

Abstract
APE1/Ref-1 is an essential DNA repair/gene regulatory protein in mammals of which intracellular level significantly affects cellular sensitivity to genotoxicants. The APE1 functions are altered by phosphorylation and acetylation. We here report that APE1 is also modified by ubiquitination. APE1 ubiquitination occurred specifically at Lys residues near the N-terminus, and was markedly enhanced by MDM2, the major intracellular p53 inhibitor. Moreover, DNA damaging reagents and nutlin-3, an inhibitor of MDM2/p53 interaction, increased APE1 ubiquitination in the presence of p53. Downmodulation of MDM2 increased APE1 level, suggesting that MDM2-mediated ubiquitination can be a signal for APE1 degradation. In addition, unlike the wild-type APE1, ubiquitin-APE1 fusion proteins were predominantly present in the cytoplasm. Therefore, monoubiquitination not only is a prerequisite for degradation, but may also alter the APE1 activities in cells. These results reveal a novel regulation of APE1 through ubiquitination.
Introduction
Oxidative DNA damage is continuously generated in cells and is mutagenic and cytotoxic (Fishel and Kelley, 2007). DNA base excision repair (BER1) is primarily responsible for repairing DNA damage generated by reactive oxygen species (ROS), environmental toxicants and cancer chemotherapeutic reagents (Fishel and Kelley, 2007). In BER, apurinic/apyrimidinic endonucleases (APEs) generate 3’-OH termini at AP sites, abnormal bases, and DNA single-strand breaks (Ischenko and Saparbaev, 2002; Izumi et al., 2003). E. coli and yeast cells defective in APE genes are inviable (Saporito et al., 1989; Guillet and Boiteux, 2002). In mammals, deficiency of APE1 alone causes the embryonic lethality and apoptotic cell death in cultured cells (Xanthoudakis et al., 1996; Ludwig et al., 1998; Meira et al., 2001; Fung and Demple, 2005; Izumi et al., 2005), clearly indicating that APEs are fundamental for cellular growth.
Post-translational modifications play important roles in modulating BER process. APE1 phosphorylation was first reported and shown to affect the repair and Ref-1 activities (Yacoub et al., 1997; Fritz and Kaina, 1999). Bhakat et al. reported that acetylation specifically occurred on two Lys residues near the N-terminus of APE1 (K6 and K7) (Bhakat et al., 2003). The acetylation enhanced APE1 binding to nCaRE (negative calcium response elements) consensus elements in DNA (Bhakat et al., 2003). The nCaRE-binding by APE1 may have a significant impact on global gene expression as many other genes have been shown to contain nCaRE elements in their promoter regions (Okazaki et al., 1994; McHaffie and Ralston, 1995; Izumi et al., 1996; Fuchs et al., 2003). Acetylation of APE1 was also found to activate the PTEN gene by a different mechanism from the nCaRE-oriented regulation (Fantini et al., 2008). Therefore, a posttranslational modification at APE1’s N-terminus can affect its DNA binding affinity and influence a variety of cellular metabolic functions.
Studies also have shown a correlation between a high level of APE1 and tumor resistance against chemotherapeutic drugs and ionizing radiation, implying that APE1 enhances repair and survival of these tumor cells (Koukourakis et al., 2001; Bao et al., 2006). Subcellular distribution of APE1 has also been linked to tumor aggressiveness (Tell et al., 2005; Di Maso et al., 2007). Colorectal adenoma and carcinoma cells showed higher concentrations of cytoplasmic APE1 than normal cells (Kakolyris et al., 1997). Therefore, understanding the mechanism of subcellular localization of APE1 may provide new strategies for the cancer therapy (Tell et al., 2008), as other studies have utilized DNA repair proteins as the target for the cancer therapy (Balusu et al., 2007; Jaiswal and Narayan, 2008).
Meira et al. reported that APE1 heterozygosity increased skin cancer onset when the XPC (Xeroderma pigmentosum group C) gene was nullizygous (Meira et al., 1997). This additive effect of APE1 on the cancer predisposition was nullified by inactivating p53, suggesting that APE1 was involved in tumor suppression regulated by p53 (Meira et al., 1997). Consistently, BER enhancement by p53 was also reported (Zhou et al., 2001). Yet the regulatory role of p53 for the APE1 functions has not been established.
In the present study, we report that APE1 was modified by ubiquitination which was increased following treatments with genotoxicants. We show that the modification was enhanced by MDM2 and was dependent on p53. We also identified the ubiquitin acceptor Lys residues at near N-terminus, namely, 24, 25, and 27K. To our knowledge, this is the first report for APE1 ubiquitination, and provides further evidence of the biological interaction between the BER and p53 signaling pathways.
Results
Decrease of APE1 during apoptosis has been reported in human cytolytic T-cells and myelolytic cells (Fan et al., 2003; Robertson et al., 1997). We examined the stability of APE1 after DNA damage generation by H2O2 using Kasumi-1, a human myeloblastic leukemia cell line (Asou et al., 1991). High-molecular weight-band (HWB) APE1 was detected in an immunoblot assay using an anti-APE1 antibody (top Fig. 1A), while the amount of the intact APE1 was observed to decrease (bottom, Fig. 1A) by about 10% compared to the control, which is consistent with the small ratio of HWB relative to intact APE1. This result implies that APE1 was ubiquitinated.
Fig. 1. APE1 ubiquitination in vivo and in vitro.
