

Abstract
Memantine, an uncompetitive NMDA receptor antagonist used for the treatment of Alzheimer's disease (AD), has been hypothesized to have neuroprotective properties. However, the similarity of its mechanism of action to other NMDA receptor antagonists has led to concerns that it may also have neurotoxic effects. To assess both the neuroprotective and neurotoxic potential of memantine in a mouse model of AD (Tg2576 mice), we used quantitative light and electron microscopy to investigate the effects of long-term (6 months) administration of memantine (5, 10 and 20 mg/kg) on plaque deposition and neuronal morphology in the hippocampus and overlying cortex. A fear-conditioning paradigm was used to evaluate the behavioral consequences of any observed changes in structure. Administration of the two higher doses of memantine (10 and 20 mg/kg) was associated with a significant decrease in β-amyloid (Aβ) plaque deposition, increases in synaptic density and the appearance of degenerating axons; the latter two effects were independent of genotype. Administration of the lowest dose of memantine (5 mg/kg) was associated with a significant decrease in Aβ plaque deposition and a significant increase in synaptic density, but not a significant increase in degenerating axons. However, memantine did not significantly improve behavioral deficits associated with genotype in a fear-conditioning paradigm at any dose. These results suggest that chronic memantine administration may have both neuroprotective and neurotoxic effects in a mouse model of AD.
Keywords: memantine, synapse density, amyloid plaque, conditioned fear, Tg2576 mice, Alzheimer's disease
Introduction
Memantine, an uncompetitive antagonist at NMDA receptors (Kornhuber et al, 1989; Chen et al, 1992), is now widely prescribed for the treatment of moderate-to-severe Alzheimer's disease (AD). Memantine is rapidly displaced from the NMDA receptor, which may avoid prolonged receptor blockade and the detrimental effects on learning and memory associated with prolonged blockade of the NMDA receptor (Lipton, 2006). Further, memantine acts as an antagonist at nicotinic acetylcholine receptors and at 5-HT receptors (Buisson and Bertrand, 1998; Aracava et al, 2005). The rationale for using memantine in AD patients is based on the hypothesis that blockade of NMDA receptor-mediated excitotoxicity can help preserve neuronal structure and function (Lipton, 2006, 2007; Wenk et al, 2006). The ability of memantine to protect neurons against NMDA- or glutamate-induced excitotoxicity has been shown in vitro (Erdö and Schäfer, 1991; Chen et al, 1992; Weller et al, 1993), and in animal models of neurodegenerative disease (Wenk et al, 1995, 1997, 2006; Miguel-Hidalgo et al, 2002). Therefore, it has been proposed that memantine can improve cognition in AD and perhaps even slow disease progression by blocking NMDA receptor-mediated excitotoxicity (Ditzler, 1991; Frankiewicz et al, 2000; Reisberg et al, 2003; Lipton, 2006). However, concerns have also been raised about the potential for memantine to damage neurons given that other NMDA receptor antagonists, such as phencyclidine and MK-801, have been shown to trigger neuronal degeneration in rodents (Li et al, 2002; Creeley et al, 2006). At present, there are no published postmortem data that can be used to assess the potential for memantine to either slow or induce neuronal degeneration in patients with AD. Thus, whether long-term memantine treatment can reverse or prevent damage to areas of the brain resulting from AD pathology remains unclear.
In the current study, we used quantitative light and electron microscopic studies of the hippocampus and overlying cortex, and contextual memory as assessed in a conditioned fear paradigm, to assess the effects of chronic administration of memantine on neuronal structure and function in the Tg2576 mouse model of AD. We focused our structural assessments on the hippocampal formation because these structures play a major role in learning and memory (Knowles, 1992; Bannerman et al, 2001) and is also a site for Aβ deposition in AD patients (Probst et al, 1983; West et al, 2004) and in several transgenic mouse models of AD (Su and Ni, 1998; Reilly et al, 2003). Synapse loss induced by Aβ remains a likely basis for the behavioral deficits observed in Tg2576 mice (Stern et al, 2004). Previously, we demonstrated spatial and temporal relationships between synapse loss and Aβ deposition in the dentate gyrus of Tg2576 mice at 6–9 months of age (Dong et al, 2007). In this study, the effects of memantine on Aβ deposition and neuronal structure in the hippocampus and overlying cortex and contextual memory were assessed in transgenic Tg2576 mice at 9–10 months of age, following 6 months of memantine or vehicle administration. In addition, we evaluated plaque deposition in the cortex overlying the hippocampus and looked for evidence of memantine-induced neuronal vacuolization in the posterior cingulate and retrosplenial cortices under light microscopy.
Materials and Methods
Animals
The Tg2576 mouse strain, created by Hsiao et al (1996), was used for these experiments. Tg2576 mice were derived from C57B6/SJL × C57B6 crosses, and contained the double mutation Lys670-Asn, Met671-Leu (K670N, M 671L). This mutation is driven by a hamster prion protein gene promoter in C57B6j × SJL. Brain levels of APP are more than four times higher, and Aβ levels are 5–14 times higher, in transgenic mice than in control mice. The presence of the human APP gene was demonstrated by post-weaning tail biopsy and DNA genotyping using primers as previously described (Hsiao et al, 1996). The breeding and maintenance of the Tg2576 mouse colony were conducted in consultation with the veterinary staff in the Department of Comparative Medicine at Washington University School of Medicine. All animal procedures were done in accordance with the National Institutes of Health and Institutional Guidelines.
Transgenic mice that express the human APP gene (Tg +) and their non-transgenic (Tg−) littermates were randomly assigned to various groups for memantine administration. A total of 89 animals of both genders (Tg + = 42; Tg− = 47) were used for this study. Eighty-four animals were used for behavioral tests (5 mg/kg (Tg + = 12, Tg− = 10); 10 mg/kg (Tg + = 17, Tg− = 10); 20 mg/kg (Tg + = 9; Tg− = 12) and vehicle (Tg + = 8, Tg− = 6)). After behavioral testing, 38 of these animals were used to assess neuronal density and ultra-structure changes (5 mg/kg (Tg + = 4, Tg− = 4); 10 mg/kg (Tg + = 4, Tg− = 4); 20 mg/kg (Tg + = 6, Tg− = 4) and vehicle (Tg + = 6, Tg− = 6)), and 22 Tg + mice were used to measure amyloid plaques (5 mg/kg (Tg + = 4), 10 mg/kg (Tg + = 8), 20 mg/kg (Tg + = 5), and vehicle (Tg + = 5)).
