca2+]i should initiate or potentiate a variety of Ca2+-dependent intracellular cascades that are associated with the mechanisms of pain hypersensitivity during inflammatory pain states, GluR2 internalization may participate in the maintenance of CFA-induced pain hypersensitivity by increasing synaptic Ca2+-permeable AMPARs in dorsal horn neurons (Fig. 10). More importantly, our behavioral study with GluR2K882A KI mice demonstrated that preventing CFA-induced dorsal horn GluR2 internalization through targeted mutation of the GluR2 PKC phosphorylation site attenuated CFA-evoked mechanical and thermal hypersensitivity during the maintenance period. This finding provides strong evidence that dorsal horn GluR2 internalization may underlie the mechanism of persistent inflammatory pain maintenance. It bears noting that the GluR2K882A KI mice, like conventional gene knock-out mice, might generate compensatory mechanisms during development. Thus, it will be necessary to confirm the role of dorsal horn GluR2 internalization in the maintenance of inflammatory pain by interfering with other mechanisms that determine GluR2 internalization (e.g., by inhibiting spinal PKCα and targeting disruption of the PICK1 gene).
Emerging evidence suggests that AMPAR subunit trafficking is critically involved in activity-induced synaptic plasticity (Liu and Zukin, 2007; Santos et al., 2009). Our study demonstrated GluR2 internalization and GluR1 membrane insertion in dorsal horn neurons after CFA-induced inflammation (Park et al., 2008). Previous studies reported that acute inflammatory insult (e.g., capsaicin) led to a significant increase in amount of GluR1, without altering GluR2 level, in the dorsal horn plasma membrane fraction (Galan et al., 2004; Larsson and Broman, 2008). We recently found that neither GluR1 nor GluR2 expression levels changed in either the cytosolic or plasma membrane fractions of dorsal horn after mouse hindpaw incision (data not shown). These findings suggest that different peripheral nociceptive insults may induce distinct changes of spinal AMPAR subunit trafficking. Thus, it will be very important to further examine whether GluR2 is internalized in dorsal horn neurons under other persistent pain conditions (e.g., neuropathic pain) and to investigate additional potential molecular mechanisms to fully understand the role of spinal AMPAR trafficking in spinal central sensitization that underlies persistent pain.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS058886 and NS057343 and the Johns Hopkins University Blaustein Pain Research Fund (Y.-X.T.), Juvenile Diabetes Research Foundation Grant 1-2004-30 and INTAS Grant 8061 (N.V.), the Intramural Research Program of National Institute on Deafness and Other Communication Disorders (R.S.P.), and NIH Grant NS036715 and the Howard Hughes Medical Institute (R.L.H.). We thank R. Dubner, K. Ren, S. N. Raja, Y. Guan, and P. Belan for their consultation, D. Isaev and J. Galik for their electrophysiological technical support, Y. X. Wang for her help with the immunogold labeling, and C. F. Levine for her editorial assistance.