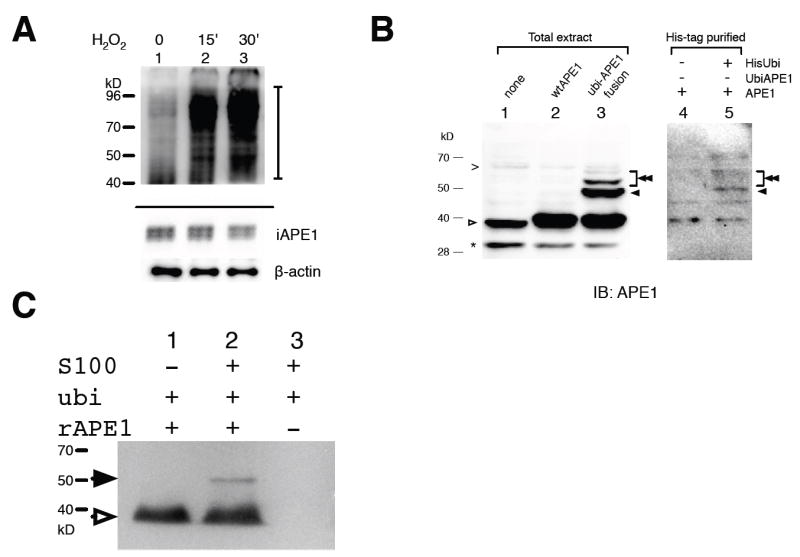
(A) AML cell line Kasumi-1 was treated with 1mM H2O2 for 15 (lane 2) or 60 min (lanes 3), and the total extracts were analyzed with APE1 immunoblot as described in Materials and Methods. (Top) HWB APE1 with intensified signals. A bar at the right side indicates the appearance of HWB APE1 (top panel). (Bottom) Intact APE1 and β-actin (re-blot) of the same sample set. (B) Detection of APE1 ubiquitination in cells. (Left) Molecular weight references for ubiquitinated APE1. Total protein extracts from HCT116 cells were blotted with anti-APE1 antibody. Cells were expressing (lane 1) none, (lane 2) wtAPE1, and (lane 3) ubiquitin-APE1 fusion (ubi-APE1). (Right) Protein extracts from HCT116 cells expressing wtAPE1 and (lane 4) the pcDNA3.1 control vector or (lane 5) His-tagged ubiquitin were purified through Ni-NTA magnet beads under the denaturing condition (Materials and Methods). (C) In vitro ubiquitination. Recombinant APE1 was incubated with ubiquitin and HeLa S100 fraction, and then analyzed with anti-APE1 in immunoblot. (A-C) Protein positions are indicated for intact APE1 (open arrow), monoubiquitinated APE1 (filled arrow), polyubiquitinated APE1 (double filled arrow), a truncated APE1 due to degradation (*), and non-specific bands (>).
To test the possibility of APE1 ubiquitination, we expressed APE1 and the His-tagged ubiquitin (His-ubi), a ubiquitin gene N-terminally tagged with the histidine hexamer, in the human colon carcinoma cell line HCT116. When the ubiquitinated proteins were purified and analyzed with an anti-APE1 antibody, distinct APE1 bands appeared at a higher position than the intact APE1 in the SDS/PAGE (lane 5, Fig. 1B). The generation of the HWB APE1 was dependent on the expression of the His-ubi (lane 5 vs 4), and the band position was consistent with that of a ubiquitin-APE1 fusion protein (lane 3). Next, we carried out an in vitro ubiquitination reaction using S100 fraction from HeLa cells supplemented with recombinant APE1 and ubiquitin proteins (Fig. 1C). A HWB identical in size to the ubiquitinated APE1 of the previous experiments was detected only when the cell-free extract, APE1, and ubiquitin were all present in the reaction (lane 2).
Ubiquitin-like modifiers, i.e., SUMO, Nedd8, and ISG15, have been demonstrated to modulate cellular factors including DNA repair proteins (Kerscher et al., 2006). Because of their biochemical and functional similarities to ubiquitin, we tested the possibility of APE1 SUMOylation, neddylation, and ISGylation, using His-tagged SUMO, Nedd8, and ISG15 in the cells with the same assay as for ubiquitin. Under these conditions, we did not detect any modification of APE1 by the ubiquitin-like modifiers (data not shown).
Upon stress, p53 activates a number of genes that result in apoptosis, cell cycle arrest, or senescence (Shmueli and Oren, 2004). Based on the genetic link between p53 and APE1 (Meira et al., 1997), we examined the effects of DNA-damaging reagents, such as etoposide (a topoisomerase II inhibitor) and H2O2, on APE1 ubiquitination, because these reagents affect the intracellular p53 level. When HCT116 (p53+/+ or p53-/-) cells expressing the His-ubi and APE1 were treated with H2O2 and etoposide, APE1 ubiquitination was significantly increased in the p53+/+ but not in the p53-/- cells (Fig. 2A). These findings suggest that APE1 ubiquitination is dependent on the level of p53 and induced in response to cellular stress.
Fig. 2. Involvement of p53 in APE1 ubiquitination.
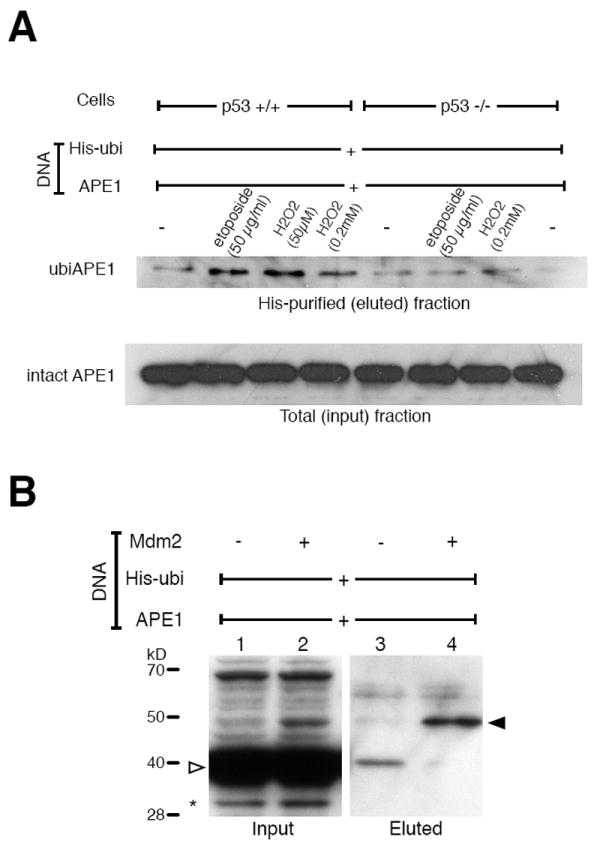
(A) p53-dependent APE1 ubiquitination. HCT116 p53+/+ and p53-/- cells were transfected with APE1 and His-ubi, and 24 h later the cells were treated for 4 h with the DNA-damaging reagents as indicated. His-ubi-enriched fractions as well as the total fractions were analyzed in immunoblot assays with anti-APE1. (B) APE1 ubiquitination by MDM2. Ubiquitination assays were performed by co-transfecting a control vector (lanes 1 and 3) or the MDM2-encoding plasmids (lanes 2 and 4) in addition to the His-ubi and APE1. The cells were lysed in a non-denaturing condition, and the total fractions (lanes 1 and 2) or the ubiquitin-enriched fractions (lanes 3 and 4) were analyzed with anti-APE1 antibody. Positions are indicated for intact APE1 (open arrow), monoubiquitinated APE1 (filled arrow), and truncated APE1 (*, due to degradation).
