

Abstract
The p53 tumour suppressor is involved in several crucial cellular functions including cell cycle arrest and apoptosis. p53 stabilisation occurs under hypoxic and DNA damage conditions. However, only in the latter scenario is stabilised p53 capable of inducing the expression of its pro-apoptotic targets. Here we present evidence that under hypoxia mimicking conditions p53 acetylation is reduced to a greater extent at K320 site targeted by PCAF than at K382 site targeted by p300/CBP. The limited amounts of acetylated p53 at K320 are preferentially recruited to the promoter of the p21WAF-1/CIP-1 gene, which appears to be unaffected by hypoxia, but are not recruited to the BID promoter and hence p53 is incapable of up-regulating pro-apoptotic BID in hypoxic conditions. Since the K320 p53 acetylation is the site predominantly affected in hypoxia the PCAF HAT activity is the key regulator of the cellular fate modulated by p53 under these conditions. In addition, we provide evidence that PCAF acetylates HIF-1α in hypoxic conditions and that the acetylated HIF-1α is recruited to a particular subset of its targets. In conclusion, PCAF regulates the balance between cell cycle arrest and apoptosis in hypoxia by modulating the activity and protein stability of both p53 and HIF-1α.
Keywords: transcription, p53, Hypoxia Inducible Factor, PCAF, apoptosis
Introduction
The transcriptional activity of the tumour suppressor protein p53 is crucial for the mediation of its physiological functions [Vousden and Prives, 2005]. Cells adapt to various forms of stress by mechanisms orchestrated by p53, which interacts with specific classes of genes, ultimately leading to differential susceptibility to diverse types of stress [Vousden and Lane 2007]. Modulation of the p53 transcriptional activity is achieved by stabilization of the protein and binding to various co-factors such as p300 and the P300/CBP Associated Factor (PCAF). These cofactors contribute to the regulation of the p53 transcriptional activity by acetylating different lysine residues in its C-terminal region [Liu et al., 1999].
During tumourigenesis, cells encounter hypoxia as a major stress and most if not all human solid tumours contain subpopulations of hypoxic cells. Among these tumour types, those showing greater levels of hypoxia show resistance to therapy, greater malignancy and increased metastatic potential [Vaupel and Harrison 2004]. One reason that hypoxic tumours are resistant to DNA damaging therapies is because of alterations in the p53 transcriptional activity [Achison and Hupp, 2003]. Under hypoxic conditions, increased p53 protein stability has been shown to occur in a HIF-1α dependent [An et al., 1998; Hansson et al., 2002] and independent manner [Pan et al., 2004]. Furthermore, under hypoxic conditions p53 is incapable of inducing the expression of several of its transcription targets including pro-apoptotic members of the Bcl-2 family, due to failure to recruit the p300/CBP co-activator proteins to their promoters [Koumenis et al., 2001], although enhanced p53 transcriptional activity in anoxic conditions has also been reported [Zhu et al., 2002].
Functional crosstalk between the HIF-1α and p53 pathways occurs at several levels, including p53 protein stability [Ravi et al., 2000], attenuated HIF-1 transcriptional activity by p53 [Blagosklonny et al., 1998] and vice versa [Chen et al., 2003]. Interference of each one of these transcription factors with the transcriptional activity of the other has been attributed to the availability of the transcriptional co-activator p300 [Schmid et al., 2004; Arany et al., 1996], since the binding site of p300 for both p53 and HIF-1α is the CH1 domain [Freedman et al., 2002]. Differential acetylation of p53 by p300 and other Histone Acetyl Transferases (HATs) mediates explicit responses to diverse signals and adjustment to the cellular requirements depending on the environmental conditions [Liu et al., 1999]. p300 mediated acetylation of p53 at K373 and K382 [Liu et al., 1999; Sakaguchi et al., 1998; Luo et al., 2004] occurs under severe and irreparable DNA damage leading to the induction of pro-apoptotic p53 targets and cellular death. Milder stress conditions stimulate acetylation of the p53-K320 by PCAF and result in the induction of the expression of the cyclin/cdk inhibitor p21WAF-1/CIP-1 and cell cycle arrest [Knights et al., 2006]. The general consensus of all the above mentioned reports is that site specific acetylation of p53 leads to selective response to various signals [Roy and Tenniswood, 2007].
Hypoxia-inducible factor-1 (HIF-1α) has a key role in cellular responses to hypoxia [Wenger, 2002]. The half life and transcriptional activity of HIF-1α is controlled by several post-translational modifications including prolyl hydroxylations, which regulate both stability and transactivation by governing the interaction of HIF-1α with the von Hippel-Lindau ubiquitin E3 ligase (pVHL), and the p300 [Kaelin, 2005; Mahon et al., 2001; Lando et al., 2002; Freedman et al., 2004]. It is clear that p300 is required for the trans-activation of both HIF-1α and p53 and there is competition for limiting amounts of this cofactor in hypoxia between HIF-1α and p53 [Freedman et al., 2002].
ARD1 mediated acetylation of HIF-1α, has also been reported [Jeong et al., 2002], although contradictory results have been presented with regard to the effect of acetylation on HIF-1α transcriptional activity and protein stability [Bilton et al., 2006]. Other post translational modifications regulating HIF-1α and p53 function by the same enzymatic cascades include phosphorylation, nitrosylation and SUMOylation [Brahimi-Horn et al., 2005].
The above observations increasingly support the notion that similar and in many cases common regulatory mechanisms exist that modulate the transcriptional activity and protein stability of both HIF-1α and p53. In order to decipher the molecular mechanisms by which p300 and PCAF regulate p53 transcriptional activity under hypoxic conditions and define the mechanisms underlying selective activation by p53 in these conditions, we studied the expression of the pro-apoptotic p53 target BID, which is known to contain in its promoter region regulatory sequences targeted by both p53 [Sax et al., 2002] and HIF-1α [Erler et al., 2004]. We asked whether p53 binds preferentially to upstream regulatory regions of genes involved in cell cycle arrest or control of apoptosis and if this involves the function of PCAF. Furthermore, in this report we present evidence that PCAF acetylates HIF-1α under hypoxic conditions and directs it selectively to a subset of its transcription targets. Our results provide an additional evidence for the importance of co-activator function in determining the cell fate under hypoxia by modulating both p53 and HIF-1α responses.