References
- Adesnik et al., 2005.Adesnik H, Nicoll RA, England PM. Photoinactivation of native AMPA receptors reveals their real-time trafficking. Neuron. 2005;48:977–985. doi: 10.1016/j.neuron.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Basbaum and Woolf, 1999.Basbaum AI, Woolf CJ. Pain. Curr Biol. 1999;9:R429–R431. doi: 10.1016/s0960-9822(99)80273-5. [DOI] [PubMed] [Google Scholar]
- Beloeil et al., 2006.Beloeil H, Ji RR, Berde CB. Effects of bupivacaine and tetrodotoxin on carrageenan-induced hind paw inflammation in rats (Part 2): cytokines and p38 mitogen-activated protein kinases in dorsal root ganglia and spinal cord. Anesthesiology. 2006;105:139–145. doi: 10.1097/00000542-200607000-00023. [DOI] [PubMed] [Google Scholar]
- Burnashev et al., 1992.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Chacur et al., 2001.Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–244. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- Chu et al., 2005.Chu YC, Guan Y, Skinner J, Raja SN, Johns RA, Tao YX. Effect of genetic knockout or pharmacologic inhibition of neuronal nitric oxide synthase on complete Freund's adjuvant-induced persistent pain. Pain. 2005;119:113–123. doi: 10.1016/j.pain.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Chung et al., 2000.Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung et al., 2003.Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Derkach et al., 2007.Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Dev et al., 2004.Dev KK, Nakanishi S, Henley JM. The PDZ domain of PICK1 differentially accepts protein kinase C-alpha and GluR2 as interacting ligands. J Biol Chem. 2004;279:41393–41397. doi: 10.1074/jbc.M404499200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong et al., 1997.Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Dong et al., 1999a.Dong H, Zhang P, Liao D, Huganir RL. Characterization, expression, and distribution of GRIP protein. Ann N Y Acad Sci. 1999a;868:535–540. doi: 10.1111/j.1749-6632.1999.tb11323.x. [DOI] [PubMed] [Google Scholar]
- Dong et al., 1999b.Dong H, Zhang P, Song I, Petralia RS, Liao D, Huganir RL. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J Neurosci. 1999b;19:6930–6941. doi: 10.1523/JNEUROSCI.19-16-06930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubner and Basbaum, 1997.Dubner R, Basbaum AI. Spinal dorsal horn plasticity following tissue or nerve injury. In: Wall PD, Melzack R, editors. The textbook of pain. London: Churchill-Livingstone; 1997. pp. 225–241. [Google Scholar]
- Galan et al., 2004.Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Granados-Soto et al., 2000.Granados-Soto V, Kalcheva I, Hua X, Newton A, Yaksh TL. Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. doi: 10.1016/S0304-3959(99)00281-X. [DOI] [PubMed] [Google Scholar]
- Greger and Esteban, 2007.Greger IH, Esteban JA. AMPA receptor biogenesis and trafficking. Curr Opin Neurobiol. 2007;17:289–297. doi: 10.1016/j.conb.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Guo et al., 2002.Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci. 2002;22:6208–6217. doi: 10.1523/JNEUROSCI.22-14-06208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley and Henley, 2005.Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann et al., 2004.Hartmann B, Ahmadi S, Heppenstall PA, Lewin GR, Schott C, Borchardt T, Seeburg PH, Zeilhofer HU, Sprengel R, Kuner R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–650. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Ho et al., 2007.Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman and Henley, 2007.Holman D, Henley JM. A novel method for monitoring the cell surface expression of heteromeric protein complexes in dispersed neurons and acute hippocampal slices. J Neurosci Methods. 2007;160:302–308. doi: 10.1016/j.jneumeth.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac et al., 2007.Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Iwakura et al., 2001.Iwakura Y, Nagano T, Kawamura M, Horikawa H, Ibaraki K, Takei N, Nawa H. N-methyl-d-aspartate-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor down-regulation involves interaction of the carboxyl terminus of GluR2/3 with Pick1. Ligand-binding studies using Sindbis vectors carrying AMPA receptor decoys. J Biol Chem. 2001;276:40025–40032. doi: 10.1074/jbc.M103125200. [DOI] [PubMed] [Google Scholar]