Although multiple ubiquitin E3 ligases for p53 are known, MDM2 is by far the most crucial regulator for p53 (Brooks and Gu, 2006). Importantly, p53 activates MDM2 transcription due to a genotoxic stress, leading to a temporal increase of MDM2 (Iwakuma and Lozano, 2003). For this reason, we tested whether MDM2 could catalyze the ubiquitination reaction on APE1. MDM2 was expressed along with the APE1 and His-ubi, and its effect on APE1 ubiquitination was examined. The amount of ubiquitinated APE1 was markedly increased by expressing MDM2, suggesting that MDM2 enhanced APE1 ubiquitination (Fig. 2B).
To determine whether the RING-finger domain of MDM2, critical for its E3 ubiquitin ligase activity, is required for the enhanced APE1 ubiquitination, a C464A MDM2 mutant lacking the proper RING conformation (Uldrijan et al., 2007) was used. The C464A-MDM2 did not show any activity for APE1 ubiquitination (Fig. 3A), indicating that the RING domain was directly involved in this process. As an E3 ligase, MDM2 requires E1 (Uba1) and E2 ubiquitin ligases (Honda et al., 1997). The In vitro ubiquitination reaction was carried out with recombinant APE1, ubiquitin, ube1 (E1), UbcH5 (E2), and MDM2 RING domain (Fig. 3B). Using only these components, the ubiquitinated APE1 was detected, indicating that no other factors were essential for APE1 ubiquitination.
Fig. 3. Mechanism of MDM2-dependent ubiquitination of APE1.
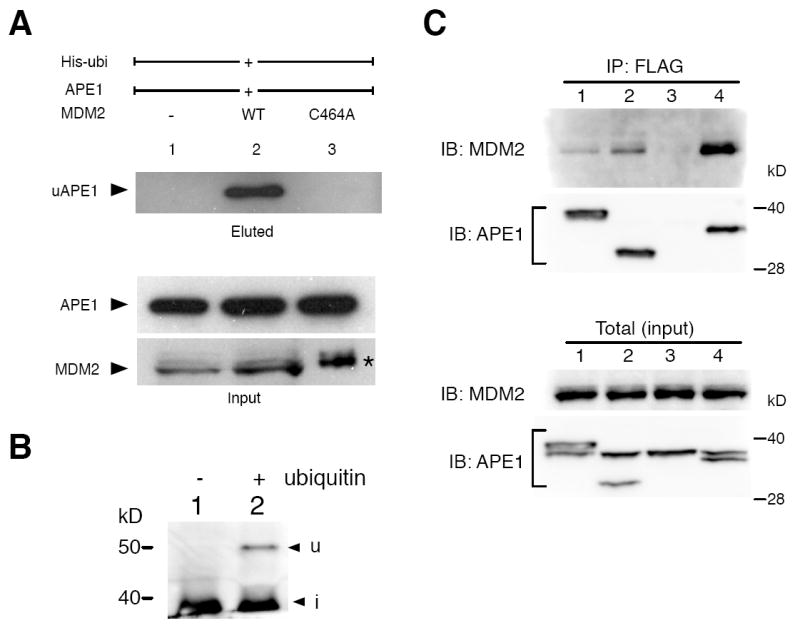
(A) Lack of APE1 ubiquitination by a mutant MDM2 C464A. The ubiquitin assay was conducted with expression of MDM2 C464A, APE1, and His-ubi. (*) The amount of the extract loaded was 15 times less than those for lanes 1 and 2 due to the high stability of C464A MDM2. (B) APE1 ubiquitination with recombinant ubiquitin and ubiquitin ligases. The reactions were carried out in the presence of 20 ng APE1, E1-activating enzyme, UbcH5b, and MDM2 RING-finger domain with (lane 2) or without (lane 1) ubiquitin. (C) Interaction of APE1 with C464A MDM2 in vivo. Cells expressing C464A MDM2 and full-length APE1-FLAG (lane 1), ND42 APE1-FLAG lacking 42 a.a. of the N-terminus (lane 2), full-length APE without FLAG fusion (lane 3), or CD20 APE1-FLAG lacking the 20 a.a. of the C-terminus (lane 4), were immunoprecipitated using anti-FLAG. The FLAG-enriched or total fractions were examined with anti-MDM2 (N-20) or APE1 antibodies as indicated.
To further probe the mechanism for APE1 ubiquitination, the possibility of the direct interaction of APE1 with MDM2 was examined. Interaction between cellular APE1 and C464A MDM2 was detected by FLAG co-immunoprecipitation assay using FLAG-tagged APE1 (lane 1, Fig. 3C), whereas APE1 without FLAG epitope did not co-precipitate the C464A MDM2 protein (lane 3). Deleting APE1’s N-terminal 41 amino acid (a.a.) residues (ND42) caused no detectable effect on the MDM2 co-immunoprecipitation (lane 2), suggesting that the N-terminal region was not required for the interaction. Interestingly, the C-terminal 20 a.a. deletion of APE1 (CD20) repeatedly resulted in much more efficient co-precipitation of MDM2 than the full-length APE1. It is likely that the C-terminal deletion exposed the presumptive interaction domain of APE1. Essentially the same results were obtained with the wild-type (wt) MDM2 and APE1 (Supplemental Fig. 1). The results indicate that the MDM2 RING structure is not essential for the interaction, and supports the idea that the MDM2 E3 ligase activity is directly involved in APE1 ubiquitination (Fig. 3A).
