

Abstract
MicroRNAs (miRNAs) are endogenously small non-coding RNAs which are capable of silencing gene expression at the posttranscriptional level. In this study, we report that miR-205 is significantly underexpressed in breast tumor compared to the matched normal breast tissue. Similarly, breast cancer cell lines including MCF-7 and MDA-MB-231 express a lower level miR-205 than the non-malignant MCF-10A cells. Of interest, ectopic expression of miR-205 significantly inhibits cell proliferation and anchorage independent growth as well as cell invasion. Furthermore, the animal model indicates that miR-205 suppresses lung metastasis. Finally, western blot combined with the luciferase reporter assays demonstrate that ErbB3 and vascular endothelial growth factor A (VEGF-A) are direct targets for miR-205 and this miR-205-mediated suppression is likely through the direct interaction with the putative miR-205 binding site in the 3’-untranslated region (3-UTR) of ErbB3 and VEGF-A. Together, these results suggest that miR-205 is a tumor suppressor in breast cancer.
Keywords: breast cancer, cell growth, ErbB3, miRNA, miR-205, posttranscriptional regulation, VEGF-A
Introduction
MicroRNAs (miRNAs) are endogenously processed non-coding RNAs which regulate gene expression by blocking the translation or decreasing the stability of mRNAs (1-3). They are initially transcribed by RNA polymerase II into larger transcripts, called pri-miRNAs (4-6). In the nucleus, the RNase III Drosha, interacting with DGCR8, cleaves pri-miRNAs to form pre-miRNAs (7). The pre-miRNAs are then exported by exportin-5 to the cytoplasm, where Dicer further processes them into mature double-stranded miRNAs with ~22 nt in length (1, 8). Finally, one strand of mature miRNA duplex is incorporated into RNA-induced silencing complex (RISC) and guides the complex to target mRNAs, leading to posttranscriptional repression (1-3). It is generally believed that base pairing between seed sequences (nucleotides 2-7) of miRNAs and target mRNAs is critical for target recognition, although there are reports that the seed sequence base pairing may not be sufficient to induce miRNA-mediated translation repression, suggesting the possible involvement of other components in miRNA:mRNA interactions (9-11).
To date, over 600 human miRNAs have been reported, and more than 1000 predicted miRNA genes remain to be experimentally confirmed (12, 13). As a new class of regulatory molecules, miRNAs exert diverse functions in a broad range of biological events, including cell growth, apoptosis and differentiation, as well as viral infection (1, 14-16). Based on computer-aided predictions, miRNAs are estimated to target about one third of the human genes (17), so it is not surprising that miRNAs could play a key role in human malignancy. The role of miRNAs in human cancers was initially demonstrated in chronic lymphocytic leukemia (CLL), where a loss of miR-15a and miR-16-1 due to the 13q14 deletion was found in over half of CLL cases (18). Since then, the deregulation of miRNA expression has been demonstrated in other types of cancers (19-21). Of interest, many miRNAs are located at fragile sites or cancer-associated regions, which may explain why there is a correlation between tumorigenesis and aberrant expression of certain miRNAs (22). Although a large number of miRNAs have been identified to date, the role for many of them in tumorigenesis and their underlying mechanisms remain to be determined.
miR-205 is a highly conserved miRNA among different species (http://microrna.sanger.ac.uk/cgi-bin/sequences/query.pl?terms=mir-205). miR-205 was first predicted by computational methods based on the conservation with mouse and Fugu rubripes sequences (23), and its expression was later validated in zebrafish and human (24, 25). Human miR-205 is located in the second intron of LOC642587 locus in chromosome 1. Tissue specific expression of miR-205 has been previously reported. In zebrafish, miR-205 is expressed in epidermis (26), and in humans, miR-205 is detected in breast, prostate and thymus, suggesting that miR-205 may play a role in the development of these organs (27).
In this study, we reported that miR-205 is downregulated in breast tumor tissues as well as breast cancer cell lines; in contrast, miR-205 is highly expressed in normal breast tissues and non-malignant breast epithelial cell line MCF-10A. These results are in good agreement with the previous report (21). Importantly, ectopic expression of miR-205 cells significantly reduces cellular proliferation, clonogenic survival and anchorage-independent growth in breast cancer MCF-7 cells. In addition, miR-205 is able to suppress invasion and metastasis of breast cancer MDA-MB-231 cells. We further demonstrated that miR-205 can specifically suppress expression of ErbB3 and VEGF-A by directly interacting with the putative miR-205 binding site at the 3’-UTR (3’-untranslated region).
Results
miR-205 is downregulated in breast tumor tissues and breast cancer cell lines
Accumulating evidence indicates that miRNAs can function as oncogenes or tumor suppressors by targeting corresponding tumor suppressor genes or oncogenes (28). To determine the role of miRNAs in breast cancer, we profiled miRNA expression in matched breast tumor specimens by using miRNA TaqMan real-time PCR. We previously showed that the oncogenic miR-21 was more highly expressed in the breast tumors than in the matched normal breast tissues (29). In contrast, we detected substantial downregulation of miR-205 in breast cancer specimens based on our initial profiling. Therefore, we used single miR-205 primer set and probe to confirm the preliminary finding. From a total of 19 pairs of matched breast tumor tissue specimens, average CT (threshold cycle) value for the normal tissue was ~25 whereas the CT value for tumors was ~28 (Fig. 1A). After conversion (30), miR-205 expression level on average was downregulated in breast tumor tissues by ~80% (Fig. 1B) compared to the matched normal tissue. We also examined the miR-205 expression in 9 pairs of matched colon cancer specimens, but found no significant difference in miR-205 expression level between tumors and the matched normal colon tissues (data not shown), suggesting that downregulation of miR-205 may be specific to breast cancer. We then determined miR-205 expression in various breast cancer cell lines along with the non-malignant breast epithelial cell lines MCF-10A. As shown in Fig. 1C, breast cancer cell lines expressed lower levels of miR-205, as compared with MCF-10A cells. These results suggest that miR-205 may function as a tumor suppressor in breast cancer.