Drug Treatment
Approximately equal groups of Tg + and Tg− mice received one of three daily doses of memantine or vehicle for 6 months beginning at 3 months of age. Three doses of memantine (equivalent to 5, 10, and 20 mg/kg/day) were dissolved in drinking water that was prepared fresh weekly. To ensure the consistency of drug dosing and that the presence of memantine did not influence daily water consumption, we measured the amount of water consumed in individual mice weekly and calculated total and mean volume of drinking water with and without drug. The volume of consumption within individual mice was significantly different (F(250,9) = 8.55, p<0.0001), but the mean amount of consumption between groups was not significantly different (F(38,1) = 0.195, p = 0.66).
Conditioned Fear Testing
The assessment of contextual and cued memory within a fear-conditioning paradigm was performed using methods previously described (Bardgett et al, 2003; Dong et al, 2005). Animals were trained and tested in two Plexiglas conditioning chambers (26 × 18 × 18 cm) (Med Associates Inc., Georgia, VT), with a metal grid floor, within a larger sound-attenuating chamber. On day 1, training took place in the first chamber which contained a cup containing mint extract placed beneath the grid floor. Freezing behavior, defined as no movement (ambulation, sniffing or stereotypy) other than respiration, was recorded every 10 s for 5 min. After the first 2 min, a 20 s, 80 dB, a 2800 Hz tone was presented, and during the last 2 s of the tone, the animals received a 1.0 mA continuous footshock. This procedure was repeated two more times at 1 min intervals. On day 2, animals were returned to the chamber and the amount of freezing behavior in response to context (ie memory for context) was recorded every 10 s for 8 min. On day 3, the animals were placed in a second chamber (scented with coconut with the grid floor covered with polyurethane). Freezing behavior was again recorded for 2 min. Then, the 80 dB, 2800 Hz tone was represented continuously for 8 min, and freezing behavior in response to the cue was recorded.
Sensitivity to the footshock was tested on the day following completion of the cued conditioning evaluation. The animals were returned to the first conditioning chamber for 2 min and exposed to a series of 2 s shocks, beginning with an intensity of 0.05 mA. The shock intensity was increased by 0.05 mA every 20–30 s until a jumping response was evoked.
Tissue Preparation
For Aβ plaque evaluation, animals were deeply anesthetized and perfused transcardially with 1% heparinized 0.01 M phosphate-buffered saline (PBS) for 2 min and then 4% paraformaldehyde for 25–30 min. Brains were removed and post-fixed overnight at 4°C using the same fixative with 30% sucrose. The brains were cut into 35 μm thick serial sections in the coronal plane using a cryostat (Leica CM 1850 UV, Nussloch, Germany). Of the six series of sections (15–20 sections each), we used one series for Thioflavine S staining to assess compact (fibrillar) Aβ plaques and plaque burden. Floating sections in a 1% Thioflavine S aqueous solution for 5 min then differentiated in 70% alcohol for 3–5 min (Guntern et al, 1992).
For studies of neuronal density and ultra-structure, animals were deeply anesthetized and perfused transcardially with 0.01 M PBS-containing heparin sodium for 2 min, followed by a 30 min perfusion with 2% paraformaldehyde, 2% glutaraldehyde and 4% sucrose in 0.1 M PBS. Sections 250 μm thick were cut in the coronal plane using a vibratome. Fifteen sections encompassing the whole hippocampus were selected from each brain and were rinsed in cold 0.1 M PBS, treated with 2% OsO4 in 0.1 M PBS for 90 min at 4°C and rinsed again in 0.1 M PBS at room temperature. The sections were then dehydrated in a graded series of ethanol solutions, followed by propylene oxide, and left overnight in a 1 : 1 mixture of propylene oxide-Polybed 812 (Electron Microscopy Sciences, Hatfield, PA). Finally, the sections were flat embedded in Polybed 812 in an oven at 60°C for 48–72 h. From the 15 embedded sections, three were selected for semi- and ultra-thin sectioning. Representative sections included the dorsal, medial and ventral hippocampus and underlying cortex.
Selected embedded sections (250 μm thick) were trimmed and sectioned again using a Reichert Ultracut E Ultra-microtome (Austria). Semi-thin (1 μm) sections that included hippocampus and underlying cortex were cut and stained with toluidine blue for studying overall histopathological changes in the posterior cingulate and retrasplenial cortices and the hippocampus. The semi-thin sections were also served as reference sections for ultra-thin cutting. The sections were then continued to be trimmed and ultra-cut the thin (75–90 nm) sections containing the outer molecular layer of the dentate gyrus (dorsal blade), which were mounted on 400-mesh grids (every mesh grid is 62 × 62 μm2, Electron Microscopy Sciences, Hatfield, PA). The sections were stained using 3% uranyl acetate for 20 min followed by lead citrate for 5 min and then were examined using a JEOL 100 CX electron microscope (Japan).
Quantitative Evaluation of β-Amyloid Plaque Deposition
Aβ plaques were measured in the hippocampus and overlying cortex using non-biased stereological methods, as described previously (Dong et al, 2004, 2007). The stereological system program (Olympus, Albertslund, Denmark) was used to measure Aβ plaque number and burden (total Aβ plaque area/brain area in coronal section). The numbers calculated from one series of sections were multiplied by six to obtain an estimate of total Aβ plaque number and burden per brain.
Quantitative Evaluation of Neuronal Morphology
Two to three semi-sections from each brain were selected to look for evidence of memantine-induced changes in neuronal morphology, and in particular vacuolization, in the hippocampus and in the posterior cingulate and retrosplenial cortices. The selected coronal sections were cut at the level of the decussation of the corpus callosum (approximately −5.30 to −5.60 mm caudal to bregma) to ensure sampling each brain at the same rostrocaudal level. Also, the brain regions selected using this sampling scheme have been reported to be particularly vulnerable to vacuolization induced by the administration of NMDA antagonists (Fix et al, 1993, 1994; Creeley et al, 2008). A total of 10 neurons in each section of the posterior cingulate and retrosplenial cortices and the hippocampus were detected (under ×100 objective lens) and evaluated for the presence and severity of interacytoplasmic vacuoles and other features of neuronal damage. Neuronal number in the retrosplenial cortex was also counted using stereological methods. Only neurons with a visible nucleolus were counted (under ×40 objective lens). The unbiased counting frame was 185 000 μm × 147 000 μm = 2.72 × 1010 μm2, the dissector height was 1 μm, and the dissector volume was 2.72 × 1010 μm3. The latter value was used to calculate the density of neurons as the quotient of the mean number of neuron counted per dissector and the mean volume of dissectors.
