

The d-2-hydroxyacid dehydrogenase from Haloferax mediterranei has been crystallized in two different forms. Diffraction data have been collected to 1.9 Å resolution for the non-productive ternary complex of the enzyme and to 2.7 Å for the selenomethionyl derivative.
Keywords: d-2-hydroxyacid dehydrogenase, Haloferax mediterranei
Abstract
d-2-Hydroxyacid dehydrogenase (D2-HDH) from Haloferax mediterranei has been overexpressed in Escherichia coli, solubilized in 8 M urea and refolded by rapid dilution. The protein was purified and crystallized by the hanging-drop vapour-diffusion method using ammonium sulfate or PEG 3350 as precipitant. Two crystal forms representing the free enzyme and the nonproductive ternary complex with α-ketohexanoic acid and NAD+ grew under these conditions. Crystals of form I diffracted to beyond 3.0 Å resolution and belonged to the monoclinic space group P21, with unit-cell parameters a = 66.0, b = 119.6, c = 86.2 Å, β = 96.3°. Crystals of form II diffracted to beyond 2.0 Å resolution and belonged to the triclinic space group P1, with unit-cell parameters a = 66.5, b = 75.2, c = 77.6 Å, α = 109.1, β = 107.5, γ = 95.9°. The calculated values for V M and analysis of the self-rotation and self-Patterson functions suggest that the asymmetric unit in both crystal forms contains two dimers related by pseudo-translational symmetry.
1. Introduction
Studying archaeal microorganisms that survive in extreme physical and chemical environments provides the opportunity to identify the mechanisms by which they have adapted to their environment and to define the limits of such biological adaptation. One type of extremophiles are the halophilic archaea that are adapted to living under extreme saline conditions, inhabiting hypersaline environments such as the Dead Sea and the Great Salt Lake. Halophilic archaea grow optimally in solutions containing 2.5–5.2 M NaCl and they are unable to grow at salinities below 1.5 M (Mevarech et al., 2000 ▶; Grant, 2004 ▶; Won-A & Chan-Wha, 2005 ▶). Different adaptive strategies have been selected by halophilic cells in order to compensate for the strong osmotic pressure of their environment. In the case of extreme halophiles from archaea, KCl is accumulated inside the cytoplasm up to concentrations close to saturation. Consequently, the enzymes and other molecules of the cellular machinery of halophilic archaea are adapted to function in this extreme high-salt environment. These enzymes are attractive biotechnology targets, as they naturally function in a low-water environment and hence offer the potential to serve as biocatalysts in non-aqueous environments (Sellek & Chaudhuri, 1999 ▶; Marhuenda-Egea et al., 2002 ▶; Won-A & Chan-Wha, 2005 ▶).
d-2-Hydroxyacid dehydrogenases catalyze the stereospecific NADH-dependent reduction of 2-ketocarboxylic acids to their corresponding 2-hydroxycarboxylic acids,
A d-2-hydroxyacid dehydrogenase (D2-HDH; EC 1.1.1.) from the halophilic archaeon Haloferax mediterranei has recently been cloned, sequenced and characterized, showing a subunit molecular weight of 33 kDa. This enzyme is the only isolated d-2-hydroxyacid dehydrogenase to exhibit dual cofactor specificity, with a marked preference for NADPH (Domenech & Ferrer, 2006 ▶). Sequence comparisons suggest that H. mediterranei D2-HDH is a member of the wider d-2-hydroxyacid dehydrogenase superfamily with, for example, 65% identity to the d-3-phosphoglycerate dehydrogenase from Haloarcula marismortui (Baliga et al., 2004 ▶) and 38% identity to the d-2-hydroxyacid dehydrogenase from Halorubrum lacusprofundi (GenBank accession number YP_002566205).