Fig. 1. Expression of miR-205 in breast cancer specimens and breast cancer cell lines.
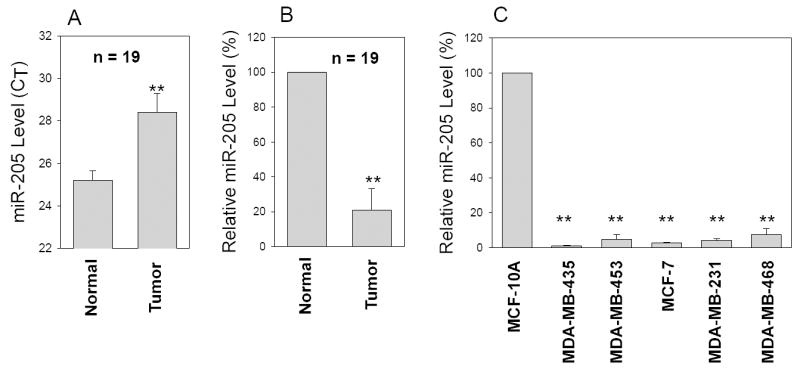
miR-205 expression was determined by TaqMan real-time PCR method as described in Materials and Method. Relative CT value (A) or relative expression level of miR-205 (B) in the matched breast tumors and normal breast tissue. C, Relative expression of miR-205 in breast cancer cell lines compared to non-malignant MCF-10A cells from three separate experiments (mean ± SE). **, p < 0.01.
Growth inhibition of MCF-7 cells by miR-205
To test this hypothesis, we performed proliferation assays in MCF-7 cells with miR-205 overexpression. MCF-7 cells were transfected with either miR-205 expression vector or control vector. Two stable clones #17 and #21 were selected out based on the high level of exogenous miR-205 level (Fig. 2A) compared to a pooled vector control (over 20 clones). MTT assays indicated that cell growth for both miR-205 expressing clones was slower than vector control. For example, clones miR-205 #17 and miR-205 #21 grew at 60% and 30% of the vector control, respectively (Fig. 2B), which appears to be negatively correlated with the exogenous miR-205 level (Fig. 2A). We also tested transiently transfected MCF-7 cells. The result was very similar to that of stable clones (Fig. 3C).
Fig. 2. Suppression of cell growth by miR-205.
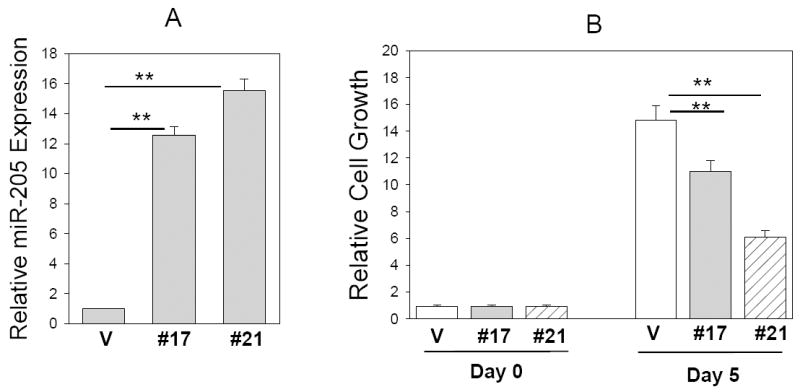
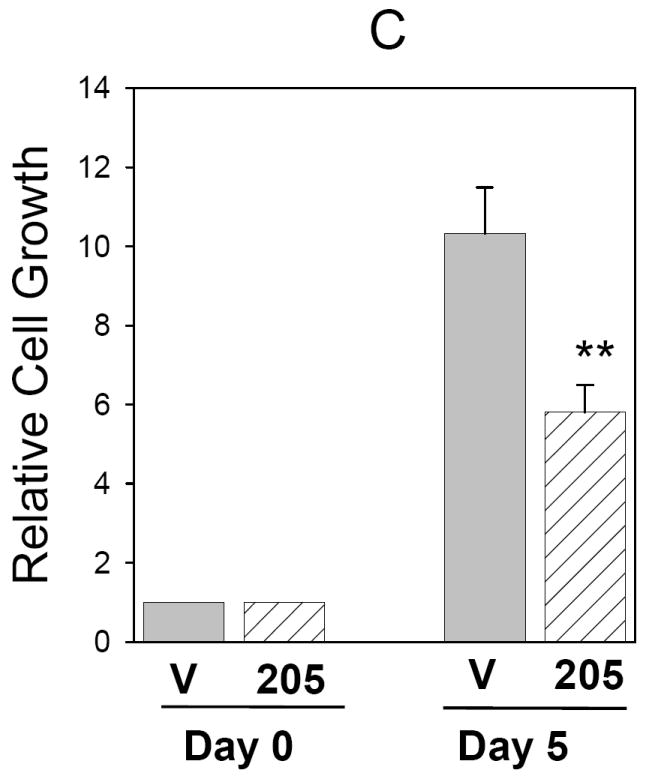
A, Relative expression of miR-205 in stable clones #17 (miR-205 #17) and #21 (miR-205 #21), as detected by TaqMan real time PCR. MTT assays reveal reduced cell growth for these two stable clones compared to vector control (V) (B) as well as for the transiently transfected MCF-7 cells compared to vector control (C). Values are means of three separate experiments ± SE. **, p < 0.01.
Fig. 3. Suppression of clonogenic survival and anchorage independent growth by miR-205.