Quantitative Evaluation of Synapse Density and Axon Morphology
At low magnification under the electron microscope, the boundaries of the outer molecular layer of the dentate gyrus were identified according to their characteristic cellular structures (Figure 1a). Then, 8–15 photographs from each electron microscope section were taken systematically at ×800 magnification using alternate grid squares (Figure 1b). Three sections from each animal, including the dorsal, medial and ventral dentate gyrus, were assessed. A total of 1140 photographs were taken for analysis. Synapses were identified under electron micrographs that were enlarged photographically to a final magnification of ×29 000. A magnification standard (grating replica) was used for each series of electron micrographs. Synapses in the molecular layer of dentate gyrus were identified on photographs by the presence of synaptic vesicles and postsynaptic densities (Figure 1c). Asymmetrical (ie excitatory glutamatergic synapses) and symmetrical synapses (ie inhibitory GABAergic synapses) (Watson, 1988; Kennedy, 2000; Lund et al, 2001) were counted separately. Degenerating axons were identified on photographs by the presence of vesicular and lamellar debris within the atrophic axon. A stereological dissector technique was used to measure the density of synapses and degenerating axons (West and Gundersen, 1990; Geinisman et al, 2000; Dong et al, 2007). Each dissector consisted of micrographs of two adjacent ultrathin sections, a reference section and a look-up section immediately above it. Only synapses or axons that occurred in the reference, but not in the look-up section, were counted. The area of the unbiased counting frame was 247 μm2, the dissector height was 0.085 μm, and the dissector volume is 20.99 μm3. The latter value was used to calculate the density of synapses or degenerating axons (synapses or axons per unit volume) as the quotient of the mean number of synapses or axons counted per dissector and the mean volume of dissectors.
Figure 1.

(Panel a) A section through the hippocampus. The rectangle indicates the ultrasection that was trimmed and cut, which included the stratum radiatum of CA1 and molecular layer of dentate gyrus. (Panel b) Semi-section through the hippocampus adjacent to the ultrasection. The section and grid schematically indicates the samplings made under electron microscopy. One photograph from each grid mesh (Red dots indicated) that covered the SR and ML of dentate gyrus was taken systematically. For this study, only ML of dentate gyrus was counted for analysis. (Panel c) EM photographs used to examine the neuronal structure and synapses. The red dots indicate the synapses that were counted. Synapses were identified by the presence of synaptic vesicles and postsynaptic densities (see high magnification inset).
Statistical Analysis
Anatomic variables were compared across groups using a two-way analysis of variance (ANOVA). Freezing behavior was analyzed using repeated measures ANOVA with minute as the repeated measure, and shock sensitivity was analyzed with factorial ANOVA. Statistical significance was accepted for p-values less than 0.05. When significant genotype effects (ie (Tg +) mice vs (Tg−)controls), drug effects, or genotype × drug interactions were found, post-hoc analyses were performed using a Fisher's Protected Least Squares Design tests.
Results
Plaque Density in the Hippocampus and Overlying Cortex
In Tg + mice at 9 months of age, small Aβ plaques (<50 μm in diameter) were observed in the hippocampus and overlying cortex, in keeping with previous findings (Figure 2). No Aβ plaques were observed in Tg− animals, and therefore Tg− mice were not included in further analyses for this variable. Memantine administration was associated with decreases in amyloid deposition in Tg + mice (Figure 2b–d). A significant effect of memantine administration was found on total plaque number (F(3,19) = 8.13, p = 0.001) (Figure 3a) and plaque burden (F(3,16) = 5.58, p = 0.008)) (Figure 3b). Post hoc tests showed that all three doses of memantine significantly reduced plaque number and burden as compared to vehicle (p<0.02). Further, the 5 mg/kg treatment group showed fewer plaques than the 10 mg/kg-treated group (p<0.03), while the difference between 10 and 20 mg/kg-treated groups was not significant (Figure 3a and b).
Figure 2.

β-Amyloid plaque stained by Thioflavine S in the brain of 9–10 months old Tg + mice treated with 5, 10 and 20 mg/kg of memantine and vehicle. Memantine administration for 6 months decreased β-amyloid plaque deposition in the entorhinal cortex (Tg + mice with 5 mg/kg (panel b), 10 mg/kg (panel c), and 20 mg/kg (panel d) as compared to vehicle Tg + mouse (panel a). Arrow heads indicate the plaques. Bar in panel d also applies to a, b and c.
Figure 3.

β-Amyloid plaque number and burden in the brain of Tg + mice treated with 5, 10 and 20 mg/kg of memantine. A significant drug effect was found on the total number of amyloid plaques in the brain (panel a) and the percentage of brain occupied by β-amyloid plaque (panel b).
Neuronal Morphology in the Posterior Cingulate, Retrosplenial Cortices and the Hippocampus
Following long-term administration of memantine at all doses to both Tg + and Tg− mice, the hippocampus and selected regions of the cortex had a generally normal appearance under light microscopy. In particular, we did not observe aberrant vacuolization in the cytoplasm of pyramidal neurons in the hippocampus, or in the posterior cingulate, retrosplenial cortices (Figure 4). However, a number of cells appeared to be darkly stained in these areas. Some of the dark cells showed features of neurodegeneration (Figure 4a, arrow indicated) but whether these cells were undergoing cell death could not be confirmed. The density of dark cells in the posterior cingulate and retrosplenial cortices was not significantly affected by either genotype drug administration (data not shown). However, there was a trend towards a drug effect (F(1,24) = 2.91; p = 0.055), but not a genotype effect (F(1,24) = 0.11, p = 0.74), nor a drug × genotype interaction (F(1,24) = 1,93; p = 0.15), on neuronal density in the retrosplenial cortex.
Figure 4.

