

The purification, crystallization and preliminary X-ray diffraction studies of vitamin D3 hydroxylase isolated from P. autotrophica are reported.
Keywords: cholecalciferol, CYP107, cytochrome P450 monooxygenase, hydroxylation, Pseudonocardia autotrophica, vitamin D3
Abstract
Vitamin D3 hydroxylase (Vdh) is a novel cytochrome P450 monooxygenase isolated from the actinomycete Pseudonocardia autotrophica and consisting of 403 amino-acid residues. Vdh catalyzes the activation of vitamin D3 via sequential hydroxylation reactions: these reactions involve the conversion of vitamin D3 (cholecalciferol or VD3) to 25-hydroxyvitamin D3 [25(OH)VD3] and the subsequent conversion of 25(OH)VD3 to 1α,25-dihydroxyvitamin D3 [calciferol or 1α,25(OH)2VD3]. Overexpression of recombinant Vdh was carried out using a Rhodococcus erythropolis expression system and the protein was subsequently purified and crystallized. Two different crystal forms were obtained by the hanging-drop vapour-diffusion method at 293 K using polyethylene glycol as a precipitant. The form I crystal belonged to the trigonal space group P31, with unit-cell parameters a = b = 61.7, c = 98.8 Å. There is one Vdh molecule in the asymmetric unit, with a solvent content of 47.6%. The form II crystal was grown in the presence of 25(OH)VD3 and belonged to the orthorhombic system P212121, with unit-cell parameters a = 63.4, b = 65.6 c = 102.2 Å. There is one Vdh molecule in the asymmetric unit, with a solvent content of 46.7%. Native data sets were collected to resolutions of 1.75 and 3.05 Å for form I and form II crystals, respectively, using synchrotron radiation. The structure solution was obtained by the molecular-replacement method and model refinement is in progress for the form I crystal.
1. Introduction
Vitamin D3 (VD3) is a steroid hormone that plays a crucial role in bone metabolism, control of cell proliferation and cell differentiation in mammals (Jones et al., 1998 ▶). VD3 is physiologically activated by sequential hydroxylation reactions that occur first at the C25 and then at the C1α positions; these reactions are catalyzed by cytochrome P450 monooxygenases (CYPs). In humans, three CYPs (CYP27A1, CYP27B1 and CYP2R1) are involved in the activation of VD3 in the liver and kidney (Prosser & Jones, 2004 ▶). 1α,25-Dihydroxy VD3 [1α,25(OH)2VD3] and its derivatives are currently used as pharmaceuticals for the treatment of osteoporosis, psoriasis, rickets and hypoparathyroidism. They can be chemically synthesized from cholesterol via approximately 20 reaction steps, but the maximum yield is only 1% (Zhu & Okamura, 1995 ▶). In contrast to the synthetic chemistry approach, a few bacterial CYPs are known to efficiently convert VD3 into 1α,25(OH)2VD3. P450SU-1 (CYP105A1) obtained from Streptomyces griseolus is one such bacterial enzyme that catalyzes the hydroxylation of VD3 (Sawada et al., 2004 ▶) and crystal structures of P450SU-1 in the substrate-free and transient product-bound forms have been reported (Sugimoto et al., 2008 ▶; Hayashi et al., 2008 ▶). The actinomycete Pseudonocardia autotrophica can also catalyze the sequential hydroxylation of VD3 (Sasaki et al., 1992 ▶) and is currently used for the production of 1α,25(OH)2VD3. Recently, we successfully identified and cloned the P. autotrophica gene that encodes the enzyme that hydroxylates VD3 to 1α,25(OH)2VD3 (Fujii et al., manuscript in preparation). This enzyme, VD3 hydroxylase (Vdh), is a novel cytochrome P450 consisting of 403 amino-acid residues and a sequence-similarity search indicates that Vdh is classified into the CYP107 family. The structures of three CYPs from the CYP107 family [P450eryF (CYP107A1), P450 PikC (CYP107L1) and P450BioI (CYP107H1)] have