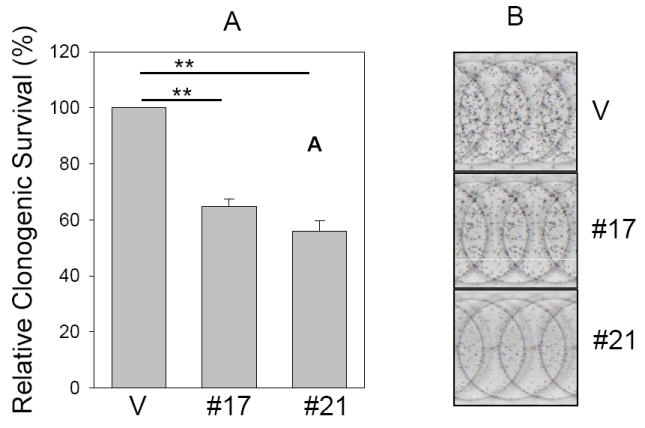
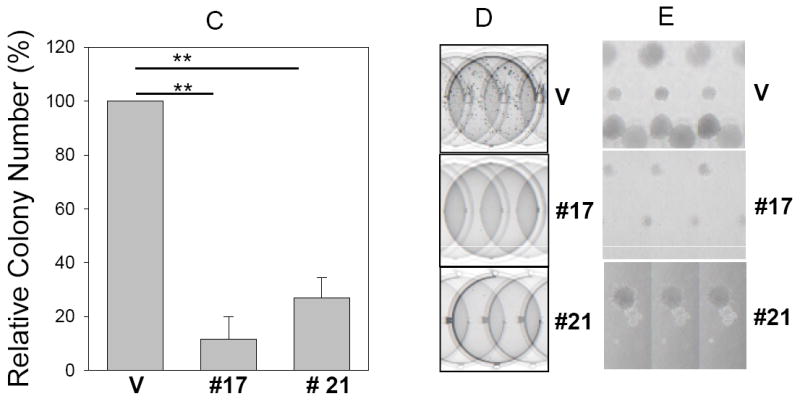
Clonogenic assay (A and B) and soft agar assay (C, D and E) were performed as described in Materials and Methods. B, Representative wells of the colonies. D, Representative fields of colonies in soft agar and E, Closeups of the formed colonies. Note that the colonies are larger in vector control than miR-205 #17 or #21. Values in B and C are means of three separate experiments ± SE. V, vector; #17, miR-205 # 17; #21, miR-205 # 21**, p < 0.01.
miR-205 suppresses clonogenic survival and anchorage-independent growth
To assess whether miR-205 inhibits clonogenic survival, colony formation assay was performed in MCF-7 stable clones. Compared to control vector, the number of colonies for miR-205 #17 and miR-205 #21 was decreased to about 63% and 56% respectively (Fig. 3A and B). Since anchorage-independent growth is strongly correlated with tumorigenicity (31), we then determined whether miR-205 inhibits anchorage-independent growth. Thus, the same MCF-7 stable transfectants were plated in soft agar and incubated for 2 weeks before counting the number of colonies. The ability of miR-205 clones to form colonies in soft agar was drastically decreased in stable transfectants with miR-205 expression compared to vector control (Fig. 3C, D and E).
miR-205 suppresses invasiveness in MDA-MB-231 cells
Based on miRNA profiling data, breast cancers without vascular invasion show high miR-205 expression compared to breast cancers with vascular invasion (21). Furthermore, a recent report indicates that miR-205 is dramatically downregulated in cells undergoing epithelial to mesenchymal transition (EMT) (32). These results suggest that miR-205 may also play a role in cell invasion. To test this hypothesis, we chose a metastatic breast cancer cell line MDA-MB-231 because MDA-MB-231 cells also expressed a low level of miR-205. Matrigel chamber assays indicated that the invasion ability of MDA-MB-231 cells was substantially reduced by miR-205 (Fig. 4 A and B). Since invasion is one of the key steps in metastasis (33), suppression of invasion by miR-205 in MDA-MB-231 cells suggests that miR-205 may also affect breast cancer metastasis. In vivo metastasis assays supported this notion (Fig. 4 C and D). For example, while average number of lung nodules in the miR-205 cells was about 24, this number was only about 2 in the vector control cells.
Fig. 4. miR-205 inhibits cell invasion and metastasis.
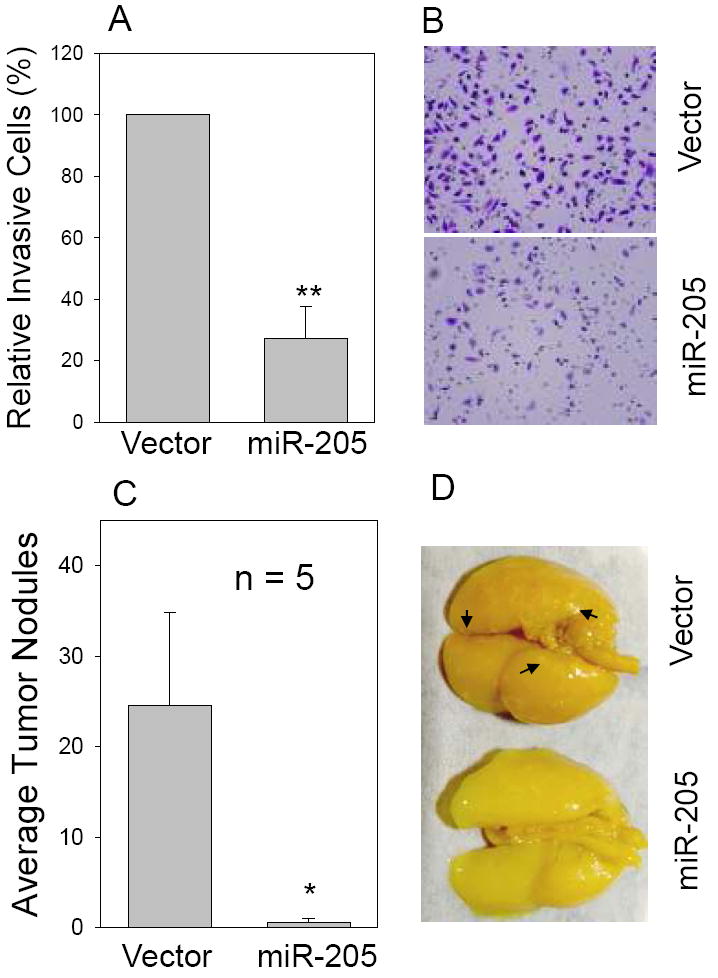
MDA-MB-231 cells were first infected with miR-205 or vector alone and then were tested for invasion ability in matrigel chambers or metastasis in nude mice as described in Materials and Methods. A and B, In vitro cell invasion assays with representative fields of invaded cells. Values in A are means of three experiments ± SE. **, p < 0.01. C, Effect of miR-205 on tumor metastasis. *, p <0.05. D, Representative lungs harvested from the nude mice. Arrows indicate some of tumor nodules.
