

Abstract
In heart, pore-forming Kv4 α channel subunits underlie the K+ transient outward current (Ito). Expression of Kv4 is greater in left ventricular epicardial (EPI) than in endocardial (ENDO) cells, resulting in larger Ito in EPI than in ENDO cells. In adult ventricular myocytes, the transcription factor NFATc3 suppresses Kv4 expression. NFATc3 activity is higher in ENDO than in EPI cells and this has been proposed to contribute to heterogeneous Kv4 expression across the left ventricular free wall. Here, we tested the hypothesis that regional activation of NFATc3 signaling dissipates the gradient of Ito density across the mouse left ventricle during chronic activation of β adrenergic signaling. [Ca2+]i, calcineurin, and NFAT activity were larger in ENDO than in EPI myocytes. Infusion of the β adrenergic receptor agonist isoproterenol increased [Ca2+]i, calcineurin, and NFAT activity in EPI, but not in ENDO myocytes, leading to equalization of these parameters in EPI and ENDO cells. This was accompanied by dissipation of the transmural gradient in Kv4.2 expression and Ito density. Unlike wild type, ENDO or EPI myocytes from β1 adrenergic receptor-null and NFATc3-null mice did not undergo changes in Ito density during isoproterenol infusion. Collectively, these data suggest that calcineurin and NFATc3 signaling contributes to the loss of heterogeneous Kv4 expression, and hence Ito density, in the mouse left ventricle during chronic β adrenergic stimulation.
Keywords: Kv4.3, Kv4.2, Hypertrophy, Heart failure, Arrhythmias, Adrenergic signaling
1. Introduction
Ito is a voltage-gated transient outward K+ current that contributes to the early repolarization phase of the ventricular action potential (AP). In mice, Ito is produced by a heterotetramer of pore-forming Kv4.2 and Kv4.3 subunits with associated accessory KChIP2 subunits. An interesting feature of the mammalian heart is that Ito density is larger in epicardial (EPI) than in endocardial (ENDO) myocytes [1–4]. This transmural gradient of Ito is important for normal ventricular repolarization [3,5,6].
Recent studies have examined the molecular mechanisms underlying differential Ito density in ENDO and EPI cells. In the mouse ventricle, where a transmural gradient in KChIP2 and Kv4.3 is absent [7], differential Kv4.2 expression presumably underlies heterogeneous Ito density [4,7,8]. Diastolic and systolic [Ca2+]i also varies across the left ventricular wall; it is higher in ENDO than in EPI myocytes [9,10]. These differences in [Ca2+]i have important implications. Indeed, it was recently demonstrated that activation of the transcription factor NFATc3 by the Ca2+-dependent phosphatase calcineurin transduces variations in [Ca2+]I into differences in Ito density in ENDO and EPI myocytes [11]. NFATc3 decreased Ito by reducing Kv4 expression [11,12]. [Ca2+]i and calcineurin/NFATc3 activity is higher in ENDO than in EPI myocytes, resulting in lower Kv4 expression and Ito density in ENDO cells [9,11]. These data were consistent with a model in which differential patterns of [Ca2+]i, calcineurin, and NFATc3 signaling contribute to regional variations in Ito density in the mouse myocardium.
A growing body of evidence suggests that changes in [Ca2+]i and NFATc3 activity as well as loss of heterogeneous Ito density are associated with the development of hypertrophy and heart failure [13–18]. One important mechanism involved in the regulation of [Ca2+]i in ventricular myocytes under physiological and pathophysiological conditions is the β adrenergic signaling pathway. Although acute activation of β adrenergic receptors (βAR) can increase heart function, multiple studies suggest that their chronic activation causes hypertrophy, electrical remodeling, and arrhythmogenesis [19,20]. Indeed, chronic activation of βAR signaling activates NFATc3 and decreases Ito in ventricular myocytes after myocardial infarction [12]. These changes in Ito density are primarily due to changes in Kv4 expression. At present, however, whether chronic βAR signaling activation alters regional variations in calcineurin and NFAT activity as well as Kv4 expression and Ito density is unknown.
In this study, we tested the hypothesis that differential activation of calcineurin/NFATc3 signaling across the mouse left ventricle contributes to the dissipation of the gradient of Ito density between ENDO and EPI during chronic activation of β1 adrenergic signaling. Consistent with this hypothesis, we found that chronic infusion of the βAR agonist isoproterenol (ISO) – which causes hypertrophy [21–23] –increased [Ca2+]i, calcineurin, and NFAT transcriptional activity in EPI, but not in ENDO cells. This resulted in the dissipation of the Kv4.2 and Ito gradient between EPI and ENDO cells. Unlike wild type, both NFATc3 knock out (NFATc3−/−) and β1 adrenergic receptor knock out (β1AR−/−) ENDO or EPI myocytes did not undergo changes in Ito density during ISO infusion. These data suggest that calcineurin and NFATc3 signaling contributes to the loss of heterogeneous Kv4 expression, and hence Ito density, in the mouse left ventricle during chronic β adrenergic stimulation.