Results
PCAF and p300 differentially occupy the promoters of p53 target genes in hypoxia
Studies by Erler et al. [Erler et al., 2004] have shown that the protein levels of several anti-apoptotic members of the Bcl-2 family remain unchanged under normoxic, hypoxic (1% O2) or anoxic (0.1% O2) conditions. In contrast, pro-apoptotic Bcl-2 family members such as Bax and BID are down regulated despite of the fact that both of these genes are p53 transcriptional targets and p53 is stabilised under anoxic and hypoxic conditions [An et al. 1998]. The altered affinity or lack of binding between p53 and the transcriptional co-activator p300 under anoxic conditions has been suggested as the reason for the inability of p53 to activate its targets under these conditions [Hammond and Giaccia, 2005].
To test whether other p53 cofactors are implicated in the determination of p53 transcriptional activity under these conditions we carried out chromatin immunoprecipitation (ChIP) experiments and monitored the occupancy of the p53 binding sites of BID, PUMA, Bax and p21WAF-1/CIP-1 promoter regions by the p300/CBP complex component PCAF in cells treated with the hypoxia mimicking agent desferrioxamine (DSFX). PCAF/chromatin complexes were precipitated and the promoter regions containing the p53 binding sites of the above mentioned genes were amplified with specific primers. PCAF recruitment to the promoters of the pro-apoptotic p53 target genes BID, PUMA and Bax was substantially lower (10 fold or more) in the cells treated with DSFX compared to the non treated cells (Figure 1A). Interestingly, the recruitment of PCAF to the p21WAF-1/CIP-1 promoter was reduced to a lesser extent, indicating preferential PCAF promoter occupancy of the cell cycle arrest p21WAF-1/CIP-1 gene rather than the pro-apoptotic p53 targets in hypoxia mimicking conditions. In contrast, recruitment of p300 to the promoters of the same p53 target genes exhibited a different occupancy profile. In the case of PUMA and p21WAF-1/CIP-1 the p300 binding efficiency was 40% lower, in the case of BID was tenfold higher and no change was observed in Bax promoter occupancy in DSFX treated U2OS cells (Figure 1C). These differences in PCAF or p300 promoter recruitment were not due to differences in the amount of the cofactors precipitated as equal quantity of PCAF and p300 were precipitated in DSFX treated or not treated cells (Figure 1B and D).
Figure 1.

PCAF and p300 differentially occupy the promoters of p53 target genes in hypoxia. U2OS cells were transfected with Flag-PCAF (A) or CMV-p300 expression plasmids (C), and processed for ChIP. Flag-PCAF (A and B) and p300 (C and D) antibodies were used to immunoprecipitate cross-linked DNA-protein complexes and primers for BID, BAX, Puma and p21WAF-1/CIP-1 promoters (Supplementary Figure 1) to amplify precipitated DNA by real-time qPCR. Standard error bars represent the deviations between at least two independent experiments. (B) Flag antibody recognises specifically the Flag-PCAF expressed protein and equal amounts of PCAF are precipitated from DSFX treated or not treated cells. (D) Equal amount of p300 are precipitated by the p300 antibody in DSFX treated or not treated cells.
BID, PUMA, Bax and p21WAF-1/CIP-1 gene expression profiles indicated that PCAF promoter occupancy of these genes was closely related to their mRNA levels followed by qRT-PCR (Supplementary Figure 2).
Acetylation of p53 at K320 by PCAF is not efficient in hypoxia
Next we investigated whether the PCAF mediated acetylation of p53 at K320 occurs with the same efficiency in normoxic and DSFX treated cells. Immunoprecipitation of total endogenous p53 from U2OS cells and blotting with a specific antibody recognising the p53 K320 acetylated isoform revealed a significant reduction (~70%) of p53 acetylation at K320 in cells exposed to DSFX versus those treated with etoposide (Figure 2, compare tracks 5 to 6). In contrast, the level of reduction of p53 acetylation at K382 by p300 was not as dramatic (~30% reduction) after 16h of DSFX treatment (Figure 2A, compare tracks 5 to 6).
Figure 2.
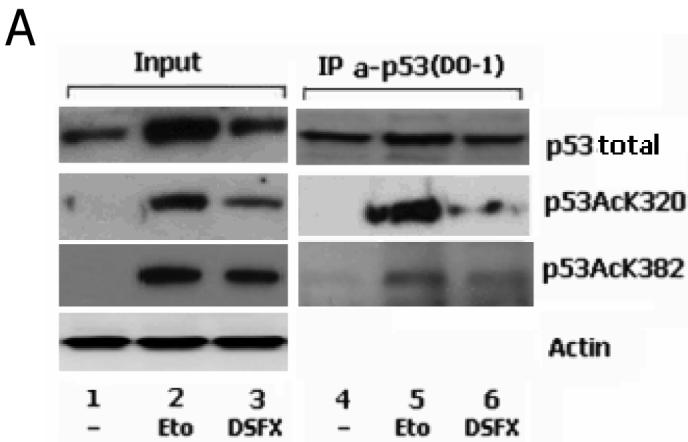

p53 K320 is not acetylated efficiently in DSFX treated cells. (A) U2OS cells were subjected to Etoposide or DSFX treatment or left untreated, and cellular extract submitted to immunoprecipitation with antibody against endogenous p53 (DO-1). Precipitates were blotted with antibodies against p53 anti-human rabbit polyclonal PC-100, p53 acetylated at K320 and p53 acetylated at K382. Actin was used as loading control and input lysates are shown in the left panel. (B) p53 acetylated at K320 is preferentially recruited to the p21WAF-1/CIP-1 promoter. U2OS cells treated as in Figure 1 were processed for ChIP using antibodies against p53 acetylated at K320 (B) or at K382 (C). Primers flanking a 200bp fragment containing the p53 binding sites of the BID and p21WAF-1/CIP-1 promoters (Supplementary Figure 1) were used to amplify precipitated DNA by real-time qPCR. Standard error bars represent the deviations between at least two independent experiments. (D) The specificity of the antibody and the amounts of the precipitated p53 protein are shown.