miR-205 directly targets ErbB3 and VEGF-A expression
To understand the molecular mechanisms by which miR-205 inhibits tumor cell growth and cell invasion, we searched for putative miR-205 targets as predicted by the commonly cited programs such as TargetScan4 (11), miRBase Target5 (http://microrna.sanger.ac.uk/targets/v5/), PicTar (34)and miRanda (http://www.microrna.org) (35). This search identified several genes that are likely associated with tumorigenicity, including K-RAS, ESRRG (estrogen-related receptor gamma), Bcl-2, eIF4E, ErbB3 and VEGF-A. Therefore, we constructed luciferase reporters carrying the 3’-UTR with the putative miR-205 binding sites for each of those genes. Luciferase assays indicated that both ErbB3 and VEGF-A gave rise to a significant reduction of the luciferase activity. The importance of ErbB3 is highlighted by the fact that ErbB3 is frequently overexpressed in breast cancer and ErbB2/ErbB3 heterodimer is the most potent oncogenic complex (36). Moreover, it has been reported that ErbB3 inactivation blocks proliferation of breast cancer cells at ErbB2-independent manner (37). As shown in Fig. 5A, the ErbB3-3’UTR carries a conserved miR-205 binding site. Luciferase assays revealed that the reduction of luciferase activity was in a dose-dependent manner (Fig. 5B). To further confirm that miR-205-mediated reduction of the luciferase activity for Luc-ErbB3-3’UTR vector is due to direct interaction between miR-205 and its putative binding site, we deleted the miR-205 binding site by side-directed mutagenesis, resulting in Luc-ErbB3-3’UTR-d. As expected, miR-205-mediated suppression of the luciferase activity was completely abolished in Luc-ErbB3-3’UTR-d (Fig. 5C), suggesting that the miR-205 binding site is critical for its suppression function. To further test the specificity of miR-205-mediated suppression, we used anti-miR-205 oligo. Since 293T cells express a low level of miR-205, we first established stable clones overexpressing miR-205 and then introduced anti-miR-205 into these stable transfectants. As shown in Fig. 5D, anti-miR-205 enhanced its activity by over a 2-fold compared to scrambled oligo.
Fig. 5. Identification of ErbB3 and VEGF-A as direct targets for miR-205.
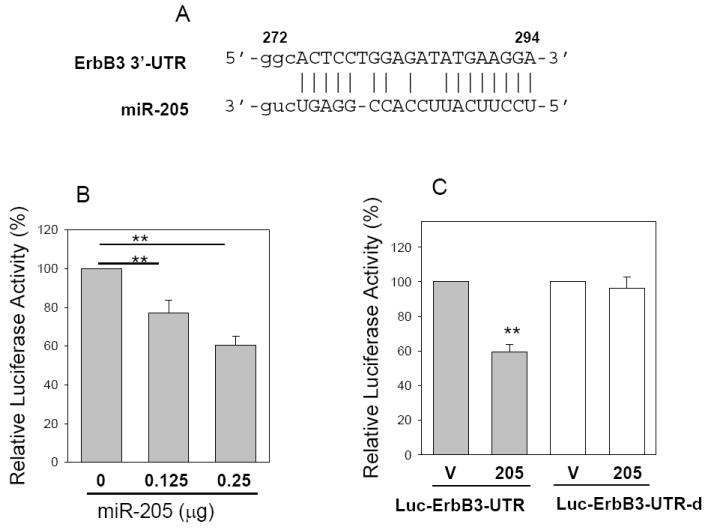
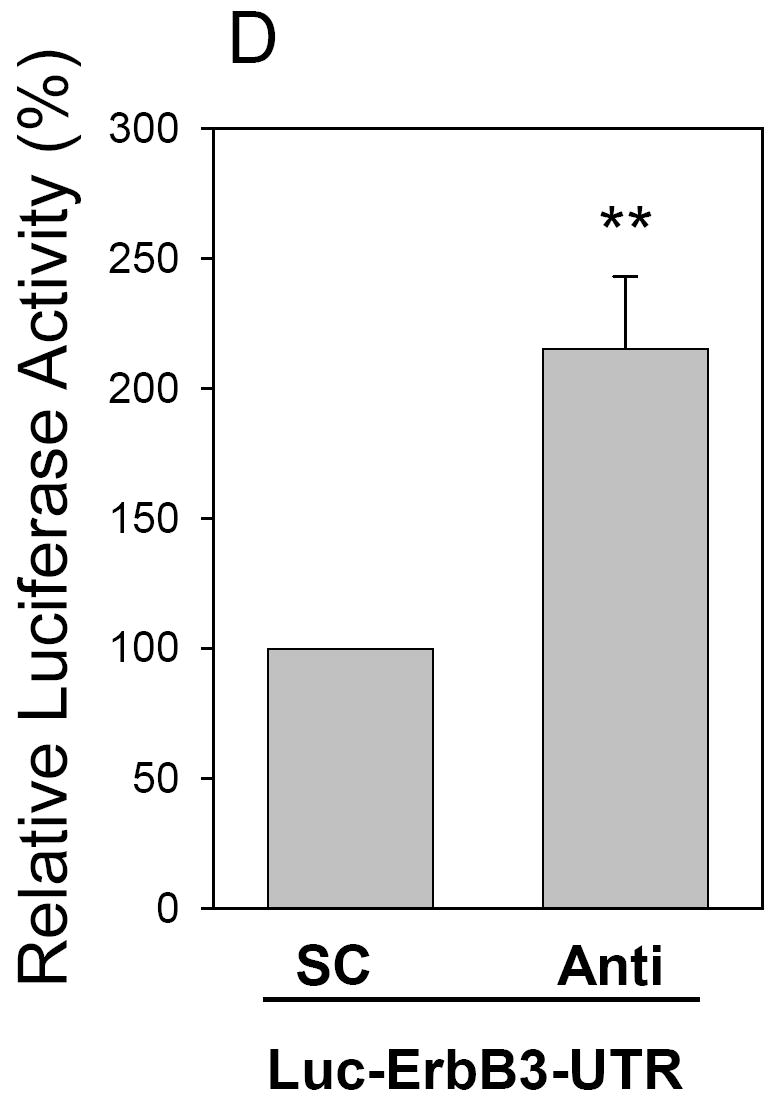
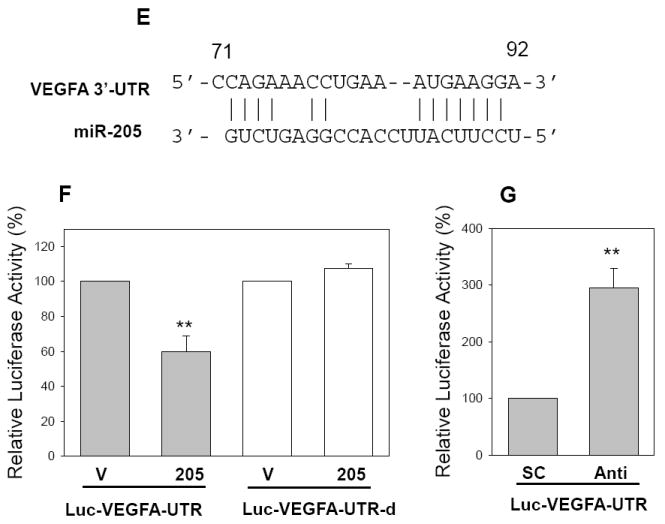