Enzymes belonging to the d-2-hydroxyacid dehydrogenase family share a similar subunit structure based on two domains of different size separated by a deep cleft, with the active site lying at its base. The larger domain contains a Rossmann fold (Rossmann et al., 1974 ▶) and is responsible for binding the dinucleotide, with the smaller domain providing the substrate-binding residues and generally showing a lower level of sequence similarity within the family. The majority of the enzymes are homodimers (Goldberg et al., 1994 ▶; Razeto et al., 2002 ▶; M. Sugahara & N. Kunishima, unpublished work), but there is some variation in their quaternary structure, with d-2-hydroxyisocaproate dehydrogenase, for example, being a homohexamer (Dengler et al., 1997 ▶). However, this hexamer is constructed from a trimer of dimers in D3 symmetry, with the dimer being the same as that of the other family members.
Previous structural studies of halophilic proteins have revealed that their molecular surfaces are highly negatively charged (Dym et al., 1995 ▶; Pieper et al., 1998 ▶; Bieger et al., 2003 ▶; Zeth et al., 2004 ▶; Besir et al., 2005 ▶; Britton et al., 2006 ▶), with the protein surface being coated in shells of ordered water molecules (Britton et al., 2006 ▶). Other comparative analyses have highlighted a significant reduction in hydrophobic surface owing to a decrease in the number of exposed lysine residues and thus the removal of their side-chain alkyl components (Britton et al., 1998 ▶, 2006 ▶). Although these trends can been seen in a genome-wide context (Satoshi et al., 2003 ▶), less than 15 halophilic protein structures have been determined and it is as yet uncertain how these sequence patterns are related to the observed tolerance to high salt.
In this paper, we report the crystallization and preliminary analysis of X-ray diffraction data from both methionine-containing (SMet) and selenomethionine-containing (SeMet) d-2-hydroxyacid dehydrogenase from H. mediterranei. Although this enzyme clearly belongs to the d-2-hydroxyacid dehydrogenase family, the closest sequence match to a member of known structure is only 29% identity (to the phosphoglycerate dehydrogenase from Lactobacillus plantarum) and, coupled with the very low level of sequence similarity in the small domain, it was anticipated that experimental phasing methods would be required. The determination of the three-dimensional structure of this enzyme will allow us to enhance our understanding of the structural factors of halophilic adaptation, to contribute to the development of enzymes that function efficiently in other dehydrating conditions such as organic solvents and to understand the molecular basis of the substrate-specificity of D2-HDH and its exploitation in the synthesis of fine chemicals.
2. Experimental
2.1. Overexpression of SMet and SeMet protein
The gene coding H. mediterranei D2-HDH (ATCC 335500) was cloned in the expression vector pET3a (Novagen) and used to transform Escherichia coli BL21 (DE3) strain. Induction of expression, refolding and purification of the protein were performed as reported previously (Domenech & Ferrer, 2006 ▶).
In order to produce SeMet-incorporated protein for use in multiwavelength anomalous dispersion (MAD) experiments, the transformed E. coli BL21 (DE3) strain was grown in minimal medium containing 10.5 g l−1 K2HPO4, 1 g l−1 (NH4)2SO4, 4.5 g l−1 KH2PO4, 0.5 g l−1 trisodium citrate.2H2O, 5 g l−1 glycerol, 0.5 g l−1 adenine, guanine, thymine and uracil, 1 mM MgSO4.7H2O, 4 mg l−1 thiamine, 40 mg l−1 selenomethionine and 100 mg l−1 of the amino acids Lys, Phe and Thr in addition to 50 mg l−1 Ile, Leu and Val. Analysis of the molecular weight of the SeMet protein by electrospray mass spectroscopy suggested that the selenium incorporation of the sample was greater than 90%.
2.2. Crystallization and preliminary X-ray analysis
Prior to crystallization, protein samples were concentrated to approximately 12 mg ml−1 and then buffer-exchanged into 20 mM Tris–HCl pH 8.0, 3 mM EDTA and 1 M NaCl using a Vivaspin concentrator (30 kDa molecular-weight cutoff; Vivascience). For crystallization of the ternary complex, the substrates were added to final concentrations of 50 mM for α-ketohexanoic acid and 5 mM for NAD+.