been reported and show that the residues that create the substrate-binding pocket are highly diversified in accordance with the size and the chemical properties of individual substrates (Cupp-Vickery & Podust, 1995 ▶; Cupp-Vickery et al., 2001 ▶; Nagano et al., 2005 ▶; Sherman et al., 2006 ▶; Cryle & Schlichting, 2008 ▶). The substrates of these CYPs are not analogous to VD3. Enzymatic characterization of recombinant Vdh coupled with spinach ferredoxin and ferredoxin reductase revealed that the kinetic parameters of Vdh are comparable to those of mammalian VD3-hydroxylating CYPs (Fujii et al., manuscript in preparation; Sawada et al., 2004 ▶; Uchida et al., 2004 ▶; Strushkevich et al., 2008 ▶). Crystallographic studies are required in order to elucidate the structural mechanisms that underlie the regiospecific sequential hydroxylation reactions of Vdh. Furthermore, the structural information is key to creating highly active Vdh mutants that are more efficient at bioconversion. Here, we report the purification, crystallization and preliminary X-ray diffraction studies of recombinant Vdh produced in Rhodococcus erythropolis.
2. Materials and methods
2.1. Overproduction and purification of recombinant Vdh
The identification and cloning of the Vdh gene will be published elsewhere (Fujii et al., manuscript in preparation). Briefly, the gene encoding Vdh was cloned from genomic DNA of P. autotrophica and inserted into pET29a expression vector using NdeI and XhoI restriction enzymes. Recombinant Vdh was obtained by expression in Escherichia coli BL21(DE3) and was used for enzymatic studies. For crystallization, recombinant Vdh was produced using R. erythropolis as a host cell (Mitani et al., 2005 ▶). The gene encoding Vdh was amplified using the polymerase chain reaction (PCR) with a pET29a-Vdh plasmid as the template DNA. PCR fragments were digested with NdeI and XhoI and cloned into the corresponding site of the pTip-QC2 expression vector for Rhodococcus (Nakashima & Tamura, 2004a ▶,b ▶). The resulting plasmid encoded full-length Vdh with a Leu-Glu-(His)6 sequence at the C-terminus, comprising a total of 411 amino-acid residues (45.4 kDa). The plasmid was transformed into R. erythropolis L88 cells by electroporation. The transformed cells were grown at 303 K in 400 ml Luria–Bertani (LB) medium containing 20 µg ml−1 tetracycline. The preculture was then inoculated in 4 l LB medium supplemented with 1 µg ml−1 thiostrepton for 24 h at 303 K in order to induce Vdh expression. The cells were harvested and resuspended in a buffer containing 50 mM Tris–HCl pH 7.5, 10% glycerol and 2 mM dithiothreitol (DTT). The cells were lysed by sonication and the homogenate was clarified by centrifugation. The supernatant was dialyzed against buffer A (50 mM Tris–HCl pH 7.5 and 10% glycerol) and then applied onto an Ni–NTA column (Qiagen) pre-equilibrated with buffer A. The column was washed with buffer A and the enzyme was eluted with a linear gradient of 0–400 mM imidazole in buffer A. The fractions containing Vdh were collected, dialyzed against buffer B (20 mM Tris–HCl pH 7.5, 10% glycerol, 1 mM DTT and 0.5 mM EDTA) and subsequently applied onto a DEAE-Sephacel anion-exchange column (GE Healthcare) pre-equilibrated with buffer B. The enzyme was eluted with a linear gradient of 0–400 mM NaCl in buffer B. The individual fractions were analyzed by SDS–PAGE. The peak fractions were pooled and concentrated to 25 mg ml−1 using a centrifugal filtration device (Millipore). A CO difference spectral assay was performed to verify that the purified Vdh was in an active form (Omura & Sato, 1964 ▶). The sample containing 10% glycerol was stored at 193 K until it was used for crystallization.