A, Alignment of miR-205 with ErbB3 at the 3’-UTR. B, Suppression of the luciferase activity by miR-205 in a dose-dependent manner. C. While miR-205 suppresses the luciferase activity for Luc-ErbB3-UTR, it has no effect on Luc-ErbB3-UTR-d. D, Anti-miR-205 increases the luciferase activity of Luc-ErbB3-UTR in 293T cells which overexpress miR-205. E, Alignment of miR-205 with VEGF-A at the 3’-UTR. F, Effect of miR-205 on the luciferase activity of Luc-VEGF-A-UTR and Luc-VEGF-A-UTR-d. G, Anti-miR-205 increases the luciferase activity of Luc-VEGF-A-UTR in 293T cells which overexpress miR-205. Values in B, C, D, F and G are means of three experiments ± SE. **, p < 0.01. SC, scrambled oligo; Anti, anti-miR-205.
Likewise, VEGF-A is a key regulator of angiogenesis and is shown to play a key role particularly in tumor metastasis. As shown in Fig. 5 F, we similarly demonstrated that miR-205 directly targets VEGF-A by interacting with the putative miR-205 binding site in its 3’-UTR. Finally, anti-miR-205 also significantly enhanced the activity of Luc-VEGF-A-UTR (Fig. 5G), suggesting the specificity of miR-205-mediated suppression.
To determine whether miR-205 suppresses the endogenous ErbB3, we performed Western blot for transiently transfected MCF-7 cells. As shown in Fig. 6A, miR-205 suppressed significantly the ErbB3 level in MCF-7 cells, compared to vector control. Moreover, we detected even more dramatic reduction of VEGF-A in miR-205 MDA-MB-231 cells compared to vector control (Fig. 6B). To better determine this suppression of ErbB3 at the cellular level, we performed immunofluorescence staining for transiently transfected MCF-7 cells. Since miR-205 expression vector was tagged with GFP, the green cells represented miR-205 expressing cells. As shown in Fig. 6C, ectopic expression of miR-205 clearly suppressed ErbB3 expression (arrows in right panel). In contrast, the vector control had no effect on its expression. Thus, these results further confirmed that miR-205 can negatively regulate the expression of ErbB3.
Fig. 6. Suppression of the endogenous ErbB3 and VEGF-A by miR-205.
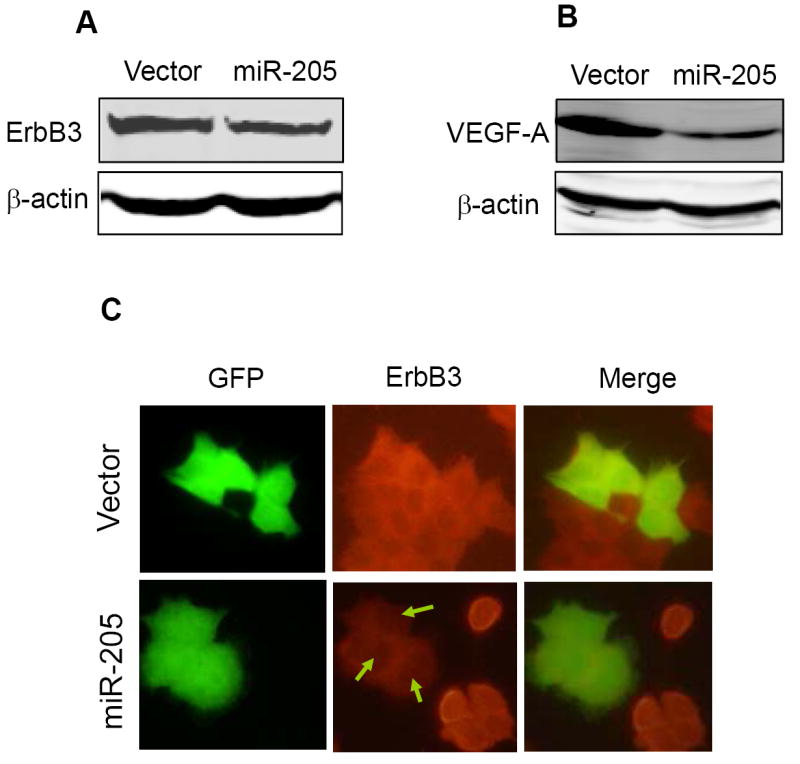
A and B, Western blot revealing reduced level of ErbB3 in miR-205 transfected MCF-7 cells and reduced VEGF-A level in miR-205 MDA-MB-231 cells. C. Detection of ErbB3 in MCF-7 cells by immunofluorescence staining. Note that the ErbB3 signal was very weak in the miR-205 transfected cells in right panel.
