

Abstract
Peptide nucleic acids (PNAs) are DNA mimics in which peptide-like linkages are substituted for the phosphodiester backbone. Homopyrimidine PNAs can invade double-stranded DNA containing the homologous sequence by displacing the homopyrimidine strand from the DNA duplex and forming a PNA/DNA/PNA triplex with the complementary homopurine strand. Among biologically interesting targets for triplex-forming PNA are (GAA/CTT)n repeats. Expansion of these repeats results in partial inhibition of transcription in the frataxin gene, 5causing Friedreich’s ataxia. We have studied PNA binding and its effect on T7 RNA polymerase transcription in vitro for short repeats (n=3) and for long repeats (n=39), placed in both possible orientations relative to the T7 promoter such that either the GAA-strand, or the CTT-strand serves as the template for transcription. In all cases PNA bound specifically and efficiently to its target sequence. For the short insert, PNA binding to the template strand caused partial transcription blockage with well-defined sites of RNA product truncation in the region of the PNA-binding sequence, whereas binding to the non-template strand did not block transcription. However, PNA binding to long repeats, whether in the template or the non-template strand, resulted in a dramatic reduction of the amount of full-length transcription product, although in the case of the non-template strand there were no predominant truncation sites. Biological implications of these results are discussed.
INTRODUCTION
Peptide nucleic acids (PNAs) are DNA mimics in which electrically neutral peptide-like linkages are substituted for the negatively charged phosphodiester backbone, while the bases are the same as in DNA or RNA (including possible base modifications) ([1]; reviewed in [2,3]). PNA has superior nucleic acid binding properties relative to those of natural nucleic acids, primarily because of the absence of electrostatic repulsion between the PNA backbone and the backbones of target nucleic acids. PNA is thus a potentially efficient tool for artificial regulation of DNA transactions. The documented effects of PNA include inhibition of DNA transcription [4], facilitation of DNA oligonucleotide invasion [5,6], mutagenesis [7,8] and creation of artificial promoters by DNA-opening [9].
Especially stable hybrids with DNA (or RNA) are formed by homopyrimidine PNAs. These PNAs can invade double-stranded DNA containing the homologous sequence by displacing the homopyrimidine strand from the DNA duplex and forming a PNA/DNA/PNA triplex with the complementary homopurine strand, where one PNA strand interacts with a DNA strand via Watson-Crick hydrogen bonding, and the other via Hoogsteen hydrogen bonding (reviewed in [2,3,10]). To form Hoogsteen bonds, cytosines must be protonated, thus DNA invasion by cytosine-containing PNA increases with acidity [11].
PNA modifications have been developed that increase the efficiency of PNA-DNA hybridization. Those include (i) making “bis PNAs”, where two PNA molecules, one interacting with DNA through Watson-Crick bonds, and the other through Hoogsteen bonds are tethered together by a flexible linker and (ii) substituting pseudoisocytosine for cytosine in the Hoogsteen-interacting half of bis PNA. Pseudoisocytosine at neutral pH contains hydrogen at the same position at which the cytosine is protonated at acidic pH; this substitution thus relaxes the requirement for an acidic pH for efficient DNA invasion [12].
Among the biologically relevant targets for triplex-forming PNA are (GAA/CTT)n repeats. Several GAA/CTT repeats are normally present in intron 1 of the frataxin gene. Expansion of these repeats results in partial inhibition of transcription in that gene, causing the progressive neurodegenerative disease called Friedreich’s ataxia. Both repeat expansion and transcription inhibition have been hypothesized to be due to an unusual DNA secondary structure formed by this sequence (reviewed in [13–15], although in the case of transcription, other mechanisms have been suggested [16]). Various DNA binding agents have been shown to alleviate the inhibitory effect on transcription [17,18]. Another approach to alleviate the deleterious effect of the expanded repeats could be through mutagenesis within the repeats to disrupt their regularity and/or make them shorter due to deletions. For example, it has been shown that interruptions in (GAA/CTT)n repeats alleviate transcription inhibition and reduce genetic instabilities [19].