PCAF enhances the expression of the p21WAF-1/CIP-1 p53 target in hypoxia
The recruitment of the two acetyl-isoforms K320 and K382 of p53 to the p53 binding sites present within the promoters of the cell cycle arrest gene p21WAF-1/CIP-1 and the pro-apoptotic p53 target BID was followed using ChIP assays (Figures 2B, C and D). BID promoter occupancy by the p53 acetylated at K320 was considerably lower (~40%) in the DSFX versus the etoposide treated cells and higher (~300%) in the case of p21WAF-1/CIP-1 (Figure 2B). In contrast, the p53 acetylated at K382 by p300 exhibited higher affinity for both the p53 binding sites of BID and p21WAF-1/CIP-1 promoters in cells treated with DSFX (Figure 2B and 2C). The anti-acetyl p53 antibodies were specific for the acetylated p53 isoforms since no p53 was precipitated in the non treated cells with these antibodies (Figure 2D, tracks 4 and 7). The amount of the precipitated p53 in Etoposide and DSFX treated cells was normalised to the input p53 protein.
The same conclusions were reached following studies using BID and p21WAF-1/CIP-1 luciferase reporter assays (Figure 3A and B respectively). Indeed, both BID-p53bs-Luc and p21-p53bs-Luc reporters were not induced to the same extent in DSFX treated (where K320 is not efficiently acetylated) versus untreated cells (Figure 3A and B, compare bars 3 to 4). Notably, the reduction in BID luciferase reporter expression in DSFX treated compared to the untreated cells was 36% (Figure 3A, compare bars 3 to 4) and in the case of p21WAF-1/CIP-1 reporter much less (19%) (Figure 3B compare bars 3 to 4). The non-acetylatable p53K320R mutant was unable to induce luciferase expression driven by either BID or p21WAF-1/CIP-1 reporters (Figure 3A and B, bars 5 and 6). In cells transfected with the constitutively acetylated K320Q mutant there was no difference in the expression of BID and p21WAF-1/CIP-1 reporters in DSFX treated or untreated cells as p53 K320 acetylation of this mutant is not affected by DSFX in this case (Figure 3A and B, bars 7 and 8).
Figure 3.

Acetylation of p53 at K320 induces preferentially the expression of the p21WAF-1/CIP-1 –Luc- reporter. SAOS2 cells transfected with either the BID-p53bs-Luc (A) or the p21WAF-1/CIP-1 –Luc (B) reporters together with CMV empty vector (bars 1, 2), pCMV-p53wt (bars 3, 4), pCMV-p53K320R (bars 5, 6), or pCMV-p53K320Q (bars 7, 8) expression plasmids were submitted to luciferase reporter assay. Relative luciferase activity was monitored as mentioned in Materials and Methods. Cells submitted to DSFX treatment for 20h are indicated with (+).
Results in Figures 2 and 3 suggest that PCAF binding and partial acetylation of p53 at K320 might explain the inability of p53 to induce the expression of its pro-apoptotic targets in cells treated with DSFX in accordance with published observations [Chao et al., 2006].
PCAF dependent acetylation of p53 regulates cell proliferation
The role of the HAT activity of PCAF on cell cycle progression and apoptosis was investigated by FACS analysis performed in DSFX treated or untreated U2OS and SAOS2 cells transfected with either empty vector, Flag-PCAFwt or Flag-PCAF-ΔHAT expression constructs as indicated in Figure 4A and B. The ratio between apoptotic versus arrested in G1 phase of the cell cycle cells is presented in Figure 4A for U2OS and in 4B for SAOS2 cells. PCAFwt transfected cells exhibited higher apoptotic potential compared to PCDNA3 or PCAF-ΔHAT overexpressing and DSFX treated cells. In agreement with previously published observations [Cohen et al., 2004], this induction of cell death was dependent on the HAT activity of PCAF, since apoptosis was not efficiently stimulated in the PCAF-ΔHAT expressing cells (Figure 4A compare bars 5 to 8). The lesser extent to which apoptosis was stimulated in SAOS2 cells under the same conditions indicates that at least in part and to the measure permitted by these correlative experiments the cell cycle events were p53 dependent (compare Figure 4A, bar 5 to 4B, bar 5). In addition, the caspase 3 assay shown in Supplementary Figure 3 confirmed that the population of cells in sub-G1 phase represented apoptotic cells.
Figure 4.

HIF-1α and p53 dependent cell cycle progression is co-ordinated by PCAF. U2OS (A) and SAOS2 cells (B) were transfected with vectors expressing either PCAFwt or PCAF-ΔHAT together with a plasmid expressing CD20. In some cases RNAi for HIF-1α was co-transfected (A and B bars 7, 8 and 9) and cells were treated with DSFX where indicated. The ratio of the fraction of cells accumulated in sub-G1 versus the percentage of cells arrested in G1 phase of the cell cycle is presented in the diagrams A and B. Results are the average of two independent experiments. (C) Western blot showing the equal expression of transfected PCAFwt and PCAF-ΔHAT in U2OS and SAOS2 cells.
To investigate the contribution of HIF-1α to the cell cycle events in U2OS (Figure 4A) and SAOS2 cells (Figure 4B) expressing the same Flag-PCAFwt, Flag-PCAF-ΔHAT, or empty (PCDNA3) expression vectors we co-transfected RNAi against HIF-1α to silence its expression. The ratio of apoptotic versus G1 arrested cells ectopically expressing PCAFwt in correlation to the respective ratio of RNAi-HIF-1α co-transfected cells was 2.9 times higher for the U2OS and 1.3 for the SAOS2 cells (compare Figure 4A, ratio of bars 5/6 with 4B, bars 5/6) indicating that p53 acetylated at K320 in the absence of HIF-1α favours cell survival and in the presence of HIF-1α apoptosis. In a similar manner HIF-1α in the presence of non acetylated p53 at K320 (PCAF-ΔHAT transfected cells) stimulated cell survival whereas the complete absence of p53 initiated apoptosis (compare Figure 4A, ratio of bars 8/9 (1.8) with 4B, bars 8/9 (2.7)).
Results shown in Figure 4 imply that the HAT activity of PCAF functions as a co-ordinating factor in the regulation of HIF-1α and p53 dependent cell cycle progression in addition to other known p53 and HIF-1α independent effects on apoptosis [Cohen et al., 2004].