Discussion
Growing evidence has indicated that miRNAs are frequently deregulated and aberrant expression of a unique number of miRNAs (miRNA signature) is directly associated with certain type of cancer. Our miRNA profiling suggests that miR-205 is specifically downregulated in breast cancer, but not colon cancer. Moreover, miR-205 expression level is also decreased in breast cancer cell lines compared to the non-malignant breast epithelial cells line MCF-10A, suggesting miR-205 as a potential tumor suppressor in breast cancer. In support of this notion, we demonstrate that ectopic expression of miR-205 suppresses proliferation, colonogenic survival and anchorage-independent growth of MCF-7 cells and invasiveness in MDA-MB-231 cells. Therefore, miR-205 modulates not only breast tumor cell growth, but also cell invasion and metastasis.
While miR-205 is downregulated in breast cancer, it has been shown to be upregulated in various types of cancers, including lung cancer, bladder cancer, ovarian cancer and head and neck cancer cell lines (38-42). In colon cancer, we detected no significant difference in miR-205 expression between tumor and the matched normal tissue. These findings along with this study may imply that miR-205 could play a dual role in tumorigenicity, depending on tissue type and specific targets. In fact, such a dual nature of miRNAs in tumorigenicity has been previously reported. For instance, miR-155 is considered as an oncogene in many cancers because of its upregulation in these cancers, but the expression of miR-155 is significantly decreased in endocrine pancreatic tumors (43). Studies on the miR-17~92 cluster have revealed that this miRNA cluster has tumor suppressive function by inhibiting E2F1 expression in human B-cell line, but the same miRNA cluster is overexpressed and acts as an oncogene by blocking apoptosis in B-cell lymphomas (44, 45). By using LNA-based microarrays and Northern blot analyses to measure miRNA levels in a collection of breast epithelial cell lines, Sempere et al demonstrated that miR-205 was consistently downregulated in tumorigenic cell lines (46), in good agreement with our finding. Given the opposite expression levels of miR-205 in different cancers, and the inhibitory functions of miR-205 to the proliferation, anchorage-independent growth and invasion ability of breast cancer cell lines, we suggest that miR-205 is a breast cancer specific tumor suppressor.
ErbB3 is a member of ErbB tyrosine kinase receptor family, which is frequently overexpressed in breast cancer. It is well known that there is a correlation between ErbB3 expression levels and tumorigenesis in breast cancer (47, 48). Because ErbB3 has an impaired tyrosine kinase activity, heterodimerization is required for ErbB3 activation and downstream signaling transduction (36, 49). By heterodimerizing with other family members, especially ErbB2, ErbB3 contributes to ErbB2-mediated proliferation and anti-cancer drug resistance of breast cancer cells (37, 50). Co-expression of ErbB2/ErbB3 heterodimer is a poor prognostic indicator. Therefore, identification of ErbB3 as a direct target for miR-205 may imply that miR-205 is a novel target for breast cancer therapy.
Apparently, ErbB3 can also be targeted by other miRNAs than miR-205. For example, a recent report indicates that both miR-125a and miR-125b can suppress ErbB3 expression by interacting with the same miR-125 binding site located in the ErbB3 3’-UTR (51), suggesting that multiple miRNAs may have an additive or synergetic effect on regulation of gene expression. In addition, lung metastasis can be enhanced by increasing ErbB3-dependent signaling in orthotopic injection models of breast cancer (52). On the other hand, since a single miRNA can have multiple targets, it is very likely that miR-205 may also regulate other genes simultaneously to inhibit breast tumor growth. For example, miR-205 also targets VEGF-A, an important invasion and metastasis factor. Although VEGF-A is well known for its role in angiogenesis and tumor metastasis, the reduced level of VEGF-A could also contribute to the observed suppression of cell invasion in vitro (Fig. 4) because a previous report indicates that VEGF-A is able to enhance cell invasion in vitro (53). In addition to these two targets we identified in this study, miR-205 may target other targets yet to be identified. Therefore, the observed miR-205-mediated inhibition of breast tumor growth and metastasis is likely due to simultaneous targeting of multiple targets.
Materials and Methods
Cell Culture
All cell lines used in this study were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). MCF-7, MDA-MB-231, MDA-MB-453 and MDA-MB-468 cells were grown in RPMI 1640 (Cambrex, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), MDA-MB-435 cells were grown in Dulbecco’s modified Eagle’s medium (Cambrex) supplemented with 10% fetal bovine serum plus 0.01 mg/ml insulin. MCF-10A cells were grown in Mammary Epithelial Basal Medium (MEBM) (Cambrex). 293T cells were grown in Dulbecco’s modified Eagle’s medium (Cambrex) supplemented with 10% FBS. All media were supplemented with 2 mM glutamine, 100 units of penicillin/ml, and 100 μg of streptomycin/ml (Cambrex). Cells were incubated at 37°C in a humidified chamber supplemented with 5% CO2.