PNA is expected to facilitate mutations upon its binding to DNA, both by creating a structural distortion (in particular, a single-stranded region) that could attract various DNA-modifying enzymes, and by creating an obstacle for enzymes acting on DNA, in particular RNA polymerase (RNAP). RNAP blockage is a signal for transcription-coupled repair (TCR), a dedicated pathway to repair lesions that interfere with transcription [20–22]. TCR might be also be induced by RNAP blockage in undamaged DNA (gratuitous TCR), and mutations could be accumulated in these DNA regions due to futile cycles of repair replication [23]. The sources of RNAP blockage in undamaged DNA could be various unusual DNA structures that interfere with transcription [24–33]. Reports that triplex-induced mutagenesis [34] and instability of CAG repeats in human cells [35] are facilitated by proteins that play a role in TCR supports the notion of TCR involvement. Thus, there could be at least two potential biological consequences of the effect of a DNA binding agent on transcription; a direct effect on yield of the gene product and a possible increase in mutagenesis in the transcribed region in the case of transcription blockage.
Here we have analyzed PNA binding and its effect on T7 RNA polymerase transcription in vitro through Friedreich’s ataxia trinucleotide repeats.
MATERIALS AND METHODS
DNA substrates
Plasmids pFR(1/2) and pFR(3/4) have a nine bp long PNA binding sequence cloned into the HindIII/EcoRI sites of pBluescript SK+ in different orientations:
pFR(1/2): ….aagcttGAAGAAGAAttc…..
pFR(3/4): ….aagCTTCTTCTTgaattc….
where the PNA-binding motive is shown in upper case, and HindIII (left) and EcoRI (right) restriction sites are underlined. Only the non-template strands are shown (5′-3′).
Plasmids pBlue-CTT39 and pBlue-iCTT39 were obtained by subcloning of the fragment from the plasmid pYES-TTC57 [36], which was a generous gift from Dr. Sergei M. Mirkin, into the multiple cloning site of the pBluescript SK(+) vector downstream from the T7 promoter. They contain (GAA/CTT)39 inserts in opposite orientations relative to the T7 promoter. (The repeats that we obtained were shorter than in the source plasmid pYES-TTC57, most likely due to spontaneous deletions during subcloning). The sequence of these plasmids (non-template strand, 5′-3′) between the Kpn I and Xba I sites (underlined) of the multiple cloning site are: ggtaccgggccccccctcgaggtcgacggtatcgataagcttgat(CTT)39catcgaatttcgagcatgcatctaga and ggtaccgggccccccctcgtctagatgcatgctcgaaattcgat(GAA)39gatcaagcttgatatcgaattcctgcagcccgggggatccactagttctaga for plasmids pBlue-CTT39 and pBlue-iCTT39, respectively. Plasmids were propagated in DH5α E. coli cells (Invitrogen), and purified using a standard maxiprep Qiagen protocol, except that the time of the cell lysis step was reduced to several seconds.
In most cases, for transcription the plasmids were linearized by AflIII and gel-purified. For the gel-purification, the Qiaquick gel purification protocol (Qiagen) was used with the following modifications: (i) To avoid ethidium bromide (EthBr) contamination or UV exposure of the transcription substrates, preparative gels were run without EthBr. The DNA bands of interest were localized and gel purified by comparison with a gel slice containing small amount of the sample that had been cut from the same gel, stained with EthBr and visualized by UV. (ii) The agarose dissolving step of purification was performed for 40 min at room temperature instead of 10 min at 50°C.
Relaxed circular closed plasmids were obtained by treatment with calf thymus topoisomerase I, followed by phenol-chloroform-isoamyl alcohol (25:24:1) extraction, chloroform extraction and ethanol precipitation. Then the samples were rinsed twice with 70 % ethanol, dried and dissolved in TE buffer (10 mM TrisHCl (pH 7.9), 1 mM EDTA). Because the pBluescript plasmid and its derivatives are phagemids, they could be recovered in single-stranded circular form. In our case with pBluescript SK(+) the recovered single-strand was a template strand for the T7 promoter. Single-stranded circular DNA was prepared using essentially a standard Stratagene protocol (Stratagene manual on line) and it was gel-purified as described above. Because single-stranded DNA migrates in the gel closer to the supercoiled DNA of the same size than to linear double-stranded plasmid, we restricted the sample with ScaI before gel loading to convert any small amounts of double-stranded plasmid DNA that could be present, into the linear form, for better separation on gels.