Ubiquitination occurs only at ε-amino groups of Lys residues, except for N-terminal ubiquitination (Bartel et al., 1990). APE1 contains 29 Lys residues in its 318 a.a.-long polypeptide, most of which are likely present on the APE1 surface and accessible by the ubiquitin ligase components. To determine whether the ubiquitination occurs at specific Lys residues of APE1, we examined its deletion mutants in the ubiquitin assay (lanes 2&3, Fig. 4). While ubiquitination of ND21 APE1 (20 a.a. N-terminal deletion) was comparable to that of the full-length APE1 (lane 2), ubiquitination of the ND42 APE1 (42 a.a. N-terminal deletion) was not detected (lane 3), indicating that the ubiquitination occurred inside of 21-42 from the APE1 N-terminus. Because there were still six Lys residues in the segment (i.e., 24K, 25K, 27K, 31K, 32K, and 35K), we mutated each individually to Arg in the ND21 background. The triple K-to-R mutant K(31/32/35)R was effectively ubiquitinated (lane 5 vs 4, Fig. 4), whereas the triple mutant K(24/25/27)R was ubiquitinated significantly less (lanes 7&8 vs 6, Fig. 4). However, none of the single point mutations (i.e., K24R, K25R, or K27R) decreased the ubiquitination level (lanes 10-12, Fig. 4). K(24/25/27)R mutations were introduced in the full-length APE1 and consistently reduced ubiquitination (lane 15 vs 13, Fig. 4). We also examined three possible dual mutations, K(24/25)R, K(24/27)R, K(25/27)R, with the ubiquitination assay. The K24/25R mutant showed reduced ubiquitination (lane 14, Fig. 4), while the other dual mutants exhibited the same level of ubiquitination as the wtAPE1 (data not shown). These results indicated that ubiquitination occurred preferentially to K24 and K25 rather than to K27. However, the fact that all the dual mutations failed to decrease APE1’s ubiquitination to the level of the triple mutant (lane 15, Fig. 4) suggest that these three Lys residues serve as backup ubiquitin acceptors when one is mutated. The ubiquitin-acceptor Lys residues are in the 6 kDa stretch which is conserved among mammalian APEs. A clustalw analysis revealed that the K24/K25/K27 residues are completely conserved among mammals (Supplemental Fig. S2). Therefore, we predict that ubiquitination reactions at these sites occur to all mammalian APE1s.
Fig. 4. Identification of ubiquitination sites in APE1.
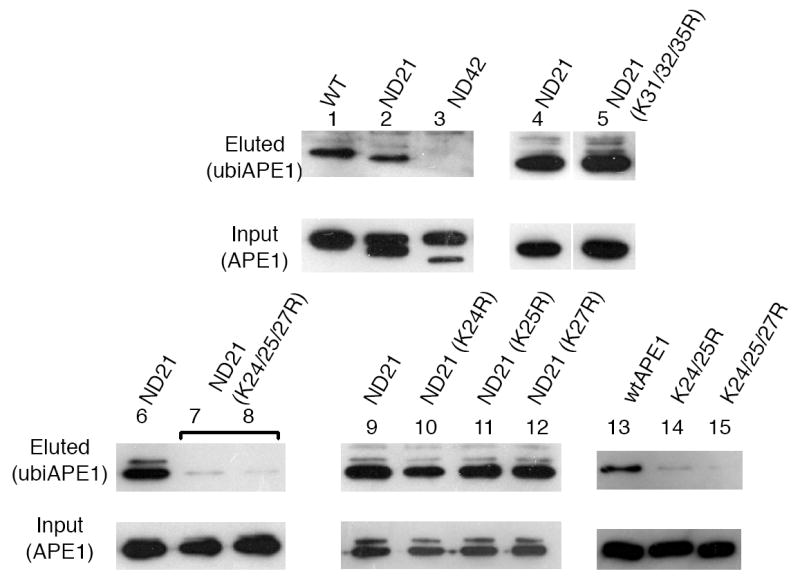
Various mutant APE1 were co-transfected with His-ubi and MDM2 for the in vivo ubiquitination assay, including wtAPE1 (lane 1), ND21 (lanes 2, 4, 6, 9), ND42 (lane 3), K31/32/35R triple mutant of ND21 (lane 5), K24/25/27R triple mutant of ND21 (lanes 7 and 8; a duplicate), K24R (lane 10), K25R (lane 11), and K27R of ND21 (lane 12). Ubiquitinated (eluted) or intact (input) APE1 were detected with anti-APE1 antibody. Lanes 13-15: wtMDM2 and His-ubi with full-length wtAPE1 (lane 13), K24/25R full-length APE1 (lane 14), K24/25/27R full-length APE1 (lane 15).
We also confirmed that mouse Mdm2 and human MDM2 were both efficient at enhancing APE1 ubiquitination (lanes 2 and 3, Fig. 5A). In Fig. 5A, while polyubiquitinated APE1 was clearly detected (lanes 2&3), monoubiquitinated APE1 appeared to be stably formed throughout our ubiquitination assays. We inferred that monoubiquitinated APE1 might exist stably and that the modification would alter APE1 localization, as monoubiquitination does to p53 (Li et al., 2003). The ubiquitin gene was thus inserted (sandwiched) in frame between the 23A and 24K residues of APE1, and was transfected into the mouse NIH3T3 cell line. The wtAPE1 was predominantly nuclear (i, Fig. 5B) as reported (Takao et al., 1998; Jackson et al., 2005), whereas the ubiquitin-APE1 fusion protein was clearly excluded from the nuclei (ii, Fig. 5B).
Fig. 5. Effect of monoubiquitination on APE1 cellular distribution.
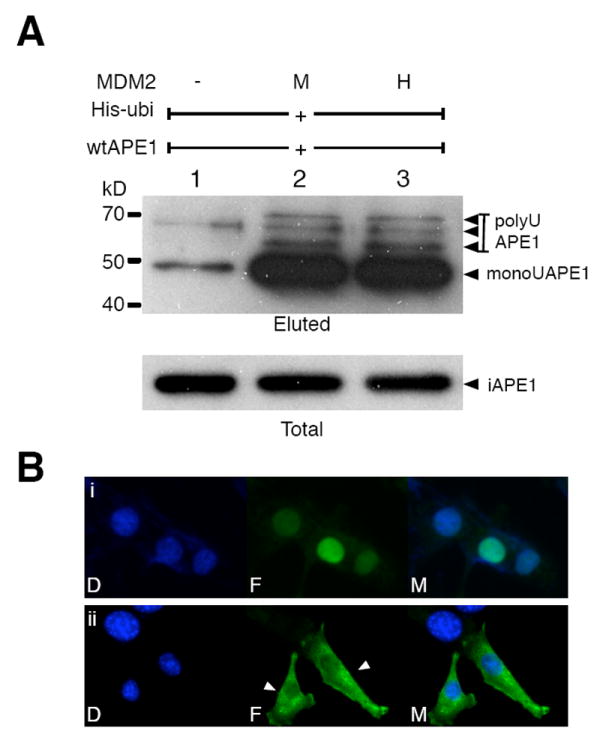
(A) APE1 ubiquitination by human MDM2 (HDM2). Ubiquitination assay was carried out with expression of His-ubi and APE1 plus the control vector (lane 1), mouse MDM2 (M, lane 2), and human MDM2 (H, lane 3). The intact APE1 (iAPE1), monoubiquitinated APE1 (monoUAPE1), and polyubiquitinated APE1 (polyUAPE1) are indicated by arrows. (B) Nuclear exclusion of ubiquitin-APE1 fusion proteins. Cells were transfected with (i) human wtAPE1 or (ii) APE1(1-23)-ubiqutin (G76A)-APE1(24-318), and human APE1 was specifically stained with FITC (mouse endogenous APE1 not detectable under the staining condition). D: DAPI (nuclei); F: FITC (human APE1); M: merged.