Constructs
To generate miR-205 expression vector, a ~600 bp fragment carrying pre-miR-205 was amplified from MCF-10A genomic DNA by the high fidelity polymerase Phusion enzyme (New England Biolabs, Ipswich, MA) using PCR primers
miR-205-5.1, 5’-GAATTCCTTATCTGGGTGGCTGTTTTG
miR-205-3.1, 5’-GGTACCGCGGTGCTTTTTCCAATCTGC
The amplified fragment was first cloned into pCR8 (Invitrogen) and then subcloned into a modified pCMV vector carrying hygromycin resistance gene. To construct miR-205 lentiviral expression vector, the same miR-205 insert was released from pCMV vector and then subcloned into pCDH-CMV-MCS-EF1-copGFP (System Biosciences, Mountain View, CA).
To generate a luciferase reporter carrying ErbB3 3’-UTR with a putative miR-205 binding site, we amplified a 638 bp ErbB3 3’-UTR region from MCF-7 cDNA using the following PCR primers,
ErbB3-UTR-5.1, 5’-CTCCTGCTCCCTGTGGCAC
ErbB3-UTR-3.1, 5’-CCCGACTTCCCTTTGTGTAAAATG
The amplified fragment was first cloned into pCR8 vector and subsequently cloned into a modified pGL3-control vector (54) at the EcoR1 site. To delete the putative miR-205 binding site, we adopted two-step PCR methods as described previously (54). Additional primers used were
ErbB3-3’UTR-5.2, 5’-CATTCCTCAGCTTCTTCACATTACTCTCCATATCCCTTCC
ErbB3-3’UTR-3.2, 5’- GGAAGGGATATGGAGAGTAATGTGAAGAAGCTGAGGAATG.
The same procedure was used to clone Luc-VEGF-A-UTR and Luc-VEGF-A-UTR-d where the putative miR-205 binding site was deleted. PCR primers were
VFGFA-UTR-5.1, 5’-TTTCGGGAACCAGATCTCTC;
VEGFA-UTR-3.1, 5’-GCGGCCGCTCTTCCCTGTCAGGATCTG;
VEGFA-UTR-Del-5.1, 5’-CCATCGACAGAACAGTCCTTAAGAGGAGACTCTGCGCAGAG
VEGFA-UTR-Del-3.1, 5’-CTCTGCGCAGAGTCTCCTCTTAAGGACTGTTCTGTCGATGG
All the PCR products were verified by DNA sequencing.
Detection of miR-205 by real-time RT-PCR
To detect mature miR-205 expression in patient specimens, we used Trizol reagent to isolate total RNA, which was then amplified by TaqMan stem-loop RT-PCR method (30). TaqMan microRNA assays used Human Panel-Early Access Kit (ABI, Forest City, CA) which includes 157 human miRNAs as well as 3 negative controls. We also used individual miR-205 specific primer sets and TaqMan probe from ABI to detect miR-205 expression in patient specimens and cell lines.
Generation of miR-205 stable clones
To establish miR-205 stable clones, MCF-7 cells were transfected with vector control or miR-205 expression vector and were then selected in the presence of hygromycin at 200 μg/ml concentration. Individual colonies were expanded and expression of the exogenous miR-205 over the endogenous counterpart (vector alone) was determined by real-time RT-PCR.
Cell proliferation assay
Either transiently transfected cells or stable clones were seeded into 96-well plates at 1000 cells/well. Relative cell growth rate was determined by MTT assay as described previously (54).
Clonogenic survival and anchorage-independent growth
To determine clonogenic survival of miR-205 stable clones, cells from either vector control or miR-205 were seeded on 6-well plates at 500 cells/well. Ten days after seeding, colonies were fixed and stained with 0.1% crystal violet. Anchorage-independent growth of miR-205 stable transfectants was tested according to a published method (55). Briefly, 1 ml culture medium with 0.4% agar was first plated into each well of a 12-well plate. After the bottom agar became solidified, each well received another 1 ml 0.3% agar in culture medium carrying 5000 cells. Colonies were stained and counted after 2-weeks of incubation.
Luciferase assay
Luciferase reporters were transfected into 293T cells along with either vector control or miR-205 expression vector by using the calcium phosphate method as described previously (56). Cells were harvested 48 hr after transfection for luciferase assay using a luciferase assay kit (Promega) according to the manufacturer’s protocol. β-galactosidase was used for normalization of transfection efficiency as described previously (54).
Western blot
Total protein was extracted from transient transfected MCF-7 cells or miR-205 stable clones by using cell lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.2, 0.1% SDS, 1.0% Triton X-100, 1% Deoxycholate, 5 mM EDTA). Protein concentration was measured with the Bio-Rad protein assay kit. The membrane was first probed with ErbB3 antibody (Cell Signaling Technology, Danvers, MA), followed by secondary antibodies labeled with IRDye800 (Rockland Immunochemicals, Gilbertsville, PA). Signals were visualized using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
Immunofluorescence microscopy
To detect the effect of miR-205 on ErbB3 expression at the cellular level, MCF-7 cells were transfected with either miR-205/pCDH-CMV-MCS-EF1-copGFP or control vector, pCDH-CMV-MCS-EF1-copGFP. Immuno detection was performed as described previously (57). In brief, transfected cells were seeded on cover slides 24 h after transfection, and further incubated for 96 h before fixing with 3% paraformaldehyde. After incubation with primary ErbB3 antibody, second antibody conjugated with Alexa Fluor 560 nm (Invitrogen) was used to reveal ErbB3 signals. Images were taken under a fluorescent microscope (Olympus, Center Valley, PA).
Invasion assay
Effect of miR-205 on the invasion ability of MDA-MB-231 cells was determined using matrigel invasion chambers (BD Biosciences). Cells infected with either miR-205 expression or control lentiviral vectors were seeded into inserts at 2 × 104 per insert in serum-free medium and then transferred to wells filled with the culture medium containing 10% FBS as a chemoattractant. After 24 h incubation, non-invading cells on the top of the membrane were removed by scraping. Invaded cells on the bottom of the membrane were fixed, followed by staining with 0.05% crystal violet. The number of invaded cells on the membrane was then counted under a microscope.