2.2. Crystallization
The sample was thawed on ice, diluted with 20 mM Tris–HCl pH 7.5 and concentrated several times in a centrifugal filtration device with a 30 kDa cutoff membrane (Millipore) to remove the glycerol. Finally, the protein concentration was adjusted to 20 mg ml−1. For crystallization in the presence of a substrate, 1 µl 100 mM 25(OH)VD3 solution dissolved in dimethyl sulfoxide (DMSO) was added to 100 µl sample and the mixture was incubated overnight at 277 K before crystallization setup. All crystallization droplets were set up by hand. The initial crystallization screening was carried out in a 96-well crystallization plate (Corning) using the sitting-drop vapour-diffusion technique at 293 K with the commercially available screening kits Crystal Screens I and II and Index Screen (Hampton Research) and Wizard I and II (Emerald Biostructures). Each sitting drop was prepared by mixing 0.8 µl sample solution and 0.8 µl reservoir solution and was equilibrated against 100 µl reservoir solution. A cluster of needle-shaped crystals grew within a week in Index Screen condition No. 53 (Hampton Research), consisting of 100 mM bis-tris pH 6.5, 50 mM CaCl2 and 30% polyethylene glycol 550 monomethylether (PEG 550 MME). This hit condition was subsequently optimized in a 24-well VDX plate (Hampton Research) by using the hanging-drop vapour-diffusion method at 293 K with variation of the buffer pH, PEG and PEG concentration and the use of various additives. Each hanging drop was prepared by mixing 2.0 µl sample solution and an equal volume of the reservoir solution and was equilibrated against 500 µl reservoir solution. Diffraction-quality crystals were obtained in two different forms (forms I and II). The form I crystals grew using reservoir solution containing 100 mM bis-tris pH 7.5, 50 mM CaCl2, 40–120 mM NaCl or KCl and 32–40% PEG 400 or PEG 550 MME. The form II crystals grew in the presence of 25(OH)VD3 using reservoir solution containing 100 mM bis-tris pH 7.5, 50 mM CaCl2 and 20% PEG 1000.
2.3. X-ray diffraction studies
Prior to the X-ray diffraction experiment, the Vdh crystals were flash-cooled at 100 K in a stream of nitrogen gas. The form II crystal was soaked in mother liquor supplemented with 15% glycerol before flash-cooling. X-ray diffraction data for the form I crystal were collected on beamline BL-5A at the Photon Factory (PF) equipped with a CCD (ADSC Quantum Q315) detector. The beam size was set to 0.1 mm (vertical) × 0.2 mm (horizontal). Each oscillation frame was taken with a rotation of 1.0° for a 7.5 s exposure; the total rotation range for data collection was 180°. X-ray diffraction data for the form II crystal were collected on PF beamline BL-17A equipped with a CCD (ADSC Quantum 4R) detector. The beam size was set to 0.1 × 0.1 mm. Each oscillation frame was taken with a rotation of 1.0° for a 10 s exposure; the total rotation range for data collection was 180°. All diffraction data were indexed, integrated and scaled using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
We observed high expression of the soluble haem-bound form of Vdh fused with a hexahistidine tag at the C-terminus in R. erythropolis. Vdh was purified by a two-step purification protocol, resulting in a single band on SDS–PAGE. This procedure yielded approximately 8 mg of purified Vdh per litre of culture. Analysis of UV–visible absorption spectra revealed that the purified Vdh was present in the ferric resting state (data not shown).