To linearize single-stranded circular DNA, 10 ng/μl (10 nM) of single-stranded circular DNA was incubated with 100 nM of the corresponding restriction oligo (EcoRI oligo: GCTTGATATCGAATTCCTGCAGCCC; AflIII oligo: GCAGGAAAGAACATGTGAGCAAAAGG, restriction sites are underlined) in 12 μl of 1.1 × NEBuffer 2 (New England Biolabs) for 10 min at 50°C and then for 3 h at 37°C. Then 0.7 μl of 20 × BSA and 1 μl of corresponding restriction enzyme were added. The restriction reaction was incubated for 4 h at 37°C, and then the enzyme was inactivated by incubation for 20 min at 80°C. To hybridize a promoter oligo, 4 μl of the obtained mixture were mixed with 2 μl of 1 μM solution of the promoter oligo, and incubated in 10 μl of NEbuffer 2 under the same conditions as for hybridization with the restriction oligo. For pBlue-iCTT39 we used the promoter oligo TAATACGACTCACTATAGGGCGAATTGGGTACCGGGCCCCCCCTCGTCTAGATGCAT GCTCGAAATTCGAT, which covers the entire length from the 5′-end of the T7 promoter to the start of the (CTT)39 insert. For the vector plasmid pBluescriptSK(+), we used the oligo TAATACGACTCACTATAGGGCGAATTGGGTACCGGGCCCCCCCTCGAGGTCGACGG TATCGATA. The T7 promoter sequence is underlined.
For extension of the annealed promoter oligo, 1.6 μl of the mixture from the previous step were mixed with 1.6 μl of solution, containing 2 × NEBuffer 2, 66 μM of dNTPs, and 0.64 units of Klenow enzyme (New England Biolabs). The reaction was incubated for 40 min at room temperature, and then the enzyme was inactivated for 20 min at 75°C. The control sample had the same components except for the Klenow enzyme. For transcription, 1μl of the sample was used.
PNA binding
PNA Lys-TTJTTJTT-OOO-TTCTTCTT-Lys was purchased from Panagene (Daejeon, Korea). Lys is lysine, J is pseudoisocytosine, O is 8-amino-3,6-dioxaoctanoic acid. The PNA was dissolved in ultrapure water (Gibco) to form a 100 μM solution and kept at − 80°C.
PNA-DNA binding (invasion) reactions were at 37°C for 2 hours. The concentrations during incubation were: target DNA, 5 nM (10 ng/μl for 3 kb plasmid); PNA, 10 μM; TrisHCl (pH 7.9) 8 mM; EDTA, 0.8 mM. In the case of negatively supercoiled DNA, the reaction mixture also contained 40 mM NaCl. Other concentrations of NaCl are indicated in the corresponding figures. For the transcription reaction (see below) 1 μl of this mixture was used.
In vitro T7 RNA polymerase transcription
Reactions were performed in 12 μl of buffer containing 33 mM TrisHCl (pH 7.9), 5 mM MgCl2, 8.3 mM NaCl, 1.7 mM spermidine, 4.2 mM DTT, 0.17 mM each ATP, CTP and UTP, 0.017 mM GTP, 10 μCi [α-32P] GTP, 20 units of T7 RNA polymerase and 16 units of RNasin (both from Promega corp, Madison, WI) for 30 min at 37°C. The reaction was stopped by adding 94 μl of buffer containing 1% SDS, 106 mM TrisHCl (pH 7.6), 13.2 mM EDTA, 160 mM NaCl, 25 μg tRNA (Invitrogen) and 10 μg Proteinase K (Invitrogen) and incubated at room temperature for 15 min. Next, 11 μl of 3 M NaOAc (pH 5.27) and 300 μl of 100 % ethanol (cooled at −20°C) were added, the mixture was incubated on dry ice for 20 min and centrifuged for 20 min at 14,000 rpm at 4°C. The supernatant was removed, then pre-cooled 75 % ethanol was added, and the mixture was centrifuged for 5 min under the same conditions. The supernatant was removed, and the pellets were dried with a SpeedVac for 10 min and dissolved in 8 μl of the formamide loading solution (94 % formamide, 2 mM EDTA, 0.05 % bromophenol blue, 0.05 % xylene cyanol). Then, 2 μl of the obtained solution were mixed with 2 μl of formamide loading solution, incubated at 85°C for 3 min, quickly chilled on ice or a pre-cooled rack and loaded on a 8 % or 10 % sequencing gel (acrylamide:bis-acrylamide 29:1) containing 8 M urea and run at 70 V/cm. As size markers, 5′-end labeled denatured DNA ladders consisting of DNA fragments of sizes increasing in steps of 100 or 10 bases were used. Then, the gel was dried and exposed to a phosphoimager screen.