PCAF is a HIF-1α co-factor
Taken together the results shown in Figure 4 with the fact that both HIF-1α and p53 share the common co-factor p300 [Freedman et al., 2002] prompted us to investigate whether PCAF as another p53 co-factor, which associates with p300, was also involved in the regulation of HIF-1α transcriptional activity. Towards this end, we performed ChIP assays monitoring the PCAF recruitment to the promoters of known HIF-1 targets such as BID [Erler et al., 2004; Supplementary Figure 5], erythropoietin (Epo) [Semenza et al., 1996] and vascular endothelial growth factor (VEGF) [Forsythe et al., 1996] in U2OS cells ectopically expressing PCAF and treated with or without DSFX. Results shown in Figure 5A confirmed that PCAF acts as HIF cofactor since it is recruited with high efficiency in both BID and VEGF promoters in hypoxic conditions. In agreement with recently published observations showing that Epo is primarily a HIF-2 specific target [Warnecke et al., 2004] PCAF was not recruited to the Epo promoter with the same efficiency as to BID and VEGF promoters in DSFX treated cells. PCAF recruitment to the BID HIF-1α Response Element (HRE) is reflected in increased BID protein levels of both the truncated and the non-truncated forms of BID in U2OS cells over-expressing PCAF and treated for 20h with DSFX (Figure 5B track 3).
Figure 5.

PCAF is recruited to the HREs of known HIF-1α targets. (A) ChIP assays were performed in U2OS cells treated as in Figure 1. Primers flanking the HREs of BID, Epo and VEGF promoters (Supplementary Figure 1) were used to amplify the cross-linked DNA by real-time qPCR. (B) Whole cell lysates from U2OS cells over-expressing PCAF and treated with DSFX for 0, 2h and 20h were submitted to western blot analysis and the protein levels of HIF-1α, Flag-PCAF, PCAF and BID were followed with specific antibodies against these proteins. Actin served as loading control.
In addition, PCAF was found to regulate the expression of other known HIF-1α transcription targets such as Carbonic Anhydrase IX (CA-IX) [Wykoff et al., 2000], VEGF [Forsythe et al., 1996], phosphoglycerate kinase-1 (PGK-1) and lactate dehydrogenase A (LDH-A) [Semenza et al., 1996]. Luciferase reporter assays shown in Figure 6 point out that PCAF functioned as HIF-1α co-activator in the case of the CA-IX-Luc and VEGF-Luc reporters (Figure 6A and B), but did not have any effect in the expression of PGK-Luc and LDH-A-Luc reporters (Figure 6C and D). In addition, PCAF HAT activity played an important role in the co-activation of CA-IX-Luc reporter (Figure 6A) but did not appear to have significant effect on the VEGF-Luc and PGK-Luc reporters (Figure 6B and C) and exerted a negative effect on the expression of LDH-A-Luc reporter (Figure 6D). The observed PCAFwt or PCAF-ΔHAT mediated effects on these HIF-1α targets were not a result of unequal HIF-1α expression levels in cells expressing Flag-PCAFwt or Flag-PCAF-ΔHAT (Figure 6E). In addition, the expression of wt or mutated cofactor was not affected by DSFX treatment (Figure 6E).
Figure 6.

PCAF directs HIF-1α to a specific subset of its targets. PCAFwt or PCAF-ΔHAT overexpressing SAOS2 cells were subjected to 20h DSFX treatment and the luciferase activity of CA-IX (A), VEGF (B), PGK-1 (C) and LDH-A (D) was analysed as described in Material and Methods. Values represent means of four experiments. Panel E shows equal levels of HIF-1α expression in cells transfected either with PCAFwt or with PCAF-ΔHAT. In addition the expression of Flag-PCAFwt and Flag-PCAF- ΔHAT proteins was not affected by DSFX treatment.
Results shown in Figures 4, 5 and 6 imply that PCAF functions as a HIF-1α co-factor, is involved in the HIF-1α mediated apoptosis and that PCAF HAT activity regulates HIF-1α transcriptional selectivity.
PCAF interacts with and acetylates HIF-1α
Next we determined whether endogenous PCAF interacts with HIF-1α in untreated (Figure 7, tracks 1 and 5), DSFX (tracks 2 and 6), etoposide (tracks 3 and 7) or both DSFX and etoposide treated U2OS cells (track 4 and 8). The interaction between the two proteins was evident in the DSFX treated cells and with lower affinity in the cells treated with both DSFX and etoposide (Figure 7, tracks 6 and 8).
Figure 7.

PCAF interacts with HIF-1α. U2OS cells were treated with DSFX (20h) and etoposide (6h) or both DSFX and etoposide or left untreated as indicated. Whole cell extracts were immunoprecipitated using an anti-PCAF antibody against the endogenous protein and blotted for HIF-1α, hDM2, PCAF and p53.
In order to study whether PCAF acetylates HIF-1α, acetylated proteins were precipitated from DSFX treated SAOS2 cells with the pan-Acetyl-lysine antibody and higher levels of acetylated HIF-1α were detected among the precipitated proteins in the extract of the cells ectopically expressing Flag-PCAF (Figure 8A track 6). A low level of HIF-1α acetylation was detected in the non transfected Flag-PCAF cells (Figure 8A, track 4) possibly representing HIF-1α acetylated by ARD-1 [Jeong et al., 2002] or other unidentified HAT(s).
Figure 8.