The initial Vdh crystals obtained from condition No. 53 of Index Screen (Hampton Research) were needle-shaped and formed clusters and were not suitable for X-ray diffraction studies (Fig. 1 ▶ a). Optimization of the initial condition by altering the pH from 6.5 to 7.5 and by the addition of ∼120 mM NaCl or KCl dramatically improved the crystal morphology. Approximately 120 optimization droplets were set up before diffraction-quality crystals were obtained. A hexagonal rod-shaped crystal (form I) appeared within a few days and grew to maximum dimensions of 100 × 100 × 1500 µm (Fig. 1 ▶ b). The X-ray diffraction quality varied according to the position at which the crystal was exposed to the X-rays. We thus looked for a position along the long side of the crystal that gave the best diffraction spots (Fig. 2 ▶). The form I crystal diffracted to a resolution of 1.75 Å and belonged to the trigonal space group P31, with unit-cell parameters a = b = 61.7, c = 98.8 Å. Assuming the presence of one Vdh molecule in the asymmetric unit, the value of the crystal volume per protein mass (V M value; Matthews, 1968 ▶) was calculated as 2.4 Å3 Da−1. This value corresponds to a solvent content of 47.6%. A needle-shaped form II crystal with typical dimensions of 30 × 30 × 1500 µm was also obtained in the presence of 25(OH)VD3 (Fig. 1 ▶ c). The form II crystal diffracted to a resolution of 3.05 Å and belonged to the orthorhombic space group P212121, with unit-cell parameters a = 63.5, b = 65.6, c = 102.2 Å. Assuming the presence of one Vdh molecule in the asymmetric unit, the V M value (Matthews, 1968 ▶) was calculated as 2.3 Å3 Da−1. This value corresponds to a solvent content of 46.7%. The crystallographic parameters and data-collection statistics are summarized in Table 1 ▶. Sequence-similarity searches revealed that Vdh is a member of the CYP107 family, with a sequence identity of around 40%. Molecular replacement was carried out by the program AMoRe (Navaza, 1993 ▶) using modified P450eryF (CYP107A1; PDB code 1jin), which has 41% amino-acid sequence identity to Vdh, as a search model. The rotation search gave a distinct peak with a correlation coefficient of 0.23 and an R factor of 54.6%. A subsequent translation search also gave a unique peak with a correlation coefficient of 0.31 and an R factor of 52.0% and there were no packing problems. The electron-density map calculated after rigid-body refinement was interpretable, indicating that the molecular-replacement solution was correct. Model building and refinement of the form I crystal are currently under way.
Figure 1.

Crystals of Vdh. (a) The initial crystals were obtained in 100 mM bis-tris pH 6.5, 30%(w/v) PEG 550 MME and 50 mM CaCl2. (b) After optimization of the crystallization conditions, a large hexagonal rod-shaped crystal was obtained in 100 mM bis-tris pH 7.5, 38%(w/v) PEG 400, 50 mM CaCl2 and 60 mM NaCl. (c) Needle-shaped crystals were also obtained in the presence of 25(OH)VD3 in 100 mM bis-tris pH 7.5, 26%(w/v) PEG 1000 and 50 mM CaCl2.
Figure 2.

Examples of the X-ray diffraction images that were obtained by exposing the form I crystal at different positions. Good (left) and poor (right) diffraction images were obtained when the edge and midpoint of the same crystal were exposed to X-rays, respectively.
Table 1. Crystallographic parameters and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Form I | Form II | |
|---|---|---|
| Beamline | PF BL-5A | PF BL-17A |
| Temperature (K) | 100 | 100 |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Resolution (Å) | 50–1.75 (1.81–1.75) | 50–3.05 (3.16–3.05) |
| Unit-cell parameters | ||
| a (Å) | 61.7 | 63.4 |
| b (Å) | 61.7 | 65.6 |
| c (Å) | 98.8 | 102.2 |
| Space group | P31 | P212121 |
| No. of reflections measured | 223054 | 30271 |
| No. of unique reflections measured | 42012 (3957) | 8366 (822) |
| Rmerge† | 0.075 (0.316) | 0.090 (0.317) |
| Data completeness (%) | 99.3 (93.2) | 97.6 (97.9) |
| Data redundancy | 5.3 (3.2) | 3.6 (3.8) |
| Average I/σ(I) | 21.9 (3.7) | 18.9 (6.0) |
| Wilson B (Å2) | 23.2 | 45.0 |
R
merge = 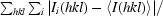
 , where 〈I(hkl)〉 is the mean intensity of a set of equivalent reflections.