RESULTS
PNA binding to the short repetitive sequence
The scheme of the hybrid formed between PNA and the target DNA is shown in Fig. 1A. To monitor PNA binding to the short repetitive sequence (GAA/CTT)3 we took advantage of the fact that PNA binding inhibits restriction enzyme cleavage, provided that the restriction site (EcoRI and HindIII in our case, see Materials and Methods) partially overlaps the PNA binding site [37]. Nearly complete inhibition of the cleavage of substrates pre-bound with PNA occurred both for negatively supercoiled and for linear substrates, indicating near-100 % PNA binding to its recognition sequence (Fig. 2).
Fig.1. PNA-DNA hybrid and DNA substrate used in the transcription experiments.
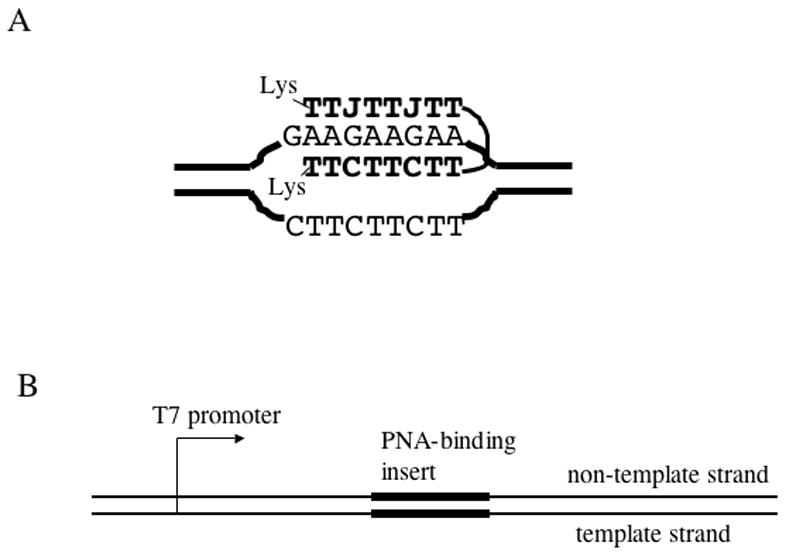
A: PNA-DNA hybrid. PNA bases are shown in bold. PNA binds the homopurine DNA strand, displacing the homopyrimidine DNA strand. The bottom PNA strand is bound to DNA via Watson-Crick hydrogen bonds, and the top PNA strand is bound to DNA via Hoogsteen hydrogen bonds. In the top strand, cytosines are replaced by pseudoisocytosines (J) which, in contrast to cytosines, do not require protonation for Hoogsteen base pairing. The top and the bottom PNA strands are connected by a flexible linker of three residues of 8-amino-3,6-dioxaoctanoic acid. B: Typical DNA substrate used in the transcription experiments. The PNA binding insert was cloned in each of the two possible orientations.
Fig. 2. Detection of PNA binding by protection from restriction enzyme cleavage.