PCAF acetylates HIF-1α and regulates its protein stability. (A) SAOS2 cells were transfected with Flag-PCAF expression vector and treated with DSFX for 20h as indicated. Whole cell extract was immunoprecipitated with pan-acetyl-Lysine antibody, resolved on PAGE and blotted for HIF-1α and PCAF antibodies. Actin was used as loading control. (B) Whole cell extract from transiently transfected with Flag-PCAFwt U2OS cells was submitted to immunoprecipitation with the anti-H3-K14 Ac-Lysine antibody and western blotted with anti-HIF-1α, anti-p53 and anti-Flag specific antibodies. 10% of the lysate was loaded as input and is shown in the left panel. Actin was used as loading control. (C) U2OS cells were transfected with Flag-PCAFwt and Flag-PCAF-ΔHAT and exposed to DSFX treatment for 20h. 48h after transfection the whole cell extract from these cells was submitted to immunoprecipitation with the pan-acetyl-Lysine antibody and immunoblotted with anti-HIF-1α antibody. U2OS transfected with empty CMV or Flag-PCAFwt expression vectors were co-transfected with plasmids expressing HA-HIF-1α(wt) (D) HA-HIF-1α(K532R) (E) or HA-HIF-1α(K389R) (F) were submitted to immunoprecipitation with HA antibody after 20h of DSFX treatment as indicated. Pan-acetyl-Lysine antibody was used to detect acetylated proteins in the immunoprecipitations, anti-HA antibody against transfected HA-HIF-1α and anti-HIF-1α to detect total HIF-1α in the inputs. Actin was used as loading control. (G) PCAF dependent acetylation of HIF-1α regulates its protein stability. Empty PCDNA3 vector, Flag-PCAFwt or Flag-PCAF-ΔHAT were transiently expressed in SAOS2 cells and incubated with DSFX for 20h and cyclohexamide for the indicated time points. Cell extracts were analysed with PAGE and subjected to immunoblot analysis with anti-HIF-1α, Flag and actin antibodies. (H) The diagram represents the ratio of the intensity of the HIF-1α bands in the cells transfected with different expression vectors (PCDNA3, PCAFwt, PCAF-ΔHAT) and the intensity of the actin bands in the respective cells. The intensity of HIF-1α divided by the intensity of actin at 0 time points was regarded as 100%. Densitometric analysis was performed using the AIDA 3.52.046 quantification software.
The same results were obtained by precipitating PCAF acetylation targets from DSFX treated U2OS cells using another antibody that recognises the histone H3 acetylated lysine, previously reported to be a specific PCAF acetylation substrate [Poux and Marmorstein, 2003]. Acetylation of HIF-1α was evident in the DSFX and both DSFX and etoposide treated cells, although in the latter case PCAF exhibited lower acetylation efficiency for HIF-1α, and slightly higher for p53 (Figure 8B, compare tracks 7 and 8), possibly due to weaker association between HIF-1α and PCAF under these conditions (Figure 7, track 8). The observed HIF-1α acetylation was PCAF dependent as the PCAF-ΔHAT mutant failed to acetylate the transcription factor (Figure 8C, compare tracks 2 to 4).
To confirm that the pan-acetyl-lysine antibody immunoprecipitated acetylated HIF-1α and not another acetylated protein that interacts with HIF-1α and co-precipitates with the transcription factor, we performed immunoprecipitation experiments in U2OS cells ectopically expressing HA-HIF-1α using an anti-HA specific antibody. Precipitated HIF-1α was recognised by the acetyl-lysine antibody (Figure 8D, tracks 2 and 4).
Furthermore, in order to identify the HIF-1α site(s) that are PCAF acetylation targets we mutated the ARD1 target K532 and K389 to arginine. Immunoprecipitation of the mutated HA-HIF-1α-K532R and HA-HIF-1α-K389R with HA antibody and immunoblotting with the acetyl-lysine antibody confirmed that HIF-1α was acetylated and revealed that K532 is not the only acetylated site in HIF-1α (Figure 8E, and 8F, tracks 2 and 4).
To test the effect of PCAF dependent acetylation of HIF-1α on its protein stability we followed HIF-1α half life in SAOS2 cells transfected with either PCAFwt or PCAF-ΔHAT expressing vectors or untransfected cells treated with cyclohexamide for different time points from 0 to 6h (Figure 8G). As shown with densitometric analysis (Figure 8H) HIF-1α in PCAFwt transfected cells could still be detected after 6h of cyclohexamide treatment and its half life was approximately 4h, whereas in PCAF-ΔHAT, or PCDNA3 transfected cells the HIF-1α half life was shorter (approximately 2h) (Figure 8G). The intensity of the HIF-1α bands in the cells transfected with different expression vectors (PCDNA3, PCAFwt, PCAF-ΔHAT) have been normalised to the intensity of the actin bands in the respective cells (Figure 8H).
Discussion
In this study, we provide insight into the molecular mechanisms regulating p53 transcriptional selectivity in the apoptotic response to treatment with the hypoxia mimicking agent DSFX. Despite the recently published analysis of the p53 transcriptional activity under hypoxic conditions by genome-wide expression profile techniques [Hammond et al., 2006] the detailed and subtle mechanisms involved in the p53 selectivity to transactivate its downstream targets in hypoxic conditions remain imprecisely defined. The most likely explanation of p53 incompetence to activate its pro-apoptotic targets under hypoxia is based on an inability to form requisite networks with specific co-activators [Hammond and Giaccia, 2005]. A precedent for this thinking is that p53 fails to induce the expression of Bnip3L under hypoxia because the binding between p53 and the p300/CBP co-activators is weak and unable to support Bnip3L expression in these conditions [Fei et al., 2004]. Considering these observations we wanted to define the composition of the transcription co-activator complexes that bind p53 and fine tune its transcriptional activity under hypoxia.
The molecular mechanisms of p53 and HIF-1α mediated apoptosis mostly operate through the activation of pro-apoptotic genes that bear binding sites for these transcription factors in their promoter such as the Bcl-2 family members that play a central role in the initiation and execution of the intrinsic pathway of apoptosis [Cory et al., 2003]. The expression and activity of anti- and pro-apoptotic members of this family is delicately balanced by several mechanisms. The expression of the pro-apoptotic Bax, PUMA, Noxa, and BID genes can be regulated by the transcriptional activity of the p53 tumour suppressor [Vousden and Lane, 2007]. Recently published observations reported that the basis of the p53 promoter selectivity lies on its differential post-translational modifications. For example, activation of the Bax promoter requires participation of p53 acetylated at Lys320 by PCAF and Lys373 by p300, while the p53-dependent induction of p21 requires the participation of p53 acetylated at either site [Roy et al., 2005]. In addition, acetylation of p53 by TIP-60 at K120 promotes apoptosis and not cell cycle arrest [Tyteca et al., 2006], whereas acetylation at p53 K373 and K382 by p300 results in the induction of pro-apoptotic p53 targets [Knights et al., 2006]. The existence of hypoxia responsive elements (HREs) in the regulatory regions of the gene promoters of several Bcl-2 family members and thus regulation of their expression by HIF-1α has also been demonstrated [Greijer and van der Wall, 2004]. These observations imply that selectivity of p53 transcriptional activity under hypoxia is regulated by upstream events such as posttranslational modifications, specific protein-protein interactions with co-activators [Demonacos et al., 2004] and other transcription factors such as HIF-1α [An et al., 1998]. To gain further mechanistic insight into apoptosis in hypoxic cells and provide details on how it is regulated we investigated the function of the known p53 co-regulator PCAF and how this factor orchestrates the coordination of regulatory events leading to the selective pattern of gene expression of both p53 and HIF-1α under hypoxia.