Neurons in the retrosplenial cortex and CA1 subregion of the hippocampus of Tg + mice following chronic administration with 20 mg/kg (panel a and b) and 5 mg/kg (panel c and d). The neurons have a normal morphological appearance without cytoplasmic vacuoles in both the retrosplenial cortex (panel a and c) and CA1 of the hippocampus (panel b and d). However, a few dark stained cells in the retrosplenial cortex showed the feature of degeneration (panel a, arrows indicated). Bar in panel d also applies to a, b and c.
Synapse Density
Increased synapse density was observed after memantine administration in the molecular layer of the dentate gyrus in both Tg + and Tg− mice (Figure 5). There was a significant effect of memantine administration on synapse density (F(3,30) = 4.86, p = 0.007), but no overall effect of genotype (F(3,30) = 0.38, p = 0.54), nor a drug × genotype interaction (Figure 6). Post hoc tests indicated that all three doses of memantine significantly increased synapse density as compared to vehicle (p<0.02), with no significant differences between doses. Further, a significant decrease in synapse density was found post hoc in vehicle-treated transgenic animals (Tg +) as compared to vehicle-treated non-transgenic animals (Tg−) (F(1,10) = 9.04, p = 0.013) (Figure 6a).
Figure 5.
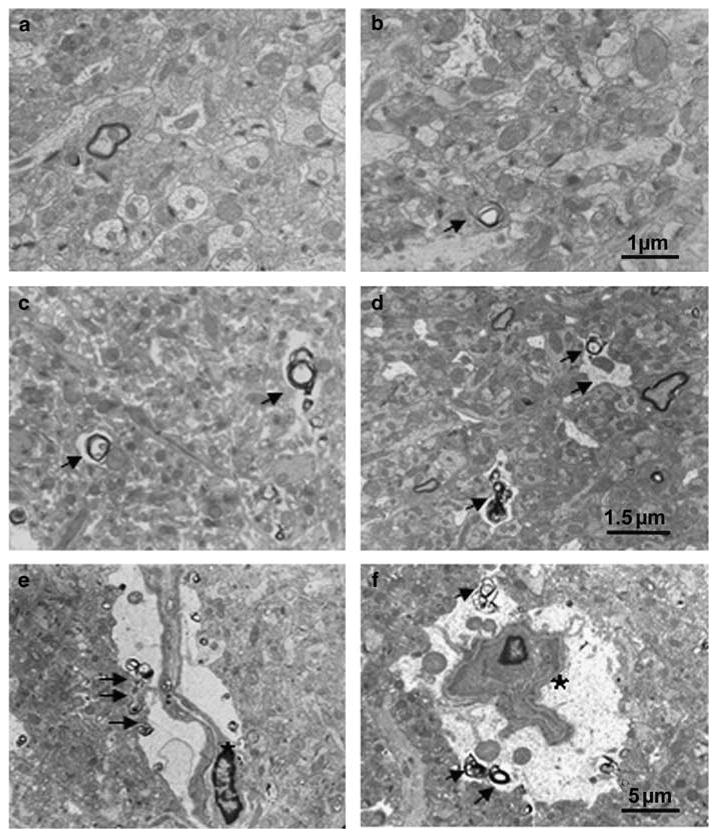
Electron microscope photographs taken in the molecular layer of the dentate gyrus following 6 months of memantine administration. (panel a) Tg + mice treated with vehicle showed decreased synapse density, but no degenerating axonal terminals. (panel b) Tg + mice with 5 mg/kg memantine demonstrated an increased synaptic density as compared to Tg + mice treated with vehicle (a), but no remarkable degenerating axons were observed. (Panel c and d) Tg + mice treated with 10 mg/kg (c) and 20 mg/kg (d) of memantine show increased degenerating axonal terminals (arrows indicated). Degenerating glial cells showing cell shrinkage with irregular space between perikarya and around neuropil (panel e and f, asterisks indicated). Bar in b applies to a; Bar in d applies to c; Bar in f applies to e.
Figure 6.

Synapse and degenerating axonal density in the molecular layer of dentate gyrus following 6 months of memantine administration. (Panel a) Synaptic density was significantly decreased in vehicle-treated Tg + mice as compared to vehicle-treated Tg− mice (p = 0.013). All three doses (5, 10 and 20 mg/kg) of memantine significantly increased synaptic density as compared to vehicle (p's<0.02). (Panel b) 10 and 20 mg/kg of memantine significantly increased the density of degenerating axon terminals as compared to 5 mg/kg and vehicle-treated groups.
The changes in synapse density related to both genotype and memantine treatment were largely confined to asymmetric synapses. In the molecular layer of the dentate gyrus, 92.5% of total synapses in Tg + mice and 90.3% of total synapses in Tg− mice were asymmetrical (ie glutamatergic synapses), and 4.8% of total synapses in Tg + mice and 5.6% of total synapses in Tg− mice were symmetrical (ie GABAergic synapses). The remaining (2.7–4.1%) synapses were labeled as unidentified because it was difficult to classify them as either asymmetrical or symmetrical synapses. In the vehicle-treated group, there was a significant genotype effect (F(1,10) = 10.96; p = 0.008) on density of asymmetrical synapses, but not on the density of symmetrical synapses (F(1,10) = 1.87; p = 0.202). In the memantine-treated groups, there was also a significant drug effect on the density of asymmetrical synapses (F(3,30) = 4.40; p = 0.01), but not the density of symmetrical synapses (F(3,30) = 2.225; p = 0.106).
Degenerating Axons
In animals treated with memantine, but not vehicle, we noted the presence of a marked increase in degenerating axon terminals or degenerated debris. Degenerating axons were identified by a variety of pathologic features including myelin degradation products, disorganization of the axoplasm, and disintegration of the internal part of the myelin, along with various vesicular and lamellar debris within the atrophic axon (Figure 5, arrows indicated). There was a significant effect of memantine administration on the density of degenerating axons (F(3,30) = 10.7, p<0.001) (Figure 6b), but no effect of genotype (F(3,30) = 0.384, p = 0.54), nor a genotype × memantine interaction (F(3,30) = 1.62, p = 0.206). Post hoc tests showed that treatment with the two higher doses of memantine (10 and 20 mg/kg) were associated with significant increases in the density of degenerating axons as compared to vehicle (p's<0.001), and the lowest dose of memantine treatment (5 mg/kg) (p's<0.02) in both Tg + and Tg− mice.