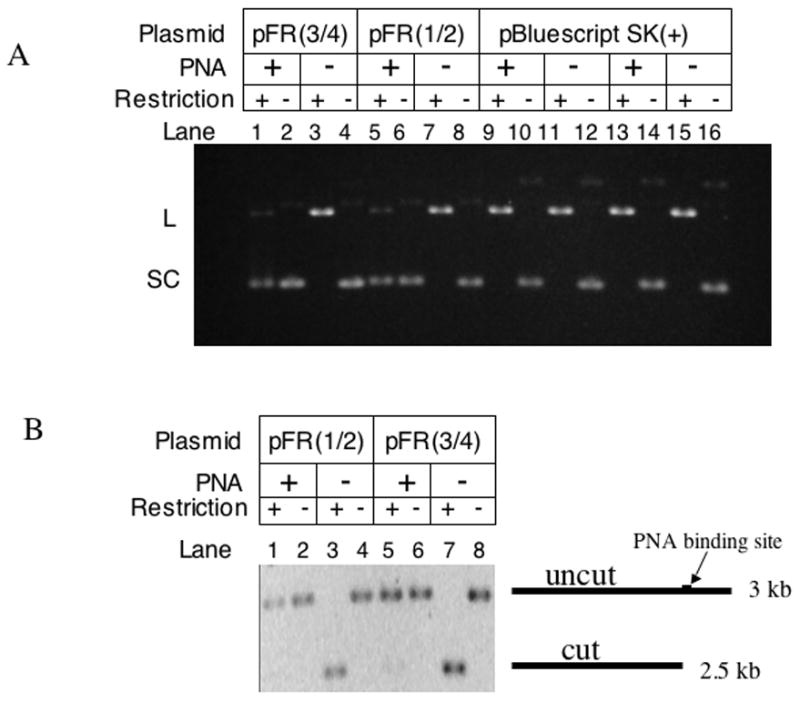
Agarose gel electrophoresis of various plasmids was carried out with and without PNA binding and restriction digestion. A: Supercoiled (SC) DNA substrates were converted to linear (L) upon cleavage by restriction digestion. In pFR(3/4) and pFR(1/2), the Hind III and EcoRI sites, respectively, are partially within the PNA binding sequence, thus HindIII and EcoRI were used for pFR(3/4) and pFR(1/2), respectively. For the vector plasmid pBluescript SK(+), both HindIII (lanes 9 and 11) and EcoRI (lanes 13, 15) demonstrate the absence of non-specific PNA effect on these enzymes. It can be seen that PNA protected the majority of its targets from cleavage (lanes 3 and 5). B: Similar experiments with linear DNA substrates.
PNA binding to the long repetitive sequence
In the case of long repeats, we monitored the shift of plasmid topoisomer distribution upon PNA binding to closed circular DNA. PNA unwinds DNA upon binding to its recognition sequence, which in the case of the circular closed target plasmid creates a compensatory overwinding in the rest of the plasmid [38]. Thus PNA binding converts relaxed circular closed DNA to positively supercoiled DNA (which would increase its electrophoretic mobility), and relaxes (partially or completely) negatively supercoiled DNA (which decreases its electrophoretic mobility). The total length of the long PNA-binding insert is 117 bp; thus its complete unwinding produces about 11 compensatory positive superturns. Fig. 3 shows that, as expected, incubation of PNA with relaxed plasmid with the long PNA-binding insert produced a faster migrating species (lane 1), consistent with positively supercoiled plasmid. Note that a smaller shift (less than one topoisomer) is also noticeable in the vector plasmid upon PNA binding (compare lanes 3 and 4). This is due to the presence of shorter and mismatched PNA binding sequences in the vector plasmid. The longest of the non-uninterrupted sequences that we found in the pBluescriptSK(+) sequence, AAGAAGA, was probably responsible for the minor topoisomer shift in lane 3. As observed previously [38], PNA invasion into relaxed DNA is inhibited by increasing the concentration of monovalent cations, and it was not observed at 100 mM NaCl (Fig. 4A). This is because cations stabilize the DNA double-helix thus making its opening (required for PNA invasion) more difficult.
Fig. 3. PNA can invade relaxed circular closed DNA generating positively supercoiled DNA.