In this study we show that the transcriptional co-activator PCAF directs p53 preferentially to the p21WAF-1/CIP-1 promoter in DSFX treated cells. In addition, we established that PCAF dependent acetylation of p53 K320 is deficient in DSFX treated cells and this correlates with the reduction of the recruitment of this acetylated p53 isoform to its pro-apoptotic targets. These observations are in agreement with recently published reports showing that K320Q prevents cell death and induces G1 arrest [Knights et al., 2006] and could explain the predominance of surviving and growth arrested versus dying cells in hypoxic tumour subpopulations [Bunz et al., 1998]. Earlier studies [Koumenis et al., 2001], have shown that hypoxia primarily induces the interaction of p53 with mSin3A, but not with p300. These data suggest that different levels of hypoxia or tissue specificity are involved in the differential interactions of p53 with specific co-activators or co-repressors modulating its transcriptional activity under diverse stress conditions.
To provide additional insight in the regulation of the cell growth program regulated by PCAF we performed FACS analysis. We observed increased number of U2OS cells expressing Flag-PCAF-ΔHAT arresting in G1 phase of the cell cycle (Figure 4A and B, compare bars 8 to 5), which could be explained by the fact that p53 was preferentially recruited to the p21WAF-1/CIP-1 promoter (Figure 3B). On the other hand the accumulation of PCAFwt expressing U2OS cells in Sub-G1 phase (Figure 4A) was at least in part a result of the upregulation of BID expression in these cells after DSFX treatment (Figure 5B). Furthermore, FACS analysis showed that p53 and HIF-1α co-operate in inducing apoptosis and they have opposing functions in the G1 cell cycle arrest in accord with previously published observations [Goda et al., 2003].
Alternative pathways involving transcription factors activated in DSFX treated cells, such as FOXO3a [You et al., 2006] mediate induction of p53 and HIF-1α independent apoptosis. PCAF as a FOXO3a HAT [Brunet et al., 2004] might be implicated in the execution of apoptosis induced by this transcription factor. Another pathway through which the PCAF HAT activity induces p53 independent apoptosis is through PCAF dependent acetylation of Ku70, which results in the disruption of the complex of this DNA damage repair protein with Bax, stimulating initiation of programmed cell death [Cohen et al., 2004].
To explore further these observations and investigate whether PCAF has any effect on HIF-1α transactivation we followed the promoter occupancy of known HIF-1α targets by PCAF. These experiments showed that PCAF is recruited to the HRE sites of two HIF-1α targets, the pro-apoptotic BID [Erler J. et al., 2004] and the angiogenic VEGF gene [Pescador et al., 2005]. Interestingly the expression of the pro-apoptotic truncated variant of BID was identified in the DSFX treated cells over-expressing PCAF, implying that PCAF is involved in the regulation of HIF-1α mediated apoptosis. Other assays showed that PCAF is a HIF-1α transcriptional co-factor and through its HAT activity it specifically and selectively directs HIF-1α to a subset of its target genes. There are several potential explanations for this distinction in the PCAF function. Firstly, differences in the structure of the promoter between the different HIF-1α targets could be responsible for preferential binding of particular subset of these targets by differently modified HIF-1α isoforms. Indeed, the gene encoding CA-IX conforms to a pattern first described among genes encoding glycolytic enzymes [Hu et al., 2003], and is specifically responsive to HIF-1α, showing no response to HIF-2α in any cell type [Raval et al., 2005]. The gene encoding VEGF is rather similar to a pattern previously reported for genes encoding proteins such as PHD3 and GLUT-1 [Raval et al., 2005]. Therefore, PCAF dependent acetylation of HIF-1α might be the mechanism by which HIF-1α subunit distinguishes between its target promoters or even between targets that are specifically responsive to HIF-1α or to HIF-2α transcription factors [Gordan et al., 2007], a possibility that we are currently investigating in our laboratory. Secondly, PCAF might help the recruitment of different transcription factors to distinct regions of the genome. In turn, these associations could allow different transcription factors to carry out specialised functions determining the cellular fate (survival or apoptosis). In line with these observations previous reports have indicated that the p300 co-activator is vital for the induction of a part of HIF responsive genes and dispensable for others [Kasper et al., 2005].
Further study of the effects of PCAF on HIF-1α revealed that the two proteins interact in DSFX treated cells (Figure 7) and this interaction results in the PCAF dependent acetylation of HIF-1α (Figure 8). To exclude the possibility that HIF-1α co-precipitated with other acetylated proteins such as other HAT(s), acetylated p53, or auto-acetylated PCAF [Liang et al., 2006] we followed two approaches. First we immunoprecipitated acetylated proteins from the extract of U2OS cells expressing either PCAFwt or PCAF-ΔHAT and immunoblotted with antibodies recognising HIF-1α. HIF-1α was more efficiently acetylated in the PCAFwt transfected cells and was acetylated to a lesser extent in the PCAF-ΔHAT expressing cells. The same conclusion was reached when we used an antibody that recognises PCAF specific acetylation targets to perform the precipitation of PCAF acetylation targets [Poux and Marmorstein, 2003]. Secondly we mutated two possible acetylation targets in HIF-1α namely K532 and K389. Precipitation of HIF-1α with HA antibodies and blot with the acetyl-lysine antibody confirmed that HIF-1α was acetylated.
Although the effect of ARD1 mediated acetylation of HIF-1α is a controversial issue, it has been linked to HIF transcriptional activity and protein stability [Bilton et al., 2006]. We tested whether PCAF mediated similar effect on HIF-1α by following its half-life and we observed that HIF-1α acetylated by PCAF was a more stable protein (Figure 8G and H). Studies to identify the possible site(s) of HIF-1α acetylated by PCAF are currently ongoing in our laboratory.