MDM2 is transcriptionally activated by p53. In turn, the accumulated MDM2 degrades p53 via polyubiquitination, forming a negative feedback loop (Vousden and Lane, 2007). Nutlin, a potent inhibitor of MDM2 for p53 ubiquitination, specifically interferes with the interaction of MDM2 with p53 (Vassilev et al., 2004). An interesting possibility was that nutlin might also inhibit APE1 ubiquitination. If indeed this were the case, the nutlin-binding domain of MDM2 would be required for APE1 ubiquitination. APE1 might thus compete with p53 for MDM2 interaction. However, when HCT116 cells (p53+/+) were treated with nutlin-3, APE1 ubiquitination was significantly increased (Fig. 6A). Therefore, nutlin-3 did not interfere with APE1 ubiquitination via MDM2. The higher ubiquitination activity in the presence of nutlin-3 can be explained by the fact that nutlin-3 causes p53 stabilization which increases the MDM2 level via transcriptional activation (Fig. 6A). Consistently, cells deficient in p53 (HCT116 p53-/-) did not respond to nutlin-3 by increasing APE1 ubiquitination (Fig. 6A). To detect ubiquitination on endogenous APE1, we transiently introduced the His-ubi alone to HCT116 which was then treated with nutlin-3. We observed the ubiquitinated form of endogenous APE1 (Fig. 6B), indicating that ubiquitination of endogenous APE1 can also occur depending on the intracellular level of MDM2.
Fig. 6. Effect of nutlin-3 on APE1 ubiquitination.
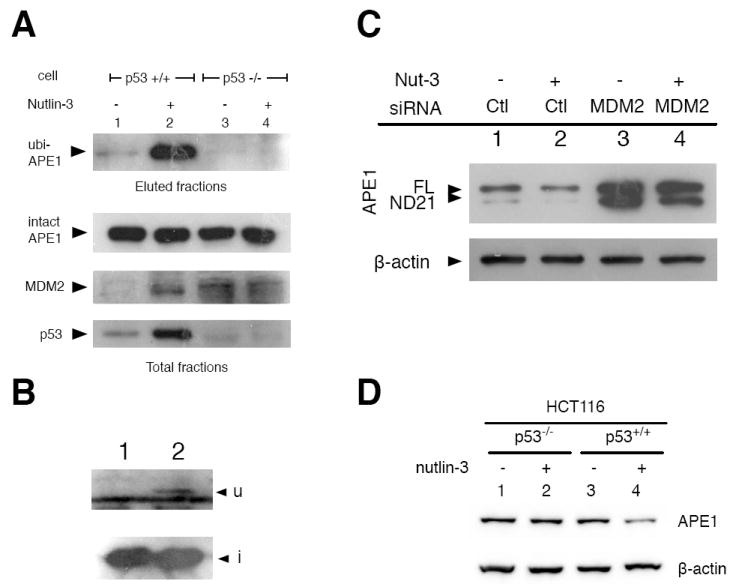
(A) Cells were treated with nutlin-3 for 16 h before the ubiquitin assay. Immunoblot assays with anti-APE1 (Top), anti-p53 (FL-393, Middle), and anti-MDM2 (Bottom). (B) Ubiquitination on the endogenous APE1. HCT116 cells were transfected with His-ubi for overnight. Cells were lysed after 4 h incubation with (lane 2) or without (lane 1) 10 μM nutlin-3, and ubiquitinated APE1 was purified by NTA resin. (C) Effect of downmodulation of MDM2 on APE1 protein levels. A control siRNA or MDM2-siRNA were co-transfected with His-ubi, APE1, and ND21 cDNA in the HCT116 cells with or without 10 μM nutlin-3. Cells were lysed after 30 h for immunoblot assay using anti-APE1 antibody. (D) Effect of nutlin-3 on the level of endogenous APE1. HCT116 (p53+/+ or p53-/-) were treated with DMSO (lanes 1 and 3) or 10 μM nutlin-3 for 6 h (lanes 2 and 4), in the presence of a deubiquitinase inhibitor mixture and 10 μM MG132 (Materials and Methods).
To further probe the significance of MDM2 on APE1 stability, human MDM2-specific siRNA was introduced with the APE1 and His-ubi genes into the HCT116 cells (Fig. 6C). The level of APE1 was significantly increased by MDM2-downmodulation, compared to that of cells treated with the control siRNA (Fig. 6C). Under the same condition, the cells that were also treated with nutlin-3 showed decreased APE1 levels (lane 2 vs 1, or 4 vs 3, Fig. 6C). Next, HCT116 cells (p53+/+ and p53-/-) were treated with nutlin-3 for 6 h. About 50% decrease in endogenous APE1 level was reproducibly observed in p53+/+ HCT116 cells, but not in the p53-/- cells (Fig. 6D). Taken together, these data indicate that the stability of APE1 was regulated by MDM2 and p53 activities.
Discussion
Multiple studies have confirmed that down-regulation of APE1 leads to cell sensitization spontaneously and by DNA damaging reagents (Izumi et al., 2005; Ono et al., 1994; Walker et al., 1994; Fung and Demple, 2005), and understanding the mechanism for regulating the APE1 level may provide new strategies for cancer therapy (Tell et al., 2008).