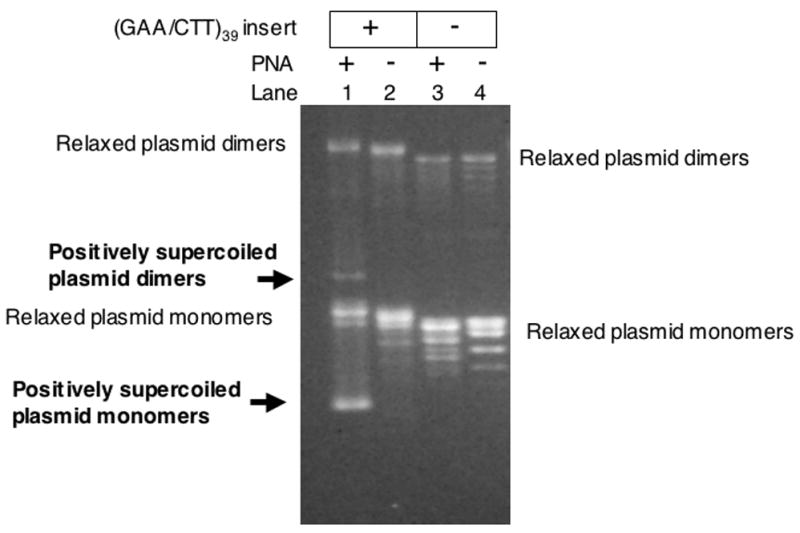
Agarose gel electrophoresis of relaxed plasmids was carried out with and without the (GAA/CTT)39 insert, and with or without PNA binding. The appearance of the faster-migrating, positively supercoiled plasmid is apparent in lane 1.
Fig.4. Effect of increasing the salt concentration on PNA-DNA invasion.
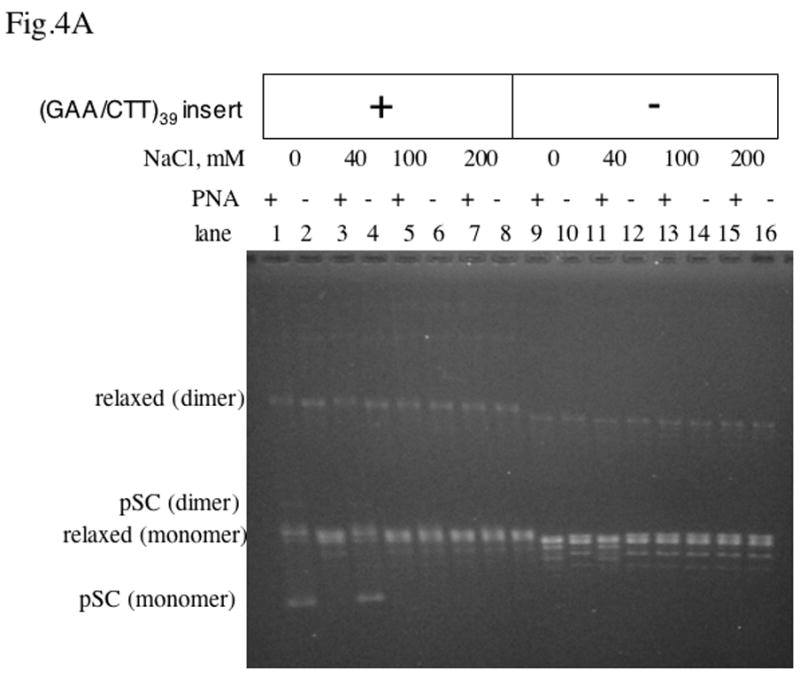
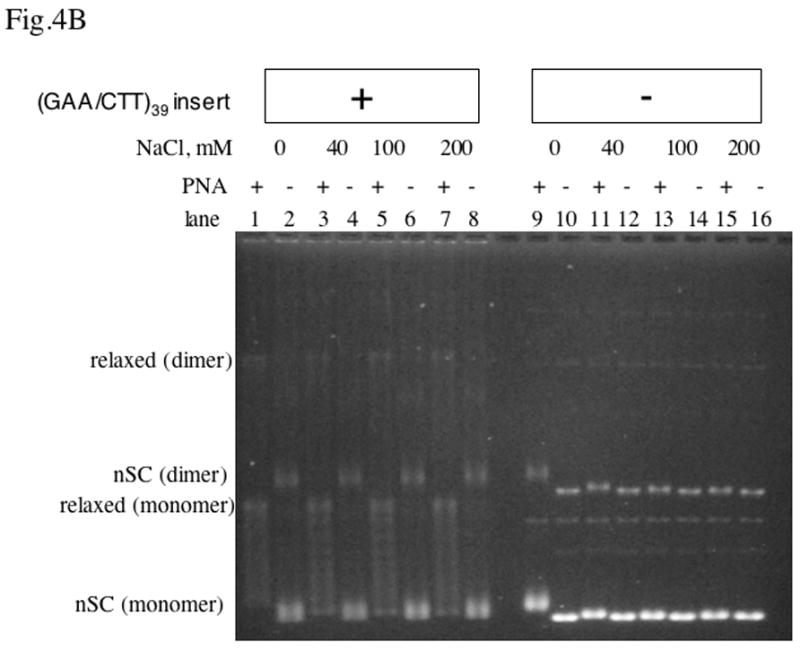
Agarose gel electrophoresis of plasmids was carried out with and without the (GAA/CTT)39 inset, and with or without PNA binding at various concentrations of NaCl.