Conditioned Fear
There was no significant effect of genotype (F(1,76) = 1.01, p = 0.32), memantine administration (F(3,76) = 2.43, p = 0.07) nor a genotype × memantine interaction (F(3,76) = 0.48, p = 0.69) on baseline freezing behavior, which suggested no differences between groups in activity level in response to the test chamber. There was a significant genotype × memantine × minute interaction (F(3,76) = 2.85, p = 0.04), and post hoc tests indicated that the Tg− 5 mg/kg memantine group froze significantly more than the Tg− 10 mg/kg memantine and the Tg + 20 mg/kg memantine groups during the second min (p's<0.05). Assessment of freezing behavior during the tone-shock presentation on day 1 revealed a significant effect of genotype (F(1,76) = 26.33, p = 0.00002), but no significant effect of memantine administration (F(3,76) = 0.89, p = 0.45), nor a genotype × memantine interaction (F(3,76) = 0.85, p = 0.47). Post hoc tests indicate that the Tg− animals froze more in response to the tone-shock pairings than the Tg + animals (p<0.01). Assessment of contextual memory on day 2 indicated a significant effect of genotype (F(1,76) = 14.99, p = 0.0002) but no effect of memantine (F(3,76) = 0.02, p = 0.99), nor a genotype × memantine interaction (F(3,76) = 0.33, p = 0.80). Evaluation of freezing behavior in response to the altered context on day 3 revealed a significant effect of genotype (F(1,76) = 8.32, p = 0.005), but no effect of memantine administration (F(3,76) = 0.71, p = 0.55), nor a genotype × memantine interaction (F(3,76) = 0.99, p = 0.40). Post hoc tests indicated that the Tg− mice froze significantly more than Tg + mice (p = 0.01). Analysis of freezing behavior during presentation of the tone on day 3 indicated a significant effect of genotype (F(1,76) = 12.82, p = 0.0006), with post hoc tests showing that Tg + mice froze less than Tg− mice (p = 0.001). The effect of memantine administration (F(3,76) = 1.31, p = 0.28) was not significant, but there was a significant genotype × memantine interaction (F(3,76) = 2.76, p = 0.048). Post hoc tests showed that the Tg + 10 mg/kg memantine group froze significantly less than both the Tg− 5 and 10 mg/kg memantine groups (p's<0.02) (Figure 7).
Figure 7.
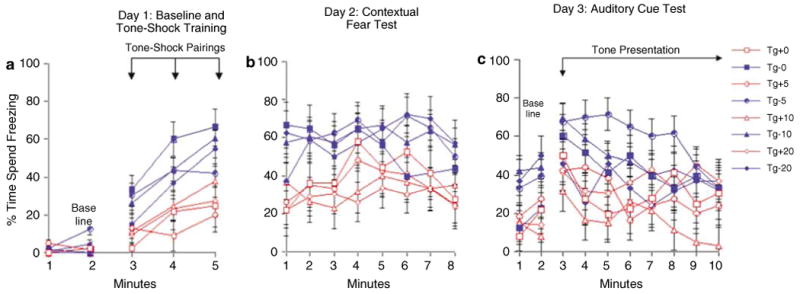
Freezing behavior following 6 months of memantine administration. Tg + mice froze significantly less than Tg− mice during the tone-shock pairings of the training phase (panel a). During the contextual fear test, Tg + animals spent significantly less time freezing than Tg− animals suggesting impaired contextual conditioning (panel b). Tg + mice froze significantly less than Tg− mice during the altered context baseline. During the auditory cue test, Tg + mice again froze significantly less than Tg− mice (panel c).
The observed power for the results of contextual fear conditioning was high for the genotype effect (0.99), but low (0.06) for the drug effect (calculated by SPSS). Although the sample sizes for our study were relative to those used in similar designs (Van Dam and De Deyn, 2006), it is possible that due to the variability in our sample sizes at each dose, our group numbers were too low to detect statistical significance.
Factorial ANOVA across groups revealed no significant differences in shock sensitivity due to genotype (F(1,76) = 0.12, p = 0.73), memantine administration (F(3,76) = 0.27, p = 0.85), nor a genotype × memantine interaction (F(3,76) = 1.37, p = 0.26).
Discussion
In this study, we evaluated the effects of chronic memantine administration on amyloid plaque deposition and neuronal morphology using quantitative light and electron microscopy in Tg2576 mice, a commonly used animal model of AD. In addition, we sought to evaluate the behavioral consequences of any observed changes in structure by assessing contextual memory using a fear-conditioning paradigm. We found that chronic memantine administration decreased Aβ plaque burden in the brain, increased synapse density and increased degenerating axon density in the molecular layer of the dentate gyrus. While we didn't observe neuronal vacuolization following memantine administration, the appearance of degenerating axons after memantine administration was dose-dependent and consistent with some of the neurotoxic changes previously observed following administration of NMDA antagonists. However, despite these changes in amyloid deposition and neuronal structure, we did not find an effect of memantine on hippocampal-dependent contextual memory in Tg + mice.
Our results are consistent with previous studies which found decreases in cortical APP levels in Tg2576 mice after 10 days of memantine treatment (Unger et al, 2006). However, the mechanism by which memantine regulates amyloid metabolism is not clear. It has been shown that increased neuronal activity leads to increased APP production, a likely precursor to Aβ plaque formation (Cirrito et al, 2005). Thus, blockade of NMDA receptors by memantine could reduce neuronal activity and subsequently lower APP production. In support of this hypothesis, we noticed that the lower dose of memantine (5 mg/kg) exerted stronger effects in reducing Aβ plaque number and burden than the two higher doses (10 mg, and 20 mg/kg) (Figure 3). Thus, it is possible that lower memantine doses may block just enough neuronal activity to lower amyloid production, while higher doses block so much neuronal activity that APP degradation and clearance mechanisms are also decreased. Our experimental design was such that the animals began treatment before the appearance of plaques would be expected to occur in the brain. Thus, our results cannot address the question whether memantine has the capacity to prevent the formation of new plaques or to reduce the number of Aβ plaques already formed.