2. Materials and methods
Myocytes were obtained from the left ventricle ENDO and EPI of wild type (WT), NFATc3−/−, β1AR−/−, and NFAT-luc as previously described [24]. Electrophysiological signals were recorded using an Axopatch 200B. For [Ca2+]i measurements cells, were loaded with the acetomethylester version of the fluorescent Ca2+ indicator fluo-4. RT-PCR and Western blot analyses were performed as described elsewhere [11,12]. Calcineurin activity was quantified using a commercially available kit (Promega). Data are presented as mean±SEM.
An expanded Materials and methods section can be found online.
3. Results
3.1. Chronic activation of β1 adrenergic receptors dissipates the Ito gradient across the left ventricular free wall
We examined [Ca2+]i, calcineurin, NFAT, and Ito in ENDO and EPI cells from control and isoproterenol (ISO)-infused mice. We selected this animal model for multiple reasons. First, chronic ISO infusion has been shown to induce reproducible cardiac hypertrophy [21,25]. Second, β adrenergic signaling activates NFAT in ventricular myocytes [12]. Third, chronic β adrenergic receptor signaling has been linked to hypertrophy, heart failure, and arrhythmogenesis in humans [18,26–29].
We recorded Ito from ENDO and EPI cells isolated from saline (control) and ISO-infused mice (Fig. 1). Normalization of Ito was performed by dividing the current amplitudes by the capacitance (an indicator of cell surface area) of the cells from which they were recorded. Analysis of these capacitance values indicated, as reported by others [30], that control ENDO cells are larger (154.2±7.7 pF, n=25; p<0.05) than EPI cells (119.5±5.6 pF, n=31). ISO infusion increased the capacitance (i.e. surface area) of EPI cells (169.5±7.8 pF, n=45) to a larger extent than ENDO (175.3±9.3 pF, n=37) cells.
Fig. 1.

Chronic ISO infusion dissipates Ito gradient across the left ventricular free wall. Voltage dependence of Ito in EPI and ENDO cells from saline (A, control) and ISO-infused mice (B). Representative Ito records (at +40 mV) from EPI and ENDO myocytes from saline (control) and ISO-infused mice are shown above the current–voltage relationships.
Ito was evoked by 1 second step depolarizations from a holding potential of −90 mV to voltages ranging from −60 to +40 mV and was defined as the difference between the peak and the sustained current measured at the end of the pulse. Ito was isolated from other voltage-gated Na+, Ca2+, and K+ currents in these cells by pharmacological means as previously described [31–33]. Experiments were performed in the absence of ISO in the external solution.
As previously reported [1,11], Ito was larger in control EPI than in ENDO cells at most voltages examined (p<0.05; Fig. 1A). Indeed, at +40 mV, Ito was 62.1±5.6 pA/pF and 21.6±6.1 pA/pF in EPI and ENDO cells, respectively. We found that the amplitude of Ito in EPI cells (30.8±5.7 pA/pF at +40 mV) isolated from ISO-infused animals was smaller than that of control EPI cells (n=7; p<0.05). Note, however, that Ito was similar in ENDO and EPI cells from ISO-infused mice and ENDO cells from control mice (p>0.05; Figs. 1A–B). Collectively, these data suggest that chronic infusion of the βAR signaling activator ISO diminishes the transmural Ito gradient by selectively decreasing this current in EPI cells.
Ventricular myocytes express β1 and β2 adrenergic receptors [34]. To determine whether ISO infusion decreased Ito in EPI cells through the activation of β1AR, we recorded this current in ENDO and EPI cells from control and ISO-infused β1AR knockout mice [35] (β1AR−/−; Fig. 2). We found that, as in control mice, Ito was larger in EPI than in ENDO cells from β1AR−/− mice at most voltages examined, suggesting that basal β1AR activity does not contribute to regional variations in Ito density in the left ventricular wall. However, unlike wild type (WT) myocytes (see Fig. 1 above), ISO infusion did not decrease Ito in β1AR−/− EPI or ENDO cells (p>0.05). These data suggest that ISO decreases Ito in EPI myocytes via β1 adrenergic receptor mediated signaling.
Fig. 2.

β1 adrenergic receptors are required for loss of Ito gradient across the left ventricular free wall during chronic ISO infusion. Voltage dependence of Ito in EPI and ENDO cells from saline (control) and ISO-infused β1AR−/− mice.