- Katano et al., 2008.Katano T, Furue H, Okuda-Ashitaka E, Tagaya M, Watanabe M, Yoshimura M, Ito S. N-ethylmaleimide-sensitive fusion protein (NSF) is involved in central sensitization in the spinal cord through GluR2 subunit composition switch after inflammation. Eur J Neurosci. 2008;27:3161–3170. doi: 10.1111/j.1460-9568.2008.06293.x. [DOI] [PubMed] [Google Scholar]
- Kerr et al., 1998.Kerr RC, Maxwell DJ, Todd AJ. GluR1 and GluR2/3 subunits of the AMPA-type glutamate receptor are associated with particular types of neurone in laminae I-III of the spinal dorsal horn of the rat. Eur J Neurosci. 1998;10:324–333. doi: 10.1046/j.1460-9568.1998.00048.x. [DOI] [PubMed] [Google Scholar]
- Larsson and Broman, 2008.Larsson M, Broman J. Translocation of GluR1-containing AMPA receptors to a spinal nociceptive synapse during acute noxious stimulation. J Neurosci. 2008;28:7084–7090. doi: 10.1523/JNEUROSCI.5749-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee and Ro, 2007.Lee J, Ro JY. Differential regulation of glutamate receptors in trigeminal ganglia following masseter inflammation. Neurosci Lett. 2007;421:91–95. doi: 10.1016/j.neulet.2007.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin et al., 2000.Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Liu and Zukin, 2007.Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lu et al., 2002.Lu CR, Hwang SJ, Phend KD, Rustioni A, Valtschanoff JG. Primary afferent terminals in spinal cord express presynaptic AMPA receptors. J Neurosci. 2002;22:9522–9529. doi: 10.1523/JNEUROSCI.22-21-09522.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu et al., 2008.Lu Y, Sun YN, Wu X, Sun Q, Liu FY, Xing GG, Wan Y. Role of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor subunit GluR1 in spinal dorsal horn in inflammatory nociception and neuropathic nociception in rat. Brain Res. 2008;1200:19–26. doi: 10.1016/j.brainres.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Matsuda et al., 2000.Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19:2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack et al., 2006.McCormack SG, Stornetta RL, Zhu JJ. Synaptic AMPA receptor exchange maintains bidirectional plasticity. Neuron. 2006;50:75–88. doi: 10.1016/j.neuron.2006.02.027. [DOI] [PubMed] [Google Scholar]
- McNally and Westbrook, 1998.McNally GP, Westbrook RF. Effects of systemic, intracerebral, or intrathecal administration of an N-methyl-d-aspartate receptor antagonist on associative morphine analgesic tolerance and hyperalgesia in rats. Behav Neurosci. 1998;112:966–978. doi: 10.1037//0735-7044.112.4.966. [DOI] [PubMed] [Google Scholar]
- Morrison et al., 1998.Morrison BM, Janssen WG, Gordon JW, Morrison JH. Light and electron microscopic distribution of the AMPA receptor subunit, GluR2, in the spinal cord of control and G86R mutant superoxide dismutase transgenic mice. J Comp Neurol. 1998;395:523–534. [PubMed] [Google Scholar]
- Osten et al., 2000.Osten P, Khatri L, Perez JL, Köhr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27:313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Park et al., 2008.Park JS, Yaster M, Guan X, Xu JT, Shih MH, Guan Y, Raja SN, Tao YX. Role of spinal cord alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in complete Freund's adjuvant-induced inflammatory pain. Mol Pain. 2008;4:67. doi: 10.1186/1744-8069-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez et al., 2001.Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia et al., 1994.Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia et al., 1997.Petralia RS, Wang YX, Mayat E, Wenthold RJ. Glutamate receptor subunit 2-selective antibody shows a differential distribution of calcium-impermeable AMPA receptors among populations of neurons. J Comp Neurol. 1997;385:456–476. doi: 10.1002/(sici)1096-9861(19970901)385:3<456::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Polgár et al., 2008.Polgár E, Watanabe M, Hartmann B, Grant SG, Todd AJ. Expression of AMPA receptor subunits at synapses in laminae I-III of the rodent spinal dorsal horn. Mol Pain. 2008;4:5. doi: 10.1186/1744-8069-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos et al., 2009.Santos SD, Carvalho AL, Caldeira MV, Duarte CB. Regulation of AMPA receptors and synaptic plasticity. Neuroscience. 2009;158:105–125. doi: 10.1016/j.neuroscience.2008.02.037. [DOI] [PubMed] [Google Scholar]