It has been reported that APE1 is down-regulated by apoptotic stimuli in certain types of blood cells (Robertson et al., 1997; Fan et al., 2003). Down-regulation of APE1 should enhance the apoptosis process, because reducing the cellular level of APE1 very likely enhances the cell death process. We have obtained multiple sources of evidence to support the existence of a ubiquitination reaction on APE1. First, the acute myeloblastic leukemia cell line Kasumi-1 showed the APE1 ladder formation. Although the amount of HWB was lower than that of the intact APE1, the appearance of the multiple HWB could not be attributed to the other types of posttranslational modifications than ubiquitination. The extent of the ladder formation appears to be cell-type specific, because different cell lines, including HCT116, A549, and U2OS, did not show detectable ubiquitination formation on APE1 by the same procedure (data not shown). It is known that multiple factors affect the level of ubiquitination, including the intracellular concentration of ubiquitin ligases and the extent of "ubiquitin trapping" by other small molecule modifier, most notably by ISG15 which is over-expressed in many tumor cell lines (Desai et al., 2006). Moreover, ubiquitination is reversible by a number of deubiquitinases (DUBs) which remove the ubiquitin moieties from the modified proteins. For example, HAUSP (USP7) is the main deubiquitinase for p53 and has a significant role in stabilizing p53 in cells (Cummins et al., 2004). Understanding the factors that regulate APE1 ubiquitination will be significant for predicting APE1 levels.
Further evidence of APE1 ubiquitination was obtained using His-tagged ubiquitin. We observed that MDM2 and its known E2 ligase, UbcH5b (Honda et al., 1997; Badciong and Haas, 2002; Saville et al., 2004), could carry out the APE1 ubiquitination reaction. MDM2 appears pivotal for the reaction, considering that the RING-finger mutant C464A MDM2 did not ubiquitinate APE1. Furthermore, this finding implies the involvement of p53, and is underscored by the enhancement of APE1 ubiquitination induced by such stimuli as H2O2, etoposide, and nutlin-3. Unlike the DNA damaging reagents, nutlin-3 disrupts the p53-MDM2 interaction and thus stabilizes p53. The enhancement of APE1 ubiquitination by nutlin-3 also indicates that APE1 interacts with MDM2 through an interface not shared with p53. It should be noted that although the direct interaction was observed between the transiently expressed APE1 and MDM2, we have not been able to detect the interaction between endogenous APE1 and MDM2. This unexpected result may be due to a technical limitation in detecting endogenous MDM2, which is notoriously unstable in vivo. It is also possible that a conformational change of APE1, probably involving the surface near APE1’s C-terminus (Fig. 3C), is required to enhance the specific interaction.
The N-terminal 6 kDa region of APE1, which contains the ubiquitin acceptor sites (K24, 25, 27), is highly conserved among mammalian APE1. Although the N-terminal 6 kDa segment is not essential for the endonucleolytic activity of APE1, the domain is known to be important for interaction with other cellular factors including XRCC1, another essential BER protein (Vidal et al., 2001). In addition, acetylation occurs at APE1’s Lys6 and 7 (Bhakat et al., 2003), thus it would be interesting to test whether ubiquitination affects the accessibility of p300 for the acetylation reaction, or vice versa.
In our ubiquitin assay, monoubiquitinated APE1 was predominant over polyubiquitinated APE1. Therefore, it is possible that, once formed, monoubiquitinated APE1 remains stable in the cells before being polyubiquitinated. However, it should be pointed out that polyubiquitinated APE1 was also detected forming a ladder (lanes 2-3 in Fig. 5A). We have yet to find a parameter to control the ratio of mono- and poly-ubiquitination on APE1. A similar dual-ubiquitination mechanism has been described by the numerous studies of p53 ubiquitination by MDM2. A critical factor for determining the ratio of the mono/poly-ubiquitination of p53 appears to be the in situ concentration of MDM2 (Li et al., 2003). A higher concentration of MDM2 would generate more polyubiquitinated p53 for its degradation. A lower amount would result in monoubiquitinated p53. The latter modification confers additional functions to p53, such as self-promoted exclusion from nuclei (Li et al., 2003) and disruption of mitochondrial membrane potential (Marchenko et al., 2007). More studies are necessary to understand whether the same parameter would apply to APE1. Alternatively, a cofactor may be required to enhance APE1 polyubiquitination, as monoubiquitinated p53 requires p300 (as an “E4 ligase”) to become fully polyubiquitinated (Grossman et al., 2003). MDM2 is also a target of down-regulation by such cellular factors as 14-3-3 sigma (Yang et al., 2003; Yang et al., 2007), which could indirectly regulate ubiquitination of APE1. Finally, involvement of other ubiquitin ligases has been reported for p53 polyubiquitination. Pirh2 and COP1 almost exclusively polyubiquitinate p53 for its degradation. The present study supports the dual role of MDM2 by invoking the mono and polyubiquitination reactions on APE1, which result in either functional alteration or degradation of APE1.
Apart from p53 ubiquitination, histone ubiquitination by MDM2 was reported (Minsky and Oren, 2004). Monoubiquitinated histone H2A/B can stably exist and modulate gene expression. Formation of stable monoubiquitinated APE1 may have a significant effect on cellular response to DNA damage, repair, and apoptosis. We have provided evidence that the monoubiquitination would result in nuclear exclusion of APE1. Whether the nuclear exclusion of APE1 is a mechanism to decrease BER activity in nuclei and enhance apoptosis, or to lead to a novel event such as the interaction of APE1 with Bcl-2 (Zhao et al., 2008) in mitochondria awaits more dedicated, future studies. Here, the identification of specific Lys residues for APE1 ubiquitination should help study the effect of the monoubiquitination on APE1. Understanding the pathway for APE1 ubiquitination should provide critical knowledge not only of its physiological role, but of the APE1 up-regulation mechanism in tumor tissues (Koukourakis et al., 2001; Bao et al., 2006; Tell et al., 2008). APE1 regulation by p53 and MDM2 is clearly a complex mechanism, and understanding the role of ubiquitin in this signaling network should reveal the significance of the APE1/p53/MDM2 interdependence in the development of tumor malignancy.
Materials and Methods
Cell culture and cDNA
All cell culture media and reagents were purchased from Invitrogen (CA USA). Tumor cell lines used in this study included human cell lines U2OS and HCT116 (p53+/+ and p53-/-, Yu et al., 2002; Bhakat et al., 2003) and mouse NIH 3T3 (gift from Dr. S. Boldogh; ATCC CRL-1658). The cells were grown in Dulbecco’s minimum essential medium with F12 modification supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin. Kasumi-1, a human myeloblastic leukemia cell line isolated by Asou et al. (Asou et al., 1991), was purchased from ATCC and grown in RPMI1640 with 20% FBS and 1% L-glutamine. Transfection experiments were carried out using lipofectamine2000 according to the vendor’s instruction.