We examined the molecular mechanisms underlying decreased Ito density in EPI cells in ISO-infused WT mice. To do this, we determined Kv4.2 and Kv4.3 transcript levels in wild type ENDO and EPI tissue (Fig. 3). As previously reported [36], Kv4.2 transcript expression levels were higher in control EPI than in ENDO (Kv4.2 ENDO/EPI=0.43±0.5, n=5). Kv4.3 transcript expressionwas similar in control and ISO-infused EPI and ENDO cells (p>0.05). Consistent with our electrophysiological data, Kv4.2 transcript was about 55% lower (n=5, p<0.05) in ISO EPI than in control EPI cells. Indeed, Kv4.2 transcript was similar in ISO EPI and control ENDO (Kv4.2 ENDO/EPI=1.0±0.3, n=5). In combination with the electrophysiological data above, our data indicate that sustained activation of β1 adrenergic signaling dissipates the Ito gradient by selectively decreasing expression of Kv4.2 transcript in EPI cells.
Fig. 3.
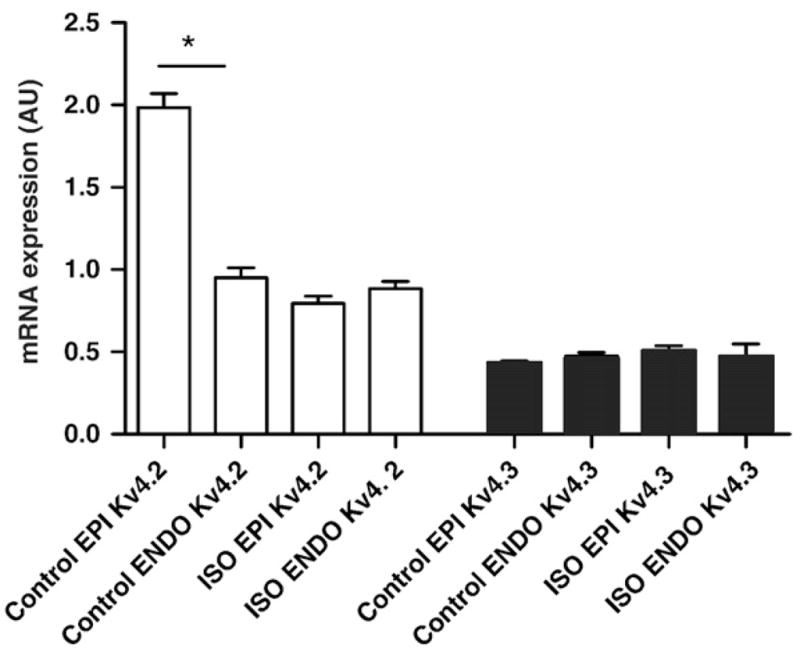
Chronic ISO infusion downregulates Kv4.2 expression in EPI cells. Real time PCR analysis of Kv4.2 and Kv4.3 transcript levels in ENDO and EPI cells from saline and ISO-infused mice. Data represents the ratio of Kv4.X transcript levels in ENDO vs. EPI. *=p<0.05.
3.2. ISO increases [Ca2+]i in EPI myocytes to a larger extent than in ENDO myocytes
Activation β adrenergic signaling increases [Ca2+]i in ventricular myocytes. Thus, we examined AP-evoked [Ca2+]i transients in ENDO and EPI myocytes loaded with the fluorescent Ca2+ indicator fluo-4 before and after the application of 100 nM ISO to activate βARs. Action potentials were activated in these cells via field stimulation (1 Hz; Fig. 4). Under control conditions, [Ca2+]i transients were larger in ENDO than in EPI cells (n=8, p<0.05). In addition, we found that the time to 50% of the amplitude (T1/2) during the decaying phase of the [Ca2+]i transient was shorter in ENDO (T1/2 =304±21 ms, n =12) than in EPI cells (T1/2 =360±13 ms, n =15; p < 0.05).
Fig. 4.

ISO increases [Ca2+]i in EPI cells to a larger extent than in ENDO cells. (A) Action potential-evoked [Ca2+]i transients from representative ENDO and EPI cells before and after the application of 100 nM ISO. (B) Bar plot of the mean±SEM of the amplitude of [Ca2+]i transients of ENDO and EPI cells under control conditions and in the presence of ISO. *=p<0.05.
Application of ISO (100 nM) increased [Ca2+]i in these cells. However, we found that ISO increased [Ca2+]i to a larger extent in EPI than in ENDO cells. Indeed, although there was a tendency for [Ca2+]i transients to be larger in ISO than in control (i.e. no ISO) ENDO cells, this difference was not statistically significant. Note also that the amplitude of the [Ca2+]i transient in control ENDO, ISO ENDO, and ISO EPI cells was similar (p>0.05). ISO also increased (p<0.05) the rate of decay of the [Ca2+]i transient in EPI (T1/2 =227±15, n =9) and ENDO myocytes (T1/2 =248 ±26 ms, n=12). Note, that in the presence of ISO, the T1/2 of the [Ca2+]i of ENDO and EPI cells was similar (p>0.05). These data suggest that β adrenergic signaling activation increases [Ca2+]i in the left ventricular wall predominantly by increasing [Ca2+]i in EPI cells.