- Santos et al., 2007.Santos SF, Rebelo S, Derkach VA, Safronov BV. Excitatory interneurons dominate sensory processing in the spinal substantia gelatinosa of rat. J Physiol. 2007;581:241–254. doi: 10.1113/jphysiol.2006.126912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder et al., 2001.Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Staudinger et al., 1997.Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J Biol Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- Steinberg et al., 2006.Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Sun et al., 2006.Sun H, Kawahara Y, Ito K, Kanazawa I, Kwak S. Slow and selective death of spinal motor neurons in vivo by intrathecal infusion of kainic acid: implications for AMPA receptor-mediated excitotoxicity in ALS. J Neurochem. 2006;98:782–791. doi: 10.1111/j.1471-4159.2006.03903.x. [DOI] [PubMed] [Google Scholar]
- Swanson et al., 1997.Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao et al., 2006.Tao F, Skinner J, Su Q, Johns RA. New role for spinal Stargazin in alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor-mediated pain sensitization after inflammation. J Neurosci Res. 2006;84:867–873. doi: 10.1002/jnr.20973. [DOI] [PubMed] [Google Scholar]
- Tao et al., 2000.Tao YX, Huang YZ, Mei L, Johns RA. Expression of PSD-95/SAP90 is critical for N-methyl-d-aspartate receptor-mediated thermal hyperalgesia in the spinal cord. Neuroscience. 2000;98:201–206. doi: 10.1016/s0306-4522(00)00193-7. [DOI] [PubMed] [Google Scholar]
- Tao et al., 2003.Tao YX, Rumbaugh G, Wang GD, Petralia RS, Zhao C, Kauer FW, Tao F, Zhuo M, Wenthold RJ, Raja SN, Huganir RL, Bredt DS, Johns RA. Impaired NMDA receptor-mediated postsynaptic function and blunted NMDA receptor-dependent persistent pain in mice lacking postsynaptic density-93 protein. J Neurosci. 2003;23:6703–6712. doi: 10.1523/JNEUROSCI.23-17-06703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigaret et al., 2006.Tigaret CM, Thalhammer A, Rast GF, Specht CG, Auberson YP, Stewart MG, Schoepfer R. Subunit dependencies of N-methyl-d-aspartate (NMDA) receptor-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor internalization. Mol Pharmacol. 2006;69:1251–1259. doi: 10.1124/mol.105.018580. [DOI] [PubMed] [Google Scholar]
- Todd and Sullivan, 1990.Todd AJ, Sullivan AC. Light microscope study of the coexistence of GABA-like and glycine-like immunoreactivities in the spinal cord of the rat. J Comp Neurol. 1990;296:496–505. doi: 10.1002/cne.902960312. [DOI] [PubMed] [Google Scholar]
- Tong and MacDermott, 2006.Tong CK, MacDermott AB. Both Ca2+-permeable and -impermeable AMPA receptors contribute to primary synaptic drive onto rat dorsal horn neurons. J Physiol. 2006;575:133–144. doi: 10.1113/jphysiol.2006.110072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth and McBain, 1998.Tóth K, McBain CJ. Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat Neurosci. 1998;1:572–578. doi: 10.1038/2807. [DOI] [PubMed] [Google Scholar]
- Vikman et al., 2008.Vikman KS, Rycroft BK, Christie MJ. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J Physiol. 2008;586:515–527. doi: 10.1113/jphysiol.2007.145581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei et al., 2007.Wei XH, Zang Y, Wu CY, Xu JT, Xin WJ, Liu XG. Peri-sciatic administration of recombinant rat TNF-alpha induces mechanical allodynia via upregulation of TNF-alpha in dorsal root ganglia and in spinal dorsal horn: the role of NF-kappa B pathway. Exp Neurol. 2007;205:471–484. doi: 10.1016/j.expneurol.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Wen et al., 2007.Wen YR, Suter MR, Kawasaki Y, Huang J, Pertin M, Kohno T, Berde CB, Decosterd I, Ji RR. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- Youn et al., 2008.Youn DH, Royle G, Kolaj M, Vissel B, Randić M. Enhanced LTP of primary afferent neurotransmission in AMPA receptor GluR2-deficient mice. Pain. 2008;136:158–167. doi: 10.1016/j.pain.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2003.Zhang B, Tao F, Liaw WJ, Bredt DS, Johns RA, Tao YX. Effect of knock down of spinal cord PSD-93/chapsin-110 on persistent pain induced by complete Freund's adjuvant and peripheral nerve injury. Pain. 2003;106:187–196. doi: 10.1016/j.pain.2003.08.003. [DOI] [PubMed] [Google Scholar]