In our study, we found that memantine administration increased synaptic density in the molecular layer of the dentate gyrus in both Tg + and Tg− mice; in fact, memantine-treated Tg + mice tended to have a greater quantity of synapses than vehicle-treated Tg− mice. The molecular layer of the dentate gyrus is one of the first regions to show amyloid deposition and synapse loss in the Tg2576 mouse model of AD (Su and Ni, 1998; Reilly et al, 2003; Dong et al, 2007). However, it is difficult to know the implications of increases in synapse density after memantine administration in Tg2576 mice. While this could represent a neuroprotective effect, it could also be compensatory to memantine-induced axonal degeneration. Since too many as well as too few synapses could adversely impact neuronal function, caution should be used in interpreting the observed changes in synapse density after memantine treatment.
We observed no neuronal vacuolization or other histopathological changes in the hippocampus, posterior cingulate and retrosplenial cortices at the light microscopy level after memantine administration as previously reported after the administration of NMDA antagonists (Fix et al, 1993, 1994; Creeley et al, 2008). However, our observation that chronic memantine administration was associated with axonal degeneration in the hippocampus may relate to other reported neurotoxic effects of NMDA antagonists (eg PCP, MK-801) (Low and Roland, 2004). The use of NMDA antagonists as therapeutic agents for neuropsychiatric disorders thought to include a component of neurodegeneration has been investigated for many years. However, their use for this purpose has been hampered by observations of adverse behavioral effects such as hallucinations and memory disturbances in humans, and reports of neuronal vacuolization, neuronal and axonal degeneration, and induction of heat-shock protein in rodents (Olney et al, 1989, 1991; Sharp et al, 1992). Memantine, at doses somewhat higher than the doses used in this study (50 mg/kg), has been shown to produce neurodegenerative effects in the rat (Creeley et al, 2006). While the axonal degeneration induced by high doses of memantine may occur because of direct effects on neurons, indirect effects on oligodendrocytes may also be possible. Notably, NMDA receptors are ubiquitous within the CNS and are expressed not only on neurons, but also on glial cells (for review see Verkhratsky and Kirchhoff, 2007). Oligodendrocytes are important for the survival as well as function of neurons (Lappe-Siefke et al, 2003; Popko, 2003), and thus, blockade of NMDA receptors on oligodendrocytes could eventually result in axonal degeneration. Our data lend preliminary support for this hypothesis in that we observed occasional degenerated oligodendrocyte somas in memantine-treated animals (Figure 5e and f).
We were surprised to find no effect of memantine on contextual memory in a fear conditioning paradigm given that we used doses similar to those previously been reported to have beneficial behavioral effects in other transgenic mouse models of AD (Minkeviciene et al, 2004; Van Dam et al, 2005; Van Dam and De Deyn, 2006). The behavior deficits we observed in Tg + mice replicated earlier findings using Tg2576 mice (Hsiao et al, 1996; Corcoran et al, 2002; Arendash et al, 2004; Barnes and Good, 2005; Dong et al, 2005) and thus imply validity of the behavioral test employed here. At present, the effects of memantine on behavior in transgenic mouse models of AD (Minkeviciene et al, 2004; Van Dam et al, 2005; Van Dam and De Deyn, 2006), as well as other models (Barnes et al, 1996; Miguel-Hidalgo et al, 2002; Lang et al, 2004; Woodruff-Pak et al, 2007; for review see Yuede et al, 2007) are inconsistent. Minkeviciene et al (2004) reported improved acquisition in water maze using APP/PS1 transgenic mice treated with 30 mg/kg memantine in drinking water for 3 weeks, whereas no effect was observed on retention. Conversely, Van Dam et al (2005) reported beneficial effects of a much lower dose (2.0 mg/kg) of memantine while higher doses (10 mg/kg) demonstrated detrimental effects in the water maze. As compared with these studies, we found that a 5 mg/kg dose was associated with better performance than a 10 mg/kg dose, even though we did not detect a significant overall effect of memantine in Tg2576 mice. Explanations for these inconsistencies include the selection of the particular mouse model, the drug doses, or the duration and route of drug dosing.
The inconsistency between our morphological and behavioral results highlights the difficulty of defining the structural basis of behavioral deficits in AD mouse models. Previous studies showed little or no correlation between the quantity of Aβ deposition and behavioral deficits (Terry et al, 1991; Arriagada et al, 1992; Berg et al, 1993; Westerman et al, 2002; Van Dam et al, 2003) in both humans with AD and animal models of AD. Increasing evidence has indicated that soluble amyloid oligomers may be directly toxic to neuronal structure and function (Gong et al, 2003; Watson et al, 2005; Lacor et al, 2007; Shankar et al, 2007). Another explanation of the inconsistency between structure and behavior observed in this study is that the animals may have developed adaptive or compensatory responses to memantine over the extended administration period. In such a scenario, the benefits of memantine on plaque burden and synapse density might have been insufficient to produce persistent behavioral changes. Finally, it is also possible that the conditioned fear paradigm used in this study was not sensitive enough, or our sample sizes were too low to detect small behavioral changes with memantine administration.
If memantine has the potential to produce both beneficial and detrimental effects in the mammalian brain, the task is to find a dose of memantine with the former but not the latter properties. We selected three doses (5, 10, and 20 mg/kg) for this study in an attempt to approach the therapeutic doses typically used in humans (Danysz and Parsons, 2003, Wenk et al, 2006). Previous studies in mice have reported that a 30 mg/kg oral dose of memantine in drinking water produces a steady-state plasma drug level of around 1 μM, which is thought to be therapeutic based on clinical studies (Kornhuber and Quack, 1995; Minkeviciene et al, 2004). However, it is difficult to select drug doses in animal studies that are equivalent to clinical doses because the composition and responses observed in the brains of rodents and humans differ (Wenk et al, 2006). Thus, our results should not be used to predict a dose of memantine in human subjects that is both safe and effective. Rather, our results suggest that in vivo neuroimaging should be combined with clinical and cognitive assessments in an effort to determine the effects of such drugs on brain structure and function as well as symptomatology. More effective treatments are greatly needed for neurodegenerative disorders, such as AD, and the NMDA receptor remains a viable pharmacological target in these efforts. However, drugs that act on the NMDA receptor have the potential to damage neurons as well as to protect them, and investigations of such drugs should be performed with appropriate attention to this possibility.