3.3. Higher β1AR expression and PKA phosphorylation of phospholamban in ENDO than in EPI
We investigated the mechanisms underlying the larger increase in the amplitude of the [Ca2+]i transient in EPI than in ENDO myocytes during activation of βAR signaling. Western blot analyses were used to determine expression β1ARs and specific PKA subunits (R1 and II α) as well as the level of phospholamban protein that is phosphorylated at the PKA-specific serine 16 (Ser16P-PLB) using a phospho-specific antibody [37] in ENDO and EPI (Fig. 5). Only hearts from wild type animals not infused (i.e. without pumps) with ISO were used in these studies.
Fig. 5.

Higher β1AR expression and PKA phosphorylation of phospholamban in ENDO than in EPI. A through D, Western blot analysis of β1AR (A), PKA RI (B), PKA RII α (C), and phospho-PLB (Ser16P-PLB, PKA-specific serine; D) in ENDO and EPI tissue. Phospho-PLB protein levels were determined in EPI and ENDO from hearts perfused with saline (control) or ISO (100 nM). The bar plots show the relative level (to β-actin) of these proteins in ENDO and EPI. *=p<0.05.
We found that β1AR protein expression was higher in ENDO than in EPI tissue (Fig. 5A; n=5 hearts, p<0.05). However, protein levels of the R1 and IIα subunits of PKA were similar in ENDO and EPI (Figs. 5B–C; n=5 hearts, p>0.05). We also investigated whether the higher β1AR protein expression in ENDO than in EPI was associated with higher PKA phosphorylation of phospholamban in ENDO than in EPI. As noted above, we used a phospho-specific antibody recognizing Ser16P-PLB to determine the levels of this protein in ENDO and EPI tissue from hearts perfused using a Langendorff system with saline (i.e. control) or 100 nM ISO for 5 min (Fig. 5D). Consistent with the β1AR protein data described above, we found that basal Ser16P-PLB levels were higher in ENDO than in EPI under control conditions (i.e. saline perfusion; p<0.05), suggesting that basal PKA activity is higher in ENDO than in EPI myocytes. In vivo perfusion with ISO (100 nM) increased Ser16P-PLB levels in ENDO and EPI (n=6 hearts, p<0.05).
3.4. Differential increases in ICa density and SR Ca2+ in EPI and ENDO during activation of βAR signaling
We investigated whether the differences in β1AR expression and PKA-dependent phosphorylation of PLB described above were linked to regional variations in L-type Ca2+ currents (ICa) and SR Ca2+ load in EPI and ENDO under control conditions and during activation of βAR signaling (Fig. 6). ICa was evoked by a 200 ms voltage step to potentials ranging from −30 to +50 mV from the holding potential of −40 mV. As previously reported [9], under control conditions, the amplitude of ICa was similar in EPI and ENDO cells (Fig. 6A). However, in agreement with our β1AR protein data described above, ISO (100 nM) increased ICa to a larger extent (≈30%) in ENDO than in EPI cells (n=6, p<0.05).
Fig. 6.

Differential increases in ICa density and SR Ca2+ in EPI and ENDO during activation of βAR signaling. (A) ICa records from representative EPI and ENDO myocyte before and after the application of 100 nM ISO. ICa was evoked by a 200 ms pulse from the holding potential of −40 mV to the test potential of +10 mV. (B) Representative field and caffeine-induced [Ca2+]i transients in EPI and ENDO cells before and after the application of 100 nM ISO. The bar plots show the mean±SEM of relative (to control) change in ICa and the caffeine-induced [Ca2+]i transient in EPI and ENDO cells after ISO treatment. *=p<0.05.
Consistent with the Ser16P-PLB data and a previous study [9] by our group, the amplitude of the [Ca2+]i transient evoked by the application of 20 mM caffeine (i.e. SR Ca2+ load) was higher in ENDO than in EPI cells under control conditions (Fig. 6B). As shown above, acute application of ISO (100 nM), increased the amplitude of the evoked [Ca2+]i transient in EPI cells to a larger extent than in ENDO cells. Note, however, that activation of βAR signaling with ISO increased SR Ca2+ load in EPI cells (1.52±0.15-fold, n=9) to a larger extent than in ENDO cells (1.04±0.08-fold, n=9, p<0.05). These data suggest that activation of β1AR signaling induces a larger increase in the [Ca2+]i transient in EPI than in ENDO, at least in part, by increasing SR Ca2+ load to a larger extent in EPI than in ENDO myocytes.