Tumor metastasis in vivo
Animal studies were conduct to determine the effect of miR-205 on breast tumor metastasis, accordance with NIH animal use guidelines and a protocol approved by SIU Animal Care Committee. The procedure involving the metastatic breast cancer cell line MDA-MB-231 cells has been previously described (58).
Patient specimens
Total RNA was isolated using Trizol reagent from freshly frozen specimens of matched breast tumors from the same patient, which were obtained from Cooperative Human Tissue Network (CHTN) Midwestern Division (Columbus, OH). The isolated RNA was directly used for TaqMan real time PCR to detect expression of miRNAs.
Acknowledgments
This study was supported by grants to Y Mo from the National Cancer Institute (CA102630) and from Department of Defense (BC052294).
Abbreviations
- EMT
epithelial to mesenchymal transition
- miRNA
microRNA
- PCR
polymerase chain reaction
- RT
reverse transcription
- UTR
untranslated region
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Mallory AC. Vaucheret H Functions of microRNAs and related small RNAs in plants. Nat Genet. 2006;38(Suppl):S31–36. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- 3.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 4.Bracht J, Hunter S, Eachus R, Weeks P, Pasquinelli AE. Trans-splicing and polyadenylation of let-7 microRNA primary transcripts. Rna. 2004;10:1586–1594. doi: 10.1261/rna.7122604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 10.Didiano D, Hobert O. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat Struct Mol Biol. 2006;13:849–851. doi: 10.1038/nsmb1138. [DOI] [PubMed] [Google Scholar]
- 11.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 14.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 15.Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, et al. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 16.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 17.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 18.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 20.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67:6130–6135. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 21.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 22.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- 24.Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, et al. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 25.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ason B, Darnell DK, Wittbrodt B, Berezikov E, Kloosterman WP, Wittbrodt J, et al. Differences in vertebrate microRNA expression. Proc Natl Acad Sci U S A. 2006;103:14385–14389. doi: 10.1073/pnas.0603529103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shingara J, Keiger K, Shelton J, Laosinchai-Wolf W, Powers P, Conrad R, et al. An optimized isolation and labeling platform for accurate microRNA expression profiling. Rna. 2005;11:1461–1470. doi: 10.1261/rna.2610405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Engl J Med. 2005;353:1768–1771. doi: 10.1056/NEJMp058190. [DOI] [PubMed] [Google Scholar]
- 29.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colburn NH, Bruegge WF, Bates JR, Gray RH, Rossen JD, Kelsey WH, et al. Correlation of anchorage-independent growth with tumorigenicity of chemically transformed mouse epidermal cells. Cancer Res. 1978;38:624–634. [PubMed] [Google Scholar]
- 32.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 33.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 34.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 35.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naidu R, Yadav M, Nair S, Kutty MK. Expression of c-erbB3 protein in primary breast carcinomas. Br J Cancer. 1998;78:1385–1390. doi: 10.1038/bjc.1998.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P, et al. Micro-RNA profiling in kidney and bladder cancers. Urol Oncol. 2007;25:387–392. doi: 10.1016/j.urolonc.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Tran N, McLean T, Zhang X, Zhao CJ, Thomson JM, O’Brien C, et al. MicroRNA expression profiles in head and neck cancer cell lines. Biochem Biophys Res Commun. 2007;358:12–17. doi: 10.1016/j.bbrc.2007.03.201. [DOI] [PubMed] [Google Scholar]
- 41.Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–8707. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 42.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 43.Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–4684. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 44.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 45.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, et al. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007;67:11612–11620. doi: 10.1158/0008-5472.CAN-07-5019. [DOI] [PubMed] [Google Scholar]
- 47.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL., 3rd Insect cell- expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A. 1994;91:8132–8136. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim HH, Vijapurkar U, Hellyer NJ, Bravo D, Koland JG. Signal transduction by epidermal growth factor and heregulin via the kinase-deficient ErbB3 protein. Biochem J. 1998;334(Pt 1):189–195. doi: 10.1042/bj3340189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lemoine NR, Barnes DM, Hollywood DP, Hughes CM, Smith P, Dublin E, et al. Expression of the ERBB3 gene product in breast cancer. Br J Cancer. 1992;66:1116–1121. doi: 10.1038/bjc.1992.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD. Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer. 2007;120:1874–1882. doi: 10.1002/ijc.22423. [DOI] [PubMed] [Google Scholar]
- 51.Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007;282:1479–1486. doi: 10.1074/jbc.M609383200. [DOI] [PubMed] [Google Scholar]
- 52.Xue C, Liang F, Mahmood R, Vuolo M, Wyckoff J, Qian H, et al. ErbB3-dependent motility and intravasation in breast cancer metastasis. Cancer Res. 2006;66:1418–1426. doi: 10.1158/0008-5472.CAN-05-0550. [DOI] [PubMed] [Google Scholar]
- 53.Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002;62:7203–7206. [PubMed] [Google Scholar]
- 54.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 55.Finlay TH, Tamir S, Kadner SS, Cruz MR, Yavelow J, Levitz M. alpha 1-Antitrypsin- and anchorage-independent growth of MCF-7 breast cancer cells. Endocrinology. 1993;133:996–1002. doi: 10.1210/endo.133.3.8365378. [DOI] [PubMed] [Google Scholar]
- 56.Mo YY, Beck WT. Association of human DNA topoisomerase IIalpha with mitotic chromosomes in mammalian cells is independent of its catalytic activity. Exp Cell Res. 1999;252:50–62. doi: 10.1006/excr.1999.4616. [DOI] [PubMed] [Google Scholar]
- 57.Wu F, Chiocca S, Beck WT, Mo YY. Gam1-associated alterations of drug responsiveness through activation of apoptosis. Mol Cancer Ther. 2007;6:1823–830. doi: 10.1158/1535-7163.MCT-06-0771. [DOI] [PubMed] [Google Scholar]
- 58.Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]