Preliminary crystallization conditions were screened by the sitting-drop vapour-diffusion method using commercial NeXtal kits and a Hydra II Plus One robot. Initial small plate-like crystals of the free enzyme (form I) grew from 3.0 M ammonium sulfate, 1%(w/v) MPD and crystals of the ternary complex grew in 0.2 M calcium acetate, 20%(w/v) PEG 3350 (form II).
Optimization of these conditions was carried out using the hanging-drop vapour-diffusion technique by mixing small volumes (1 µl) of the enzyme with different volumes of precipitant. The best form I crystals were obtained by mixing 1 µl protein solution with 2 µl well solution containing 2.0–3.5 M ammonium sulfate, 0.1 M MES pH 6.0 and 1%(w/v) MPD. For crystal form II, the protein–substrate complex was mixed with 2 or 3 µl of precipitant solution containing 14–26% PEG 3350, 0.1 M Tris–HCl pH 8.0 and 0.5 M magnesium acetate. The mixtures were allowed to equilibrate against reservoirs of precipitant solution at 290 K. After 24–48 h, clusters of crystals with a plate-like morphology with maximum overall dimensions of 150 × 100 × 15 µm grew for both the methionine and selenomethionine apoenzyme samples (Fig. 1 ▶ a). In contrast, for the ternary complex the crystals obtained were single and appreciably larger and thicker (maximum overall dimensions of 250 × 150 × 50 µm; Fig. 1 ▶ b).
Figure 1.

Photomicrographs of the two crystal forms of d-2-hydroxyacid dehydrogenase. (a) Crystal form I, SeMet apo enzyme; (b) crystal form II, ternary complex of SMet enzyme with 50 mM α-ketohexanoic acid and 5 mM NAD+.
For data collection, a single form I crystal was transferred to a cryoprotectant solution consisting of 0.1 M MES pH 6.0, 3.0 M ammonium sulfate, 1%(w/v) MPD and 15% glycerol for 10 s prior to cooling to 100 K using an Oxford Cryosystems cryostream. For crystals of form II the same process was utilized but with a cryoprotectant solution consisting of 0.1 M Tris–HCl pH 8.0, 0.5 M magnesium acetate, 20% PEG 3350 and 25% ethylene glycol.
Multiple anomalous dispersion data were collected from a single selenomethionine form I crystal to 2.7 Å resolution on ID29 at the ESRF synchrotron source (Table 1 ▶). Three wavelengths were chosen near the selenium-absorption edge based on the fluorescence absorption spectrum of the crystal in order to maximize the f′′ component (λ1, peak), to minimize the f′ component (λ2, inflection) and to maximize Δf′ (λ3, remote). A total of 212 images with 0.85° rotation per image were collected at all three wavelengths.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| SeMet apoenzyme (form I) | ||||
|---|---|---|---|---|
| Ternary complex (form II) | Peak (λ1) | Inflection (λ2) | Remote (λ3) | |
| Space group | P1 | P21 | ||
| Unit-cell parameters | ||||
| a (Å) | 66.5 | 66.0 | ||
| b (Å) | 75.2 | 119.6 | ||
| c (Å) | 77.6 | 86.2 | ||
| α (°) | 109.1 | 90.0 | ||
| β (°) | 107.5 | 96.3 | ||
| γ (°) | 95.9 | 90.0 | ||
| Matthews coefficient (Å3 Da−1) | 2.56 | 2.54 | ||
| Molecules per ASU | 4 | 4 | ||
| Temperature (K) | 100 | 100 | ||
| X-ray source | Rigaku MM007 | ESRF ID29 | ||
| Detector | MAR 345 | ADSC Q315 | ||
| Resolution (Å) | 21.7–1.86 (1.96–1.86) | 38.0–2.70 (2.85–2.70) | ||
| Energy (keV) | 8.041 (Cu Kα) | 12.660 | 12.658 | 12.710 |
| Mosaicity (°) | 0.75 | 0.8 | 0.8 | 0.8 |
| Unique observations | 101260 (12178) | 36498 (5339) | 36474 (5340) | 36512 (5346) |
| Rmerge† (%) | 0.04 (0.21) | 0.12 (0.37) | 0.17 (0.53) | 0.14 (0.57) |
| Completeness (%) | 90.7 (74.5) | 99.7 (100) | 99.6 (100) | 99.7 (100) |
| Anomalous completeness (%) | — | 97.0 (98.3) | 96.9 (99.0) | 97.2 (98.9) |
| Multiplicity | 2.4 (2.3) | 3.5 (3.6) | 3.6 (3.7) | 3.6 (3.7) |
| Anomalous multiplicity | — | 1.8 (1.8) | 1.8 (1.8) | 1.8 (1.8) |
| Mean (I)/σ(I) | 14.9 (4.6) | 10.6 (3.0) | 8.2 (2.1) | 9.2 (1.8) |
R
merge = 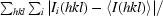
 , where 〈I(hkl)〉 is the mean intensity of the reflection.