In conclusion, our study provides an additional molecular mechanism explaining the inability of p53 to activate its pro-apoptotic targets in hypoxia and implicates PCAF in the regulation of the fine-tuning of the transcriptional activity of both p53 and HIF-1α in hypoxic conditions as well as regulating protein stability of both transcription factors [Linares et al., 2007; Jin et al., 2002].
Material and Methods
Cell lines, culture conditions and constructs
Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% foetal calf serum (FBS) and 1% 10U/ml penicillin and streptomycin was used to maintain human osteosarcoma U2OS (p53+/+) and SAOS2 (p53−/−) cell lines at 37°C and 5% CO2. Cells were collected 20h after 250μM DSFX treatment (Sigma) and 16h after incubation with 10μM etoposide (Sigma) unless otherwise indicated.
The p53K320R expression vector was a gift from Dr Halazonetis, the pCiFlag-PCAF(wt) and pCiFlag-PCAF(ΔHAT) from Dr Talianidis. Human BID luciferase reporter comprising the consensus p53-binding site has been provided by Dr El-Deiry and is described in [Sax et al., 2002]. The p21WAF-1/CIP-1 [El-Deiry et al., 1993] and CA-IX-HRE-luc [Wykoff et al. 2000] have been previously described. Silencing of HIF-1α with RNAi was performed as described previously [Erler et al., 2004].
Site-directed mutagenesis was performed by amplifying the HA-HIF-1α(wt) plasmid with two sets of primers to achieve the desired mutations. The mutagenesis was performed using the QuickChange® Site Directed Mutagenesis Kit (Stratagene) following the manufacturer's instructions.
The calcium phosphate method [Demonacos et al., 2001] and the polyfect transfection system (QIAGEN) were used to transfect cells. Luciferase reporter assays were carried out as described previously [Demonacos et al., 2001].
Immunoprecipitation and immunobloting
Total cellular protein extract was immunoprecipitated with the following antibodies: mouse monoclonal anti-p53 (DO-1, Santa Cruz), anti-human rabbit polyclonal p53 (2381-PC-100, Trevigen), mouse monoclonal anti-Flag (M2, Sigma), mouse monoclonal anti-acetyl-Lysine (4G12, Upstate), rabbit polyclonal anti-acetyl-Histone H3 at Lys14 (Upstate), anti-PCAF rabbit polyclonal (H-369, Santa Cruz), Anti-HIF-1α (H1α67, Calbiochem) and goat polyclonal anti BID (R&D). For detection of acetylated forms of p53, cells were treated with Na− butyrate (1mM) for 2h before lysis. IP extracts were analyzed accordingly by Western blotting with anti-acetyl K320 p53 (Upstate) and anti-acetyl-p53 at Lys382 (#2525, Cell Signaling). Proteins were visualised using ECL (Pierce) according to the manufacturer's instructions.
Chromatin immunoprecipitation
ChIP analysis was performed in U2OS cells using the Active Motif ChIP express kit. Briefly, after chromatin cross-linking with 1% formaldehyde chromatin complexes were immunoprecipitated from DSFX treated or not treated cells with antibodies against p53 acetylated at K320 and K382, anti-p300 NM-11 (Santa Cruz), anti-Flag M-20 (Sigma), or irrelevant (HRP-conjugated anti-rabbit). Quantitative PCR analysis was performed with the oligonucleotides described in Supplementary Figure 1. Values obtained from the ChIP assay analysis (Opticon Monitor 3 software) were normalised to the background obtained from the precipitation with a non specific antibody. Figures 1A, 1C, 2B, 2C and 5A indicate values obtained by subtracting the amount of the non specifically precipitated immunocomplexes from the amount of the specifically precipitated ones.
Flow Cytometry
Cells were transfected with the indicated expression vectors, treated with DSFX as specified in Figure 4, harvested 48h after transfection and fixed in 50% ethanol. Propidium iodide (PI) and RNase A were added to final concentrations of 50 μg/ml and 100 μg/ml, respectively and transfected cells were stained with a CD-20-FITC antibody for CD20 expression. Cell cycle profile was determined by FACScan flow cytometry and analyzed by CellQuest software (Becton Dickinson) as previously described (Demonacos et al., 2001). Cell count represents the cells exhibiting the CD20 antigen used as a transfection efficiency control. The average transfection efficiency was 30% for U2OS and 12% for SAOS2 cells. Only the CD20 tranfected population of cells (sorted by CD20-FITC conjugated antibody) has been taken into account for the FACS analysis. The average of two independent FACS experiments, is shown in Figure 4A and B and the cell cycle profiles from one experiment in Supplementary Figure 3.
Supplementary Material
Acknowledgements
We are grateful to W. El-Deiry for providing the BID-p53-Luc reporter construct, I. Talianidis for the Flag-PCAF and Flag-PCAF-ΔHAT constructs, T. Halazonetis for the p53K320R expression vector, K. Williams for the HIF-1α responsive reporters of VEGF-Luc, CA-IX-Luc, PGK-1-Luc and LDH-A-Luc, M. Blaylock and the Paterson Institute for Cancer Research for assistance with FACS analysis. Our research is supported by the School of Pharmacy, University of Manchester (CD), Cancer Research UK (CDive), MRC program grant (G0500366) to IJS and Wellcome Trust (069024) to MKD.