Acknowledgments
This work was supported by MH060883 (JGC), AG 025824 (JGC), and 5P50AG0561 (HD).
Footnotes
Disclosure: Dr Csernansky has received research grants from the NIMH, NIA, and royalties from Medtronic for a patent held jointly with Washington University School of Medicine, has been a paid consultant for Eli Lilly and Sanofi-Aventis, and has received speakers' honoraria from Janssen Pharmaceutica, Eli Lilly and Bristol-Myers Squibb. The other authors report no conflicts of interest.
References
- Aracava Y, Pereira EF, Maelicke A. Memantine blocks alpha7 nicotinic acetylcholine receptors more potently than N-methyl-d-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312:1195–1205. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, et al. Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer's disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 2004;1012:29–41. doi: 10.1016/j.brainres.2004.02.081. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Yee BK, Lemaire M, Wilbrecht L, Jarrard L, Iversen SD, et al. The role of the entorhinal cortex in two forms of spatial learning and memory. Exp Brain Res. 2001;141:281–303. doi: 10.1007/s002210100868. [DOI] [PubMed] [Google Scholar]
- Bardgett ME, Boeckman R, Krochmal D, Fernando H, Ahrens R, Csernansky JG. NMDA receptor blockade and hippocampal neuronal loss impair fear conditioning and position habit reversal in C57Bl/6 mice. Brain Res Bull. 2003;15:131–142. doi: 10.1016/s0361-9230(03)00023-6. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Danysz W, Parsons CG. Effects of the uncompetitive NMDA receptor antagonist memantine on hippocampal long-term potentiation, short-term exploratory modulation, and spatial memory in awake, freely moving rats. Er J Neurosci. 1996;8:565–571. doi: 10.1111/j.1460-9568.1996.tb01241.x. [DOI] [PubMed] [Google Scholar]
- Barnes P, Good M. Impaired Pavlovian cued fear conditioning in Tg2576 mice expressing a human mutant amyloid precursor protein gene. Behav Brain Res. 2005;157:107–117. doi: 10.1016/j.bbr.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Berg L, McKeel DW, Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer's disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–358. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- Buisson B, Bertrand D. Open-channel blockers at the human α4β2 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998;53:555–563. doi: 10.1124/mol.53.3.555. [DOI] [PubMed] [Google Scholar]
- Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, et al. Open-channel block of N-methyl-d-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;11:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Lu Y, Turner RS, Maren S. Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer's disease. Learn Mem. 2002;9:243–252. doi: 10.1101/lm.51002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley C, Wozniak DF, Labruyere J, Taylor GT, Olney JW. Low doses of memantine disrupt memory in adult rats. J Neurosci. 2006;26:3923–3932. doi: 10.1523/JNEUROSCI.4883-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley C, Wozniak DF, Nardi A, Farber NB, Olney JW. Donepezil markedly potentiates memantine neurotoxicity in the adult rat brain. Neurobiol Aging. 2008;29:153–167. doi: 10.1016/j.neurobiolaging.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer's disease: preclinical evidence. Int J Geriatr Psychiatry. 2003;18:S23–S32. doi: 10.1002/gps.938. [DOI] [PubMed] [Google Scholar]
- Ditzler K. Efficacy and tolerability of memantine in patients with dementia syndrome. A double-blind, placebo controlled trial. Arzneimittelforschung. 1991;41:773–780. [PubMed] [Google Scholar]
- Dong H, Csernansky CA, Martin MV, Bertchume A, Vallera D, Csernansky JG. Acetylcholinesterase inhibitors ameliorate behavioral deficits in the Tg2576 mouse model of Alzheimer's disease. Psychopharmacology (Berl) 2005;181:145–152. doi: 10.1007/s00213-005-2230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG. Effects of isolation stress on hippocampal neurogenesis, memory, and amyloid plaque deposition in APP (Tg2576) mutant mice. Neuroscience. 2004;127:601–609. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Dong H, Martin MV, Chambers S, Csernansky JG. Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J Comp Neurol. 2007;500:11–321. doi: 10.1002/cne.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdö SL, Schäfer M. Memantine is highly potent in protecting cortical cultures against excitotoxic cell death evoked by glutamate and N-methyl-d-aspartate. Eur J Pharmacol. 1991;198:215–217. doi: 10.1016/0014-2999(91)90625-z. [DOI] [PubMed] [Google Scholar]
- Fix AS, Horn JW, Truex LL, Smith R, Gomez E. Neuronal vacuole formation in the rat posterior cingulate/retrosplenial cortex after treatment with the N-methyl-d-aspartate (NMDA) antagonist MK-801 (dizocilpine maleate) Acta Neuropathol. 1994;88:519–551. doi: 10.1007/BF00296487. [DOI] [PubMed] [Google Scholar]
- Fix AS, Horn JW, Wightman KA, Johnson CA, Long GG, Storts RW, et al. Neuronal vacuolization and necrosis induced by the noncompetitive N-methyl-d-aspartate (NMDA) antagonist MK(+)801 (dizocilpine maleate): a light and electronmicroscopic evaluation of the rat retrosplenial cortex. Exp Neurol. 1993;123:204–215. doi: 10.1006/exnr.1993.1153. [DOI] [PubMed] [Google Scholar]
- Frankiewicz T, Pilc A, Parsons CG. Differential effects of NMDA-receptor antagonists on long-term potentiation and hypoxic/hypoglycaemic excitotoxicity in hippocampal slices. Neuropharmacology. 2000;39:631–642. doi: 10.1016/s0028-3908(99)00168-9. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Disterhoft JF, Gundersen HJ, McEchron MD, Persina IS, Power JM, et al. Remodeling of hippocampal synapses after hippocampus-dependent associative learning. J Comp Neurol. 2000;417:49–59. [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntern R, Bouras C, Hof PR, Vallet PG. An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer's disease. Experientia. 1992;48:8–10. doi: 10.1007/BF01923594. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, A elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- Knowles WD. Normal anatomy and neurophysiology of the hippocampal formation. J Clin Neurophysiol. 1992;9:252–263. [PubMed] [Google Scholar]