3.5. Chronic ISO infusion decreases the calcineurin/NFAT activity gradient across the left ventricular wall
In adult ventricular myocytes, changes in [Ca2+]i can alter the expression of Kv4 potassium channels via activation of the calci-neurin-NFATc3 transcriptional signaling pathway [11,12]. Thus, we examined whether chronic ISO infusion activated this signaling pathway in ENDO or EPI cells. First, we determined calcineurin activity in ENDO and EPI cells from control and ISO-infused mice. Consistent with our [Ca2+]i data, calcineurin activity was higher in ENDO than in EPI from control animals (Fig. 7A). Calcineurin activity was higher in ISO-infused than in control EPI cells (p<0.05). Indeed, calcineurin activity was similar in EPI and in ENDO from ISO-infused mice (p<0.05). Interestingly, ISO infusion did not increase calcineurin activity in ENDO cells (p>0.05).
Fig. 7.

Loss of calcineurin and NFAT activity gradient across the left ventricular free wall during chronic ISO infusion. Bar plots of the mean±SEM of the calcineurin activity (A) and luciferase transcript level (B) in EPI and ENDO tissue from saline- and ISO-infused mice. The images in panel B show transcript amplification products for luciferase (510 bp). *=p<0.05.
Next, we examined NFAT transcriptional activity in control and ISO-infused ENDO and EPI tissue (Fig. 7B). In these experiments, we used a transgenic mouse in which luciferase gene expression is driven by multiple NFAT binding elements [38]. We quantified NFAT activity by using RT-PCR to measure luciferase transcript in ENDO and EPI from saline (control) or ISO-infused mice. Consistent with the calcineurin data above, we measured higher luciferase transcript levels (i.e. NFAT activity) in control ENDO than EPI. Note, however, that while ISO infusion increased NFAT activity (i.e. luciferase expression) in EPI, it failed to do so in ENDO cells. These data suggest that chronic activation of βAR adrenergic signaling decreases regional differences in calcineurin and NFAT activity across the left ventricular wall, by selectively increasing the activity of this signaling pathway in EPI cells.
3.6. NFATc3 is required for loss of the Ito gradient across the left ventricular free wall during ISO infusion
We tested the hypothesis that NFATc3 is required for down-regulation of Ito in EPI cells during chronic ISO infusion. To test this hypothesis, we examined Ito in ENDO and EPI cells from NFATc3 null (NFATc3−/−) mice infused with saline (control) or ISO (Fig. 8). As previously reported [11], and unlike wild type mice, Ito is similar in NFATc3−/− EPI (56.91±4.21 pA/pF at +40 mV) and ENDO cells (57.61±3.73 pA/pF at +40 mV; p>0.05). Consistent with our hypothesis, EPI myocytes showed no decrease in Ito following chronic ISO (49.33±7.64 pA/pF) in comparison to control (55.61±5.73 pA/pF). Likewise, chronic ISO infusion did not decrease Ito in NFATc3−/− ENDO cells (at +40 mV 56.91±4.21 pA/pF in control cells vs. 54.21±9.44 pA/pF in ISO-infused cells). These data suggest that NFATc3 is required for Ito downregulation in EPI cells during chronic βAR signaling activation.
Fig. 8.

NFATc3 is required for loss of Ito gradient across the left ventricular free wall during chronic ISO infusion. Voltage dependencies of Ito (lower row) in EPI and ENDO cells from saline (A) and ISO-infused (B) NFATc3−/− mice. Representative Ito records (at +40 mV) from EPI and ENDO myocytes isolated from saline and ISO-infused NFATc3−/− mice are shown above each current–voltage relationship.
4. Discussion
Our data suggest that there are important variations in βAR signaling across the mouse left ventricular wall and that these differences contribute to heterogeneous changes in Ito density during chronic β1 adrenergic signaling activation in EPI and ENDO. We found significant differences in [Ca2+]i signaling across the left ventricular wall under control conditions and during activation of βAR signaling. Not only is the amplitude of the AP-evoked [Ca2+]i transient larger in ENDO than in EPI myocytes [9], but it also increases to a larger extent in EPI than in ENDO myocytes during βAR signaling activation. Our data suggest a mechanism for these differences in [Ca2+]i in ENDO and EPI myocytes under control conditions and during βAR signaling. β1AR protein expression and Ser16P-PLB levels (an indicator of PKA activity) are higher in ENDO than in EPI. Furthermore, SR Ca2+ load is higher in ENDO than in EPI cells under control conditions. Together, these data suggest that basal PKA activity is higher in ENDO than in EPI cells, which increases PLB phosphorylation and hence SERCA pump activity. Higher SERCA pump activity likely contributes to higher SR Ca2+ load and SR Ca2+ release during EC coupling in ENDO than in EPI cells.
As expected, we found that ICa increases in ENDO and EPI during ISO treatment. However, if as noted above, basal PKA activity is higher in ENDO than in EPI why is ICa amplitude similar in ENDO and EPI [9]? Although our data do not provide an answer to this difficult question, one intriguing possibility is that the activity of a protein phosphatase that opposes PKA is higher near Ca2+ channels in ENDO than in EPI, which would increase the threshold for PKA-dependent modulation of ICa in ENDO compared to EPI. Two recent studies support this hypothesis. Calcineurin activity is higher in ENDO than in EPI [11]. Furthermore, calcineurin opposes PKA actions on ICa: calcineurin inhibition increases ICa in ventricular myocytes [39]. Future experiments should examine the mechanisms underlying differential modulation of ICa in ENDO and EPI by PKA.