, where 〈I(hkl)〉 is the mean intensity of the reflection.
For crystal form II, data were collected from a single methionine crystal to 1.9 Å resolution (Table 1 ▶) using a Rigaku MM007 copper rotating-anode generator with Varimax confocal optics and a MAR Research image-plate desktop beamline. A total of 230 images with 1.0° rotation per image were measured.
3. Results and discussion
Analysis of the diffraction data from the form I crystals using the autoindexing routine in MOSFLM (Leslie, 1992 ▶) and scaling in SCALA (Evans, 1997 ▶) from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) suggested that they belonged to space group P21, with unit-cell parameters a = 66.0, b = 119.6, c = 86.2 Å, β = 96.3°. The corresponding unit-cell volume is 6.7 × 105 Å3, which gives V M values of 5.1, 3.4 and 2.5 Å3 Da−1 for two, three and four subunits in the asymmetric unit, respectively, all of which are within the range observed by Matthews for protein crystals (Matthews, 1997 ▶). The form I crystals exhibited a varying degree of anisotropy in the diffraction pattern, with weaker intensities normal to the short crystal dimension, perhaps indicating some degree of disorder within the crystal lattice. Analysis of a self-Patterson calculated with 25–6 Å data showed a peak with a height 46% of the origin at 0.5, 0.14, 0.5, suggesting that the asymmetric unit contains molecules related by a translational pseudo-symmetry. The self-rotation function also has a significant peak (95% of the origin) on the κ = 180 Å section at ω = 90.9°, ϕ = 0°, showing the presence of a noncrystallographic twofold axis lying close to the crystallographic a axis. These results are consistent with the asymmetric unit containing two dimers of D2-HDH related by a translation of 0.5, 0.14, 0.5.
Calculation of a selenium substructure and preliminary protein phases from the three-wavelength MAD data collected from the form I crystals using the SHELX suite of programs (Sheldrick, 2008 ▶) produced electron-density maps that could not be interpreted and which had layers of alternate weak and strong density in the c cell direction, which is perhaps related to the anisotropy of the data and the presumed disorder of the crystal lattice. We have therefore switched our attention to the form II crystals, which exhibited isotropic diffraction to a much higher resolution. Analysis of the preliminary data collected from a methionine crystal on a rotating-anode source showed that these crystals belonged to the triclinic space group P1, with unit-cell parameters a = 66.5, b = 75.2, c = 77.6 Å, α = 109.1, β = 107.5, γ = 95.9°. Similar to the form I crystals, the self-Patterson calculated using 25–6 Å data has a peak 86% of the origin at 0, 0.5, 0.5, with the κ = 180° section of the self-rotation function containing a peak 86% of the origin at ω = 138.9°, ϕ = 301.7°, again indicating that the asymmetric unit contains two dimers related by translational pseudo-symmetry, giving a V M value of 2.5 Å3 Da−1.
Selenomethionine form II crystals have recently been obtained and a full structure determination is now under way. Given the high-resolution analysis possible with the form II crystals, we will be able to carry out a detailed comparison with other enzymes of the d-2-hydroxyacid dehydrogenase family as a contribution towards understanding the molecular basis of the salt tolerance of halophilic enzymes as well as understanding the molecular basis of the specificity of H. mediterranei D2-HDH.