- Kornhuber J, Bormann J, Retz W, Hübers M, Riederer P. Memantine displaces (3H)MK-801 at therapeutic concentrations in postmortem human frontal cortex. Eur J Pharmacol. 1989;166:589–590. doi: 10.1016/0014-2999(89)90384-1. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Quack G. Cerebrospinal fluid and serum concentrations of the N-methyl-d-aspartate (NMDA) receptor antagonist memantine in man. Neurosci Lett. 1995;195:137–139. doi: 10.1016/0304-3940(95)11785-u. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Mood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang UE, Muhlbacher M, Hesselink MB, Zajaczkowski W, Danysz W, Danker-Hopfe H, et al. No nerve growth factor response to treatment with memantine in adult rats. J Neural Trans. 2004;111:181–190. doi: 10.1007/s00702-003-0090-y. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Li Q, Clark S, Lewis DV, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci. 2002;22:3070–3080. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Reviews. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr Drug Targets. 2007;8:621–632. doi: 10.2174/138945007780618472. [DOI] [PubMed] [Google Scholar]
- Low SJ, Roland CL. Review of NMDA antagonist-induced neurotoxicity and implications for clinical development. Int J Clin Pharmacol Ther. 2004;42:1–14. doi: 10.5414/cpp42001. [DOI] [PubMed] [Google Scholar]
- Lund JS, Griffiths S, Rumberger A, Levitt JB. Inhibitory synapse cover on the somata of excitatory neurons in macaque monkey visual cortex. Cereb Cortex. 2001;11:783–795. doi: 10.1093/cercor/11.9.783. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuroprotection by memantine against neurodegeneration induced by beta-amyloid (1–40) Brain Res. 2002;958:210–221. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Banerjee P, Tanila H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2004;311:677–682. doi: 10.1124/jpet.104.071027. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA. NMDA antagonist neurotoxicity: mechanism and prevention. Science. 1991;254:1515–1518. doi: 10.1126/science.1835799. [DOI] [PubMed] [Google Scholar]
- Popko B. Myelin: not just a conduit for conduction. Nat Genet. 2003;33:327–328. doi: 10.1038/ng0303-327. [DOI] [PubMed] [Google Scholar]
- Probst A, Basler V, Bron B, Ulrich J. Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res. 1983;268:249–254. doi: 10.1016/0006-8993(83)90490-0. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Games D, Rydel RE, Freedman S, Schenk D, Young WG, et al. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci USA. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Butman M, Wang S, Koistinaho J, Graham SH, Sagar SM, et al. Haloperidol prevents induction of the hsp70 heat shock gene in neurons injured by phencyclidine (PCP), MK801, and ketamine. J Neurosci Res. 1992;33:605–616. doi: 10.1002/jnr.490330413. [DOI] [PubMed] [Google Scholar]
- Stern EA, Bacskai BJ, Hickey GA, Attenello FJ, Lombardo JA, Hyman BT. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J Neurosci. 2004;24:4535–4540. doi: 10.1523/JNEUROSCI.0462-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Ni B. Selective deposition of amyloid-protein in the entorhinal-dentate projection of a transgenic mouse model of Alzheimer's disease. J Neurosci Res. 1998;53:177–186. doi: 10.1002/(SICI)1097-4547(19980715)53:2<177::AID-JNR6>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Unger C, Svedberg MM, Yu WF, Hedberg MM, Nordberg A. Effect of subchronic treatment of memantine, galantamine, and nicotine in the brain of Tg2576 (APPswe) transgenic mice. J Pharmacol Exp Ther. 2006;317:30–36. doi: 10.1124/jpet.105.098566. [DOI] [PubMed] [Google Scholar]
- Van Dam D, Abramowski D, Staufenbiel M, De Deyn PP. Symptomatic effect of donepezil, rivastigmine, galantamine and memantine on cognitive deficits in the APP23 model. Psychopharmacology. 2005;180:177–190. doi: 10.1007/s00213-004-2132-z. [DOI] [PubMed] [Google Scholar]
- Van Dam D, De Deyn PP. Cognitive evaluation of disease-modifying efficacy of galantamine and memantine in the APP23 model. Eur Neuropsychopharmacol. 2006;16:59–69. doi: 10.1016/j.euroneuro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–396. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kirchhoff F. NMDA receptors in glia. Neuroscience. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- Watson AH. Antibodies against GABA and glutamate label neurons with morphologically distinct synaptic vesicles in the locust central nervous system. Neuroscience. 1988;26:3–44. doi: 10.1016/0306-4522(88)90125-x. [DOI] [PubMed] [Google Scholar]
- Watson D, Castaño E, Kokjohn TA, Kuo YM, Lyubchenko Y, Pinsky D, et al. Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer's disease. Neurol Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- Weller M, Finiels-Marlier F, Paul SM. NMDA receptor-mediated glutamate toxicity of cultured cerebellar, cortical and mesencephalic neurons: neuroprotective properties of amantadine and memantine. Brain Res. 1993;613:143–148. doi: 10.1016/0006-8993(93)90464-x. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Danysz W, Mobley SL. MK-801, memantine and amantadine show neuroprotective activity in the nucleus basalis magnocellularis. Eur J Phramacol. 1995;293:267–270. doi: 10.1016/0926-6917(95)00028-3. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Parsons CG, Danysz W. Potential role of N-methyl-d-aspartate receptors as executors of neurodegeneration resulting from diverse insults: focus on memantine. Behav Pharmacol. 2006;17:411–424. doi: 10.1097/00008877-200609000-00007. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Zajaczkowski W, Danysz W. Neuroprotection of acetylcholinergic basal forebrain neurons by memantine and neurokinin B. Behav Brain Res. 1997;83:129–133. doi: 10.1016/s0166-4328(97)86056-1. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer's disease. Neurobiol Aging. 2004;25:1205–1212. doi: 10.1016/j.neurobiolaging.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff-Pak DS, Tobia MJ, Jiao X, Beck KD, Servatius RJ. Preclinical investigation of the functional effects of memantine and memantine combined with galantamine or donepezil. Nueropsychopharmacology. 2007;32:1284–1294. doi: 10.1038/sj.npp.1301259. [DOI] [PubMed] [Google Scholar]
- Yuede CM, Dong H, Csernansky JG. Anti-dementia drugs and hippocampal-dependent memory in rodents. Behav Pharmacol. 2007;18:347–363. doi: 10.1097/FBP.0b013e3282da278d. [DOI] [PMC free article] [PubMed] [Google Scholar]