Activation of βAR signaling increased ICa and Ser16P-PLB levels in ENDO and EPI. Yet, activation of this signaling pathway increased SR Ca2+ load in EPI, but not in ENDO myocytes. Consistent with this, acute application of ISO evoked a larger increase in the amplitude of the transient in EPI than in ENDO cells. Indeed, the relatively smaller increase in the [Ca2+]i transient observed in ENDO cells is likely due to the larger increase in ICa induced by ISO in these cells than in EPI cells. Higher β1AR expression in ENDO than in EPI could contribute to this differential effect of ISO on ICa.
On the basis of these findings, we propose a model for differential [Ca2+]i signaling in ENDO and EPI during βAR signaling. In this model, basal SR Ca2+ load in ENDO cells is high – compared to EPI – and at a level similar to one observed during activation of βAR signaling in EPI and ENDO cells. PKA-dependent phosphorylation of PLB at serine 16 does not translate into an increase in SR Ca2+ load in EPI cells. Although the exact mechanisms underlying this are unclear, it is intriguing to speculate that in ENDO cells activation of βAR signaling increases SERCA pump activity and SR Ca2+ so that there is no net change in SR Ca2+ load. In this model, SR Ca2+ load and release during βAR signaling is similar in EPI and ENDO cells. Our observation that the amplitude of the [Ca2+]i transient in ISO EPI and ENDO cells is similar suggests this assumption is reasonable.
Our data indicate that chronic activation of βAR signaling has important consequences on [Ca2+]i and Ito across the left ventricular wall. Not only did it dissipate transmural differences in [Ca2+]i, but it did also eliminate the Ito gradient across the left ventricular wall, presumably by selectively decreasing Kv4.2 expression in EPI cells. We also found that NFATc3 is required for Ito downregulation during sustained activation of βAR signaling. These findings are consistent with recent studies indicating that the calcineurin/NFATc3 signaling pathway regulates the expression of K+ channels in heart under physiological and pathophysiological conditions [11,12]. Indeed, a gradient in [Ca2+]i and calcineurin/NFATc3 signaling has been suggested to underlie differential Kv4 expression across the mouse left ventricular free wall. Our data suggest that chronic ISO infusion dissipates this Kv4.2 and Ito gradient by increasing [Ca2+]i as well as calcineurin and NFAT activity in EPI, but not in ENDO cells.
NFATc3 downregulates Kv4.2 and Kv4.3 channel expression in adult ventricular myocytes [11,12]. This observation raises an important question: why does chronic ISO infusion only result in the downregulation of Kv4.2 in EPI myocytes? A recent study by our group examining the mechanisms by which NFATc3 regulates Kv4 expression in control EPI and ENDO suggests an answer to this conundrum. In this study [11], we found that Kv4.2 and Kv4.3 genes, both of which have putative NFAT binding sites in their promoters, have different thresholds for NFAT-dependent suppression. At relatively low levels of NFAT activity (i.e., similar to those in control EPI cells), expression of Kv.4.2 and Kv4.3 is high. Increasing NFAT activity by about 1.6-fold (to about the same level observed in ENDO cells) downregulated Kv4.2, but not Kv4.3 expression in mouse ventricular myocytes. Higher elevations (presumably 3-fold) in NFAT activity are required for downregulation (60%) of both Kv4.2 and Kv4.3 genes [11,12]. Thus, the level of NFAT activity observed in ISO-infused EPI (i.e. similar to control ENDO) is sufficient to downregulate Kv4.2, but not Kv4.3 expression.
The experiments in this study were performed in mice to take advantage of available genetically engineered animals. Unlike mice and rats, Ito in canine and human hearts is produced by Kv4.3 [40]. A recent study suggests that NFATc3 modulates Kv4.3 expression and hence Ito in canine ventricular myocytes [41]. Thus, it is intriguing to speculate that chronic activation of β1AR could decrease Ito density via an NFATc3-dependent downregulation of Kv4.3 expression in larger mammals. Note, however, that the signaling pathways underlying regional variations of Kv4.3 expression in canine are still unclear. Experiments need to be performed to establish the relationship between β1AR, calcineurin/NFAT, and Kv4.3 expression in human and canine hearts.
To conclude, our data clearly indicate regional variations in β1AR signaling in the left ventricular free wall. Furthermore, our findings support the view that β1AR signaling and calcineurin/NFATc3 signaling play a critical role in the modulation of Ito across the left ventricular wall and hence modulates the ventricular excitability under physiological pathophysiological conditions.