A: relaxed DNA; B: negatively supercoiled DNA.
In the case of negatively supercoiled DNA with the long PNA binding insert (Fig. 4B, lanes 1–8), partially and completely relaxed topoisomers appear upon PNA binding. The expected number of negative superturns in the 3 kb-long vector plasmid pBluescript SK(+) with native superhelical density (about −0.05) is distributed around 15; thus, since PNA relaxes about 11 negative superturns, the presence of partially relaxed topoisomers was expected. In contrast to relaxed DNA, for negatively supercoiled DNA, PNA invaded the long PNA binding insert in the presence of NaCl concentration up to 200 mM, because negative supercoiling facilitates DNA “breathing” [39]. Note that at low salt (Fig. 4B, lane 9) a smaller partial relaxation was observed also for the vector plasmid, due to PNA invasion into mismatched and shorter versions of the recognition sequences. However, upon increasing the salt concentration, this partial relaxation became undetectable. This is because increasing the salt concentration makes PNA invasion more difficult and consequently more stringent, thus preventing invasion into most of the shorter or mismatched target sequences.
Transcription through PNA/DNA hybrids
The general scheme of the transcription experiment is shown in Fig. 1B. Both long and short PNA binding inserts were cloned downstream of the T7 promoter in two orientations: in one, PNA binds the template strand, and in the other PNA binds the non-template strand. Unless otherwise indicated, DNA substrates linearized by Afl III restriction 0.5 kb downstream from the PNA-binding insert were used in the transcription assay. PNA-DNA hybrids were pre-formed, usually under low salt conditions (see Materials and Methods, and legends to figures), and then aliquots of the samples were taken for in vitro transcription. Results for the linear DNA templates are shown in Fig. 5A. In the case of a short PNA-binding insert, PNA binding to the template strand (lane 1) produced several closely localized truncated transcription products, for which the positions in the gel were consistent with partial transcription blockage within the PNA-binding insert, although we did not determine their positions at nucleotide resolution. The probability of transcript truncation was roughly 40–60 %, estimated both from the molar ratios of truncated and run-off products, and from the reduction in run-off products compared to the PNA minus control (Note that in our transcription assay, the radioactive labeling is roughly proportional to the length of the transcript. Thus, to calculate the molar ratio of transcripts with different lengths one must normalize the radioactive signal from the transcript to its corresponding length. For example, in Fig. 5 the lengths of the truncated products were about one tenth of the lengths of the run-off products; therefore, the truncated products would produce about a ten-fold weaker radioactive signal than the same molar amount of the run-off products.) Since PNA binding was about 100 % according to the restriction-protection assay, our results suggest that T7 RNAP can displace PNA from its binding site and continue transcription with around 50 % probability. For the short PNA-binding insert, we did not detect any effect of PNA on transcription when PNA was bound to the non-template strand (lanes 3, 4), as well as for the vector plasmid (lanes 9, 10). These results for short PNA-binding inserts, including the appearance of several closely located truncation products, are in accordance with the results reported previously for other PNAs [4].
Fig.5. Effect of PNA on T7 transcription.