Acknowledgments
This work was supported by grants from the BBSRC, the Wellcome Trust, The Woolfson Foundation and the University of Alicante (VIGROB046). Financial support to JD from Generalitat Valenciana is gratefully acknowledged. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation and we thank Christoph Mueller-Dieckmann for assistance in using beamline ID29.
References
- Baliga, N. S., Bonneau, R., Facciotti, M. T., Pan, M., Glusman, G., Deutsch, E. W., Shannon, P., Chiu, Y., Weng, R. S., Gan, R. R., Hung, P., Date, S. V., Marcotte, E., Hood, L. & Ng, W. V. (2004). Genome Res.14, 2221–2234. [DOI] [PMC free article] [PubMed]
- Besir, H., Zeth, K., Bracher, A., Heider, U., Ishibashi, M., Tokunaga, M. & Oesterhelt, D. (2005). FEBS Lett.579, 6595–6600. [DOI] [PubMed]
- Bieger, B., Essen, L. O. & Oesterhelt, D. (2003). Structure, 11, 375–385. [DOI] [PubMed]
- Britton, K. L., Baker, P. J., Fisher, M., Ruzheinikov, S., Gilmour, D. J., Bonete, M. J., Ferrer, J., Pire, C., Esclapez, J. & Rice, D. W. (2006). Proc. Natl Acad Sci. USA, 103, 4846–4851. [DOI] [PMC free article] [PubMed]
- Britton, K. L., Stillman, T. J., Yip, K. S. P., Forterre, P., Engel, P. C. & Rice, D. W. (1998). J. Biol. Chem.273, 9023–9030. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dengler, U., Niefind, K., Kiess, M. & Schomburg, D. (1997). J. Mol. Biol.267, 640–660. [DOI] [PubMed]
- Domenech, J. & Ferrer, J. (2006). Biochim. Biophys. Acta, 1760, 1667–1674. [DOI] [PubMed]
- Dym, O., Mevarech, M. & Sussman, J. L. (1995). Science, 267, 1344–1346. [DOI] [PubMed]
- Evans, P. R. (1997). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.33, 22–24.
- Goldberg, J. D., Yoshida, T. & Brick, P. (1994). J. Mol. Biol.236, 1123–1140. [DOI] [PubMed]
- Grant, W. D. (2004). Philos. Trans. R. Soc. Lond. B, 359, 1249–1267. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Marhuenda-Egea, F. C., Piere-Velazquez, S., Cadenas, C. & Cadenas, E. (2002). J. Biotechnol.93, 159–164. [DOI] [PubMed]
- Matthews, B. W. (1997). The Proteins, Vol. 3, edited by H. Neurath & R. L. Hill, pp. 468–477. New York: Academic Press.
- Mevarech, M., Frolow, F. & Gloss, L. M. (2000). Biophys. Chem.86, 155–164. [DOI] [PubMed]
- Pieper, U., Kapadia, G., Mevarech, M. & Herberg, O. (1998). Structure, 6, 75–88. [DOI] [PubMed]
- Razeto, A., Kochhar, S., Hottinger, H., Dauter, M., Wilson, K. S. & Lamzin, V. S. (2002). J. Mol. Biol.318, 109–119. [DOI] [PubMed]
- Rossmann, M. G., Moras, D. & Olsen, K. W. (1974). Nature (London), 250, 194–199. [DOI] [PubMed]
- Satoshi, F., Kazuaki, Y., Mamoru, W., Mitsuaki, M. & Ken, N. (2003). J. Mol. Biol.327, 347–357.
- Sellek, G. A. & Chaudhuri, J. B. (1999). Enzyme Microb. Technol.25, 471–482.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Won-A, J. & Chan-Wha, K. (2005). J. Chromatogr. B, 815, 237- 250.
- Zeth, K., Offermann, S., Essen, L. O. & Oesterhelt, D. (2004). Proc. Natl Acad Sci. USA, 101, 13780–13785. [DOI] [PMC free article] [PubMed]