Supplementary Material
Acknowledgments
This work was supported by NIH grant HL085686. Drs. Laurie Glimcher and Brian Kobilka provided NFATc3-null and β1 adrenergic receptor-null mice, respectively. Dr. Jeff Molkentin provided the NFAT reporter mice.
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yjmcc.2008.10.016.
References
- 1.Clark RB, Bouchard RA, Salinas-Stefanon E, Sanchez-Chapula J, Giles WR. Heterogeneity of action potential waveforms and potassium currents in rat ventricle. Cardiovascular research. 1993 Oct;27(10):1795–9. doi: 10.1093/cvr/27.10.1795. [DOI] [PubMed] [Google Scholar]
- 2.Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE, et al. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001 May 15;533(Pt 1):119–25. doi: 10.1111/j.1469-7793.2001.0119b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, et al. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of Ito and confers susceptibility to ventricular tachycardia. Cell. 2001 Dec 14;107( 6):801–13. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 4.Brunet S, Aimond F, Guo W, Li H, Eldstrom J, Fedida D, et al. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J Physiol. 2004 Jun 11;559(1):103–20. doi: 10.1113/jphysiol.2004.063347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res. 1998 Sep 7;83(5):560–7. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- 6.Guo W, Li H, London B, Nerbonne JM. Functional consequences of elimination of i (to,f) and i(to,s): early afterdepolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 alpha subunit. Circ Res. 2000 Jul 7;87(1):73–9. doi: 10.1161/01.res.87.1.73. [DOI] [PubMed] [Google Scholar]
- 7.Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W, et al. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell. 2005 Oct 21;123(2):347–58. doi: 10.1016/j.cell.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo W, Li H, Aimond F, Johns DC, Rhodes KJ, Trimmer JS, et al. Role of heteromultimers in the generation of myocardial transient outward K+ currents. Circ Res. 2002 Mar 22;90(5):586–93. doi: 10.1161/01.res.0000012664.05949.e0. [DOI] [PubMed] [Google Scholar]
- 9.Dilly KW, Rossow CF, Votaw VS, Meabon JS, Cabarrus JL, Santana LF. Mechanisms underling variations in excitation-contraction coupling across the mouse left ventricular free wall. J Physiol. 2006 Jan 19;572(1):227–41. doi: 10.1113/jphysiol.2005.102020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurita KR, Katra R, Wible B, Wan X, Koo MH. Transmural heterogeneity of calcium handling in canine. Circ Res. 2003 Apr 4;92(6):668–75. doi: 10.1161/01.RES.0000062468.25308.27. [DOI] [PubMed] [Google Scholar]
- 11.Rossow CF, Dilly KW, Santana LF. Differential calcineurin/NFATc3 activity contributes to the Ito transmural gradient in the mouse heart. Circ Res. 2006 Apr 13;98:1306–13. doi: 10.1161/01.RES.0000222028.92993.10. [DOI] [PubMed] [Google Scholar]
- 12.Rossow CF, Minami E, Chase EG, Murry CE, Santana LF. NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ Res. 2004 May 28;94( 10):1340–50. doi: 10.1161/01.RES.0000128406.08418.34. [DOI] [PubMed] [Google Scholar]
- 13.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, et al. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res. 2008 Apr 25;102(8):959–65. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 14.Kaprielian R, Sah R, Nguyen T, Wickenden AD, Backx PH. Myocardial infarction in rat eliminates regional heterogeneity of AP profiles, Ito K+ currents, and [Ca2+]i transients. Am J Physiol Heart Circ Physiol. 2002 Sep;283(3):H1157–1168. doi: 10.1152/ajpheart.00518.2001. [DOI] [PubMed] [Google Scholar]
- 15.Volk T, Nguyen TH, Schultz JH, Faulhaber J, Ehmke H. Regional alterations of repolarizing K+ currents among the left ventricular free wall of rats with ascending aortic stenosis. J Physiol. 2001 Feb 1;530(Pt 3):443–55. doi: 10.1111/j.1469-7793.2001.0443k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Cheng J, Chen G, Rob F, Naseem RH, Nguyen L, et al. Remodeling of outward K+ currents in pressure-overload heart failure. J Cardiovasc Electrophysiol. 2007 Aug;18(8):869–75. doi: 10.1111/j.1540-8167.2007.00864.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, et al. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004 Oct 15;95(8):798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 18.Osadchii OE. Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart failure reviews. 2007 Mar;12( 1):66–86. doi: 10.1007/s10741-007-9007-4. [DOI] [PubMed] [Google Scholar]
- 19.Zhang LM, Wang Z, Nattel S. Effects of sustained beta-adrenergic stimulation on ionic currents of cultured adult guinea pig cardiomyocytes. Am J Physiol Heart Circ Physiol. 2002 Mar;282(3):H880–889. doi: 10.1152/ajpheart.01138.2000. [DOI] [PubMed] [Google Scholar]