, where 〈I(hkl)〉 is the mean intensity of a set of equivalent reflections.
Acknowledgments
The authors would like to thank all the beamline scientists at the Photon Factory (Tsukuba, Japan). This work was supported by the Development of Basic Technologies for Advanced Production Methods Using Microorganism Functions project of the New Energy and Industrial Technology Development Organization (NEDO).
References
- Cupp-Vickery, J. R., Garcia, C., Hofacre, A. & MacGee-Estrada, K. (2001). J. Mol. Biol.311, 101–110. [DOI] [PubMed]
- Cupp-Vickery, J. R. & Podust, T. L. (1995). Nature Struct. Biol.2, 144–153. [DOI] [PubMed]
- Cryle, M. J. & Schlichting, I. (2008). Proc. Natl Acad. Sci. USA, 105, 15696–15701. [DOI] [PMC free article] [PubMed]
- Hayashi, K., Sugimoto, H., Shinkyo, R., Yamada, M., Ikeda, S., Ikushiro, S., Kamakura, M., Shiro, Y. & Sakaki, T. (2008). Biochemistry, 47, 11964–11972. [DOI] [PubMed]
- Jones, G., Strugnell, S. A. & DeLuka, H. F. (1998). Physiol. Rev.78, 1193–1231. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mitani, Y., Meng, X., Kamagata, Y. & Tamura, T. (2005). J. Bacteriol.187, 2582–2591. [DOI] [PMC free article] [PubMed]
- Nagano, S., Cupp-Vickery, J. R. & Podust, T. L. (2005). J. Biol. Chem.280, 22102–22107. [DOI] [PubMed]
- Nakashima, T. & Tamura, T. (2004a). Appl. Environ. Microbiol.780, 5557–5568. [DOI] [PMC free article] [PubMed]
- Nakashima, T. & Tamura, T. (2004b). Biotechnol. Bioeng.86, 136–148. [DOI] [PubMed]
- Navaza, J. (1993). Acta Cryst. D49, 588–591. [DOI] [PubMed]
- Omura, T. & Sato, R. (1964). J. Biol. Chem.239, 2370–2378. [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Prosser, D. E. & Jones, G. (2004). Trends Biochem. Sci.29, 664–673. [DOI] [PubMed]
- Sasaki, J., Miyazaki, A., Saito, M., Adachi, T., Mizoue, K., Hanada, K. & Omura, S. (1992). Appl. Microbiol. Biotechnol.38, 152–157. [DOI] [PubMed]
- Sawada, N., Sasaki, T., Yoneda, S., Kusudo, T., Shinkyo, R., Ohta, M. & Inoue, K. (2004). Biochem. Biophys. Res. Commun.320, 156–164. [DOI] [PubMed]
- Sherman, D. H., Li, S., Yermalitskaya, L. V., Kim, Y., Smith, J. A., Waterman, M. R. & Podust, L. M. (2006). J. Biol. Chem.281, 26289–26297. [DOI] [PMC free article] [PubMed]
- Strushkevich, N., Usanov, S. A., Plotnikov, A. N., Jones, G. & Park, H. W. (2008). J. Mol. Biol.380, 95–106. [DOI] [PubMed]
- Sugimoto, H., Shinkyo, R., Hayashi, K., Yoneda, S., Yamada, M., Kamakura, M., Ikushiro, S., Shiro, Y. & Sasaki, T. (2008). Biochemistry, 47, 4017–4027. [DOI] [PubMed]
- Uchida, E., Kagawa, N., Sakaki, T., Urushino, N., Sawada, N., Kamakura, M., Ohta, M., Kato, S. & Inouye, K. (2004). Biochem. Biophys. Res. Commun.323, 505–511. [DOI] [PubMed]
- Zhu, G. D. & Okamura, W. H. (1995). Chem. Rev.95, 1877–1952.