The human Mdm2 homolog (MDM2) was purchased from MGC image clone library (Invitrogen, CA USA). The human MDM2 gene was then cloned into Invitrogen’s pcDNA3.1 Zeo(+) at the BamHI and XhoI sites by PCR. APE1 cDNA (Izumi et al., 2005) was sublconed into the pcDNA3.1 by PCR cloning to generate missense mutations and the ubiquitin fusions. The human ubiquitin gene was cloned into the pcDNA3.1 with introduction of N-terminal histidine hexamer. The G76A mutation in the ubiquitin gene was introduced by PCR. The primers used in this study are listed in the supplemental Table 1. All the DNA sequences amplified by PCR were confirmed for their predicted sequences.
His-ubiquitin assay
Cells were transfected with indicated DNA and incubated for 24 h. A typical transfection involves APE1 (wt or mutant) and His-tagged ubiquitin (His-ubi) with the C-terminal point mutation G76A, unless otherwise mentioned. When necessary, chemicals such as nutlin-3 (Cayman), etoposide (Sigma), and H2O2 (Fisher) were added prior to harvest. Crude cell lysates were then prepared in ice-cold “NTA buffer” containing 20 mM Tris (pH 8.0), 500 mM NaCl, 0.05 % Tween 20, 10 mM imidazole, MG132 (Boston Biochem/Sigma), proteinase inhibitor cocktail (EDTA-free, Roche), and PMSF. After centrifugation, a small volume of the supernatant was taken as total fractions, and the rest was incubated with Ni-NTA magnet beads (Qiagen) for 15 min at 4°C. The beads were washed with the NTA buffer containing 10 mM and 20 mM imidazole and eluted with the NTA buffer with 200 mM imidazole.
Co-immunoprecipitation assay for APE1-FLAG using FLAG antibody
HCT116 cells were transfected with APE1-FLAG and C464A mutant MDM2, and lysed after 24 h in the IP buffer containing 50 mM Tris (pH 7.5), 100 mM NaCl, 15 mM EDTA, 0.1% TritonX-100, 1 mM DTT, proteinase inhibitor cocktail (Roche), and 1 mM PMSF. After centrifugation, the supernatant was incubated with anti-FLAG antibody immobilized on agarose resin (M2-agarose, Sigma) for 1 h at 4°C. After supernatant was removed from the resin by centrifugation at 1,000 rpm for 30 sec, the proteins remaining with the resin were analyzed by immunoblot assays using the anti-MDM2 (N-20, Santa Cruz) and anti-APE1 (Izumi et al., 1996) antibodies.
Co-transfection of DNA and MDM2 siRNA
Final concentration of 80nM of control (UAGCGACUAAACACAUCAAUU, Dharmacon) or MDM2-specific siRNA (Santa Cruz), along with 0.25 μg of APE1 (full-length and ND21) and the His-ubi (G76A), were mixed with lipofectamin (Invitrogen) and added on to the HCT116 cells in the absence of fetal bovine serum. After 6 h, DMEM/F12 with 10% FBS was added, and cells were lysed for immunoblot detection after further incubation for 30 h.
In vitro ubiquitination reaction
Purified ubiquitin ligases, i.e., E1, UbcH5b, and RING finger domain of MDM2, were purchased from Biomol. HeLa S100 fraction was purchased from Boston Biochem. Ubiquitin was a gift from Dr. A. Haas (LSUHSC, New Orleans). The in vitro ubiquitination reaction was carried out essentially in the same way as previously published (Badciong and Haas, 2002). For the reaction using the recombinant ubiquitin ligases, purified APE1 200 ng was incubated with APE1 (20 ng), ubiquitin (20 ng), uba1 (10 ng), UbcH5b (20 ng), and MDM2 RING domain (20 ng) in 50 mM Tris 7.6, 10 mM MgCl2, 1 mM DTT, 2 mM ATP, 10 mM creatine phosphate and kinase. The reaction using the S100 fraction was carried out based on the vendor’s protocol. The reactions were carried out at 37°C for 1 hr, and then analyzed in immunoblot assay using anti-APE1 antibody as was done for above assays.
Cellular localization of ubiquitin-APE1 fusion protein
Plasmid DNA for the “sandwiched” ubiquitin-APE1 fusion was generated by inserting the ubiquitin gene between APE1’s 23A and 24K amino acids. The Gly residue at the ubiquitin C-terminus was changed to Ala (G76A) that is resistant to cellular deubiquitinase activity. NIH3T3 cells were transfected with the above-described DNA, and fixed with 3.7% formaldehyde after 24 h. The cells were then permeabilized with 0.2% Triton-X solution, and stained with anti-APE1 antibody and DAPI. The localization of the fusion proteins were observed using Nikon TE2000 microscope.
Other reagents
When necessary, cells were incubated with DUB inhibitors, namely, 0.1 μM N-ethylmaleimide (NEM, Sigma), 10 μM LDN-57444 and 10 μM 4,5,6,7-Tetrachloroindan-1,3-dione (Calbiochem).
Supplementary Material
Acknowledgments
We thank K.A. Kerlec, K.M. Lane, M.W. Lake, M.K. Rogoszewicz, and C. Bulkin for their technical assistance, and Mr. Lake’s editorial help. We are also grateful for Drs. S. Desai and A. Haas at LSU New Orleans for their critical discussion and experimental guidance. Dr. Davis’s excellent editorial help is much appreciated. This research was supported by NCI grant CA98664 (TI) and Louisiana Cancer Research Consortium funding.