- 20.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003 Nov 14;93(10):896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 21.Ulucan C, Wang X, Baljinnyam E, Bai Y, Okumura S, Sato M, et al. Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am J Physiol Heart Circ Physiol. 2007 Sep;293(3):H1662–1672. doi: 10.1152/ajpheart.00159.2007. [DOI] [PubMed] [Google Scholar]
- 22.Zhang GX, Ohmori K, Nagai Y, Fujisawa Y, Nishiyama A, Abe Y, et al. Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. J Mol Cell Cardiol. 2007 Apr;42(4):804–11. doi: 10.1016/j.yjmcc.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Iwase M, Bishop SP, Uechi M, Vatner DE, Shannon RP, Kudej RK, et al. Adverse effects of chronic endogenous sympathetic drive induced by cardiac GS alpha overexpression. Circ Res. 1996 Apr;78(4):517–24. doi: 10.1161/01.res.78.4.517. [DOI] [PubMed] [Google Scholar]
- 24.Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. 2001;276(30):28197–203. doi: 10.1074/jbc.M102548200. [DOI] [PubMed] [Google Scholar]
- 25.Zhu Z, Zhang SH, Wagner C, Kurtz A, Maeda N, Coffman T, et al. Angiotensin AT1B receptor mediates calcium signaling in vascular smooth muscle cells of AT1A receptor-deficient mice. PG - 1171–7. Hypertension. 1998 May;31(5) doi: 10.1161/01.hyp.31.5.1171. [DOI] [PubMed] [Google Scholar]
- 26.Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Mechanisms of Disease: ryanodine receptor defects in heart failure and fatal arrhythmia. Nature clinical practice. 2006 Jan;3(1):43–52. doi: 10.1038/ncpcardio0419. [DOI] [PubMed] [Google Scholar]
- 27.Mohamed U, Napolitano C, Priori SG. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007 Jul;18(7):791–7. doi: 10.1111/j.1540-8167.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- 28.Francis J, Sankar V, Nair VK, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2005 May;2(5):550–4. doi: 10.1016/j.hrthm.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 29.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002 Jan;190(1):1–6. doi: 10.1002/jcp.10031. [DOI] [PubMed] [Google Scholar]
- 30.Gerdes AM, Callas G, Kasten FH. Differences in regional capillary distribution and myocyte sizes in normal and hypertrophic rat hearts. Am J Anat. 1979 Dec;156( 4):523–31. doi: 10.1002/aja.1001560406. [DOI] [PubMed] [Google Scholar]
- 31.DuBell WH, Lederer WJ, Rogers TB. K+ currents responsible for repolarization in mouse ventricle and their modulation by FK-506 and rapamycin. Am J Physiol Heart Circ Physiol. 2000;278(3):H886–897. doi: 10.1152/ajpheart.2000.278.3.H886. [DOI] [PubMed] [Google Scholar]
- 32.Ufret-Vincenty CA, Baro DJ, Santana LF. Differential contribution of sialic acid to the function of repolarizing K+ currents in ventricular myocytes. Am J Physiol Cell Physiol. 2001;281(2):C464–474. doi: 10.1152/ajpcell.2001.281.2.C464. [DOI] [PubMed] [Google Scholar]
- 33.Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J Gen Physiol. 1999 May;113( 5):661–78. doi: 10.1085/jgp.113.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002 Jan 10;415(6868):206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 35.Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP, Jr, Barsh GS, et al. Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Acad Sci U S A. 1996 Jul 9;93(14):7375–80. doi: 10.1073/pnas.93.14.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S, Nerbonne JM. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. 2008 Jun 6;102( 11):1406–15. doi: 10.1161/CIRCRESAHA.107.170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drago GA, Colyer J. Discrimination between two sites of phosphorylation on adjacent amino acids by phosphorylation site-specific antibodies to phospholamban. J Biol Chem. 1994 Oct 7;269(40):25073–7. [PubMed] [Google Scholar]
- 38.Bueno OF, Lips DJ, Kaiser RA, Wilkins BJ, Dai YS, Glascock BJ, et al. Calcineurin Abeta gene targeting predisposes the myocardium to acute ischemia-induced apoptosis and dysfunction. Circ Res. 2004 Jan 9;94(1):91–9. doi: 10.1161/01.RES.0000107197.99679.77. [DOI] [PubMed] [Google Scholar]
- 39.Santana LF, Chase EG, Votaw VS, Nelson MT, Greven R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol. 2002 Oct 1;544(Pt 1):57–69. doi: 10.1113/jphysiol.2002.020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, et al. Role of the Kv4.3 K + channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996 Oct;79(4):659–68. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- 41.Xiao L, Coutu P, Villeneuve LR, Tadevosyan A, Maguy A, Le Bouter S, et al. Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res. 2008;103:733–42. doi: 10.1161/CIRCRESAHA.108.171157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.