References
- An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature. 1998;392:405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- Achison M, Hupp TP. Hypoxia attenuates the p53 response to cellular damage. Oncogene. 2003;22:3431–3440. doi: 10.1038/sj.onc.1206434. [DOI] [PubMed] [Google Scholar]
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilton R, Trottier E, Pouyssegur J, Brahimi-Horn MC. ARDent about acetylation and deacetylation in hypoxia signalling. Trends Cell Biol. 2006;16:616–621. doi: 10.1016/j.tcb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, An WG, Romanova LY, Trepel J, Fojo T, Neckers L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J Biol Chem. 1998;273:11995–11998. doi: 10.1074/jbc.273.20.11995. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn C, Mazure N, Pouysségur J. Signalling via the hypoxia-inducible factor-1α requires multiple posttranslational modifications. Cell Signalling. 2005;17:1–9. doi: 10.1016/j.cellsig.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, et al. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol. 2006;26:6859–6869. doi: 10.1128/MCB.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li M, Luo J, Gu W. Direct interactions between HIF-1α and Mdm2 modulate p53 function. J Biol Chem. 2003;278:13595–13598. doi: 10.1074/jbc.C200694200. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- Cory S, Huang DCS, Adams JM. The Bcl-2 family: roles in cell cycle survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- Demonacos C, Krstic-Demonacos M, La Thangue NB. A novel TPR- motif co-factor contributes to p300 activity in the p53 response. Mol Cell. 2001;8:71–84. doi: 10.1016/s1097-2765(01)00277-5. [DOI] [PubMed] [Google Scholar]
- Demonacos C, Krstic-Demonacos M, Smith L, Xu D, O'Connor DP, Jansson M, et al. A new effector pathway links ATM kinase with the DNA damage response. Nat Cell Biol. 2004;6:968–976. doi: 10.1038/ncb1170. [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Erler JT, Cawthorne CJ, Williams KJ, Koritzinksy M, Wouters B, Wilson C, et al. Hypoxia mediated down regulation of Bid and Bax in tumours occurs via HIF-1 dependent and independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24:2875–2889. doi: 10.1128/MCB.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei P, Wang W, Kim S, Wang S, Burns TF, Sax JK, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 6:597–609. doi: 10.1016/j.ccr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman SJ, Sun Z-YJ, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia-inducible factor-1α by CITED2. Nat Struct Biol. 2004;10:504–512. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun Z-YJ, Poy F, Kung AL, Livingston DM, Wagner G, et al. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1α. Proc Natl Acad Sci USA. 2002;99:5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda N, Dozier S, Johnson RS. HIF-1 in cell cycle regulation, apoptosis, and tumor progression. Antioxidants & Redox Signaling. 2003;5:467–473. doi: 10.1089/152308603768295212. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Bertout JA, Hu C-J, Diehl JA, Simon MC. HIF-2a promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson LO, Friedler A, Freund S, Rudiger S, Fersht AR. Two sequence motifs from HIF-1α bind to the DNA-binding site of p53. Proc Natl Acad Sci USA. 2002;99:10305–10309. doi: 10.1073/pnas.122347199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond EM, Giaccia AJ. The role of p53 in hypoxia-induced apoptosis. Biochem Biophys Res Comm. 2005;331:718–725. doi: 10.1016/j.bbrc.2005.03.154. [DOI] [PubMed] [Google Scholar]
- Hammond EM, Mandell DJ, Salim A, Krieg AJ, Johnson TM, Shirazi HA, et al. Genome-wide analysis of p53 under hypoxic conditions. Mol Cell Biol. 2006;26:3492–3504. doi: 10.1128/MCB.26.9.3492-3504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J-W, Bae M-K, Ahn M-Y, Kim S-H, Sohn T-K, Bae M-H, et al. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- Jin Y, Zeng SX, Dai M-S, Yang X-J, Lu H. MDM2 Inhibits PCAF-mediated p53 acetylation. J Biol Chem. 2002;277:30838–30843. doi: 10.1074/jbc.M204078200. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–128. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, et al. Two transactivation mechanisms cooperate for the bulk of HIF-1 responsive gene expression. EMBO J. 2005;24:3846–3858. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovani S, Muratoglu S, Perez R, Swartzbeck A, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Alarcon R, Hammond E, Sutphin P, Hoffman W, Murphy M, et al. Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol Cell Biol. 2001;21:1297–1310. doi: 10.1128/MCB.21.4.1297-1310.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Liang D, Kong X, Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle. 2006;5:2430–2435. doi: 10.4161/cc.5.21.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares LK, Kiernan R, Triboulet R, Chable-Bessia C, Latreille D, Cuvier O, et al. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol. 2007;9:331–338. doi: 10.1038/ncb1545. [DOI] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Ogrysko PR, Arsham AM, Koch CJ, Simon MC. p53 cannot be induced by hypoxia alone but responds to the hypoxic microenvironment. Oncogene. 2004;23:4975–4983. doi: 10.1038/sj.onc.1207657. [DOI] [PubMed] [Google Scholar]
- Pescador N, Cuervas Y, Naranjo S, Alcaide M, Villar D, Landazuri MD, et al. Identification of a functional hypoxia-responsive element that regulates the expression of egl nine homologue 3 (egln/phd3) gene. Biochem J. 390:189–197. doi: 10.1042/BJ20042121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poux AN, Marmorstein R. Molecular basis for Gcn5/PCAF histone acetyltransferase selectivity for histone and non-histone substrates. Biochemistry. 2003;42:14366–14372. doi: 10.1021/bi035632n. [DOI] [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von-Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Packman K, Jeffrey R, Tenniswood M. Histone deacetylase inhibitors differentially stabilise acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Diff. 2005;12:482–491. doi: 10.1038/sj.cdd.4401581. [DOI] [PubMed] [Google Scholar]
- Roy S, Tenniswood M. Site-specific acetylation of p53 directs selective transcription complex assembly. J Biol Chem. 2007;282:4765–4771. doi: 10.1074/jbc.M609588200. [DOI] [PubMed] [Google Scholar]
- Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by ER contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- Sakaguchi KJ, Herrera S, Saito T, Miki M, Bustin A, Vassilev A, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid T, Zhou J, Köhl R, Brüne B. p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1) Biochem J. 2004;380:289–295. doi: 10.1042/BJ20031299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Jiang B-H, Leung SW, Passantino R, Concordet J-P, Maire P, et al. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Tyteca S, Legube G, Trouche D. To die or not to die: A HAT trick. Mol Cell. 2006;24:807–812. doi: 10.1016/j.molcel.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004;9(Suppl 5):4–9. doi: 10.1634/theoncologist.9-90005-4. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. p53 and prognosis; new insights and further complexity. Cell. 2005;120:7–10. doi: 10.1016/j.cell.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Warnecke C, Zaborowska Z, Kurreck J, Erdmann VA, Frei U, Wiesener M, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1a and HIF-2a (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 2004;18:1462–1464. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- Wykoff CC, Beasley NJP, Watson PH, Turner KJ, Pastorek J, Sibtain A, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–7083. [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Mao XO, Sun Y, Xia Z, Greenberg DA. p38 mitogen-activated protein kinase mediates hypoxic regulation of Mdm2 and p53 in neurons. J Biol Chem. 2002;277:22909–22914. doi: 10.1074/jbc.M200042200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.