Footnotes
Abbreviations: AP, apurinic/apyrimidinic; APE1, AP endonuclease 1; BER, base excision repair; MDM2, mouse double minute 2; His-ubi, x6 histidine-tagged ubiquitin; H2O2, hydrogen peroxide; wt, wild-type; XPC, Xeroderma pigmentosum complementation group C; XRCC1, X-ray cross complementing group 1
References
- Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991;77:2031–2036. [PubMed] [Google Scholar]
- Badciong JC, Haas AL. MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J Biol Chem. 2002;277:49668–49675. doi: 10.1074/jbc.M208593200. [DOI] [PubMed] [Google Scholar]
- Balusu R, Jaiswal AS, Armas ML, Kundu CN, Bloom LB, Narayan S. Structure/function analysis of the interaction of adenomatous polyposis coli with DNA polymerase beta and its implications for base excision repair. Biochemistry. 2007;46:13961–13974. doi: 10.1021/bi701632e. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature S. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bartel B, Wunning I, Varshavsky A. The recognition component of the N-end rule pathway. EMBO J. 1990;9:3179–3189. doi: 10.1002/j.1460-2075.1990.tb07516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428:1–486. doi: 10.1038/nature02501. [DOI] [PubMed] [Google Scholar]
- Desai SD, Haas AL, Wood LM, Tsai YC, Pestka S, Rubin, et al. Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 2006;66:921–928. doi: 10.1158/0008-5472.CAN-05-1123. [DOI] [PubMed] [Google Scholar]
- Di Maso V, Avellini C, Croce LS, Rosso N, Quadrifoglio F, Cesaratto L, et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol Med. 2007;13:89–96. doi: 10.2119/2006-00084.DiMaso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, et al. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol. 2003;4:145–153. doi: 10.1038/ni885. [DOI] [PubMed] [Google Scholar]
- Fantini D, Vascotto C, Deganuto M, Bivi N, Gustincich S, Marcon G, et al. APE1/Ref-1 regulates PTEN expression mediated by Egr-1. Free Radic Res. 2008;42:20–29. doi: 10.1080/10715760701765616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007 doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Fritz G, Kaina B. Phosphorylation of the DNA repair protein APE/REF-1 by CKII affects redox regulation of AP-1. Oncogene. 1999;18:1033–1040. doi: 10.1038/sj.onc.1202394. [DOI] [PubMed] [Google Scholar]
- Fuchs S, Philippe J, Corvol P, Pinet F. Implication of Ref-1 in the repression of renin gene transcription by intracellular calcium. J Hypertens. 2003;21:327–335. doi: 10.1097/00004872-200302000-00024. [DOI] [PubMed] [Google Scholar]
- Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–344. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- Guillet M, Boiteux S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J. 2002;21:2833–2841. doi: 10.1093/emboj/21.11.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature. 2002;415:183–187. doi: 10.1038/415183a. [DOI] [PubMed] [Google Scholar]
- Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000. [PubMed] [Google Scholar]
- Izumi T, Brown DB, Naidu CV, Bhakat KK, Macinnes MA, Saito H, et al. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci U S A. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Henner WD, Mitra S. Negative regulation of the major human AP-endonuclease, a multifunctional protein. Biochemistry. 1996;35:14679–14683. doi: 10.1021/bi961995u. [DOI] [PubMed] [Google Scholar]
- Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, et al. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- Jackson EB, Theriot CA, Chattopadhyay R, Mitra S, Izumi T. Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1) Nucleic Acids Res. 2005;33:3303–3312. doi: 10.1093/nar/gki641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal AS, Narayan S. A novel function of adenomatous polyposis coli (APC) in regulating DNA repair. Cancer Lett. 2008 doi: 10.1016/j.canlet.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakolyris S, Kaklamanis L, Engels K, Turley H, Hickson ID, Gatter KC, et al. Human apurinic endonuclease 1 expression in a colorectal adenoma-carcinoma sequence. Cancer Res. 1997;57:1794–1797. [PubMed] [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- Koukourakis MI, Giatromanolaki A, Kakolyris S, Sivridis E, Georgoulias V, Funtzilas G, et al. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int J Radiat Oncol Biol Phys. 2001;50:27–36. doi: 10.1016/s0360-3016(00)01561-3. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery, et al. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHaffie GS, Ralston SH. Origin of a negative calcium response element in an ALU-repeat: implications for regulation of gene expression by extracellular calcium. Bone. 1995;17:11–14. doi: 10.1016/8756-3282(95)00131-v. [DOI] [PubMed] [Google Scholar]
- Meira LB, Cheo DL, Hammer RE, Burns DK, Reis A, Friedberg EC. Genetic interaction between HAP1/REF-1 and p53. Nat Genet. 1997;17:145. doi: 10.1038/ng1097-145. [DOI] [PubMed] [Google Scholar]
- Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, et al. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001;61:5552–5557. [PubMed] [Google Scholar]
- Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell. 2004;16:631–639. doi: 10.1016/j.molcel.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Chung U, Nishishita T, Ebisu S, Usuda S, Mishiro, et al. A redox factor protein, ref1, is involved in negative gene regulation by extracellular calcium. J Biol Chem. 1994;269:27855–27862. [PubMed] [Google Scholar]
- Ono Y, Furuta T, Ohmoto T, Akiyama K, Seki S. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Robertson KA, Hill DP, Xu Y, Liu L, Van Epps S, Hockenbery DM, et al. Down-regulation of apurinic/apyrimidinic endonuclease expression is associated with the induction of apoptosis in differentiating myeloid leukemia cells. Cell Growth Differ. 1997;8:443–449. [PubMed] [Google Scholar]
- Saporito SM, Gedenk M, Cunningham RP. Role of exonuclease III and endonuclease IV in repair of pyrimidine dimers initiated by bacteriophage T4 pyrimidine dimer-DNA glycosylase. J Bacteriol. 1989;171:2542–2546. doi: 10.1128/jb.171.5.2542-2546.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville MK, Sparks A, Xirodimas DP, Wardrop J, Stevenson LF, Bourdon JC, et al. Regulation of p53 by the ubiquitin-conjugating enzymes UbcH5B/C in vivo. J Biol Chem. 2004;279:42169–42181. doi: 10.1074/jbc.M403362200. [DOI] [PubMed] [Google Scholar]
- Shmueli A, Oren M. Regulation of p53 by Mdm2: fate is in the numbers. Mol Cell. 2004;13:4–5. doi: 10.1016/s1097-2765(03)00529-x. [DOI] [PubMed] [Google Scholar]
- Takao M, Aburatani H, Kobayashi K, Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–2922. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26:102–112. doi: 10.1038/sj.emboj.7601469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Walker LJ, Craig RB, Harris AL, Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoub A, Kelley MR, Deutsch WA. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res S. 1997;57:5457–5459. [PubMed] [Google Scholar]
- Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol. 2003;23:7096–7107. doi: 10.1128/MCB.23.20.7096-7107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Wen YY, Lin YI, Pham L, Su CH, Yang H, et al. Roles for negative cell regulator 14-3-3sigma in control of MDM2 activities. Oncogene. 2007;26:7355–7362. doi: 10.1038/sj.onc.1210540. [DOI] [PubMed] [Google Scholar]
- Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002;295:1526–1528. doi: 10.1126/science.1068327. [DOI] [PubMed] [Google Scholar]
- Zhao J, Gao F, Zhang Y, Wei K, Liu Y, Deng X. Bcl2 inhibits abasic site repair by down-regulating APE1 endonuclease activity. J Biol Chem. 2008 doi: 10.1074/jbc.M708345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. EMBO J. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.