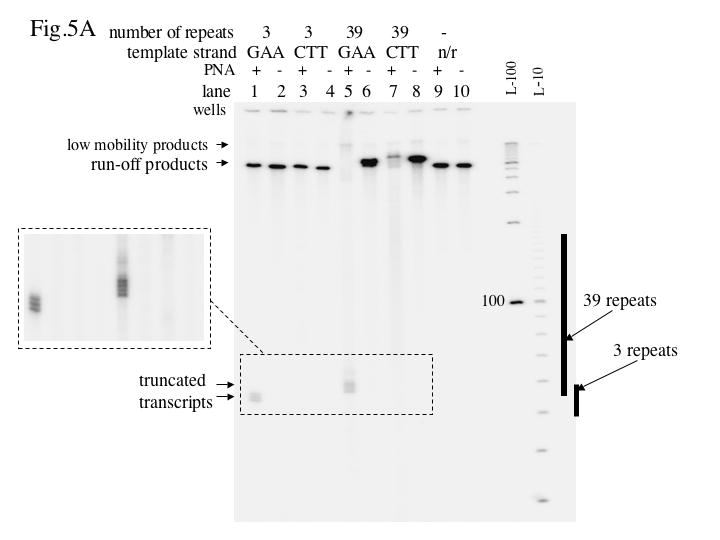
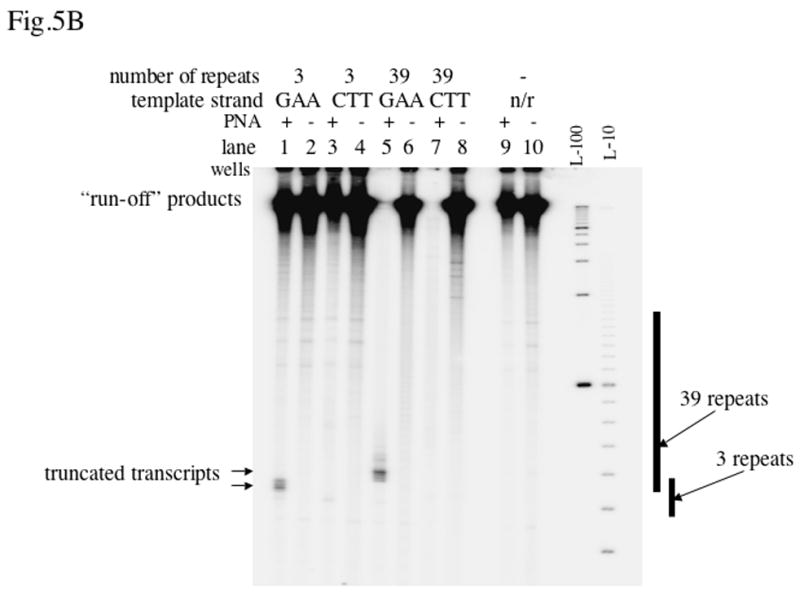
The autoradiograph shows the transcription products resolved by denaturing acrylamide gel electrophoresis. The designation “n/r” (“no repeats”) indicates the vector plasmid pBluescript SK(+). L-100 and L-10 are 100 nt and 10 nt denatured DNA ladders, respectively. The positions of the inserts are shown at right. A: Linear DNA substrates. B: Negatively supercoiled DNA substrates.
In the case of the long insert, PNA binding to the template strand also produced clearly defined truncated products, though in this case PNA binding led to nearly complete disappearance of the normal run-off product (lane 5). (The very small fraction of low mobility product that increases upon PNA addition will be discussed in the last subsection of the Results). This result was expected: the long insert could bind up to 13 PNA molecules, and if for each of them the probability of transcription truncation was around 50 % (or perhaps more, if PNA binding was cooperative), no detectable run-off product would be expected, and most of the transcripts would be terminated close to the promoter-proximal flank of the insert. However, unexpectedly, PNA binding to the non-template strand in the case of the long insert also led to a strong (about 10-fold) decrease of the yield of run-off product (lane 7). Note that in lane 7 the “smear” of shorter transcription products within and some distance downstream from the insert, is more pronounced than that in the control lane 8. This reproducible trend is more clearly seen in the images with a longer exposure in Fig. 8, lanes 9 and 10. We suggest that these products indicate an increased probability of transcription truncation within and some distance downstream from the PNA binding insert, and that this could explain the decrease in the normal run-off product upon PNA binding. A rough estimation of the increase, upon PNA binding, in the total molar amount of truncated products distributed within the PNA binding insert (considering the product distribution as uniform and using the average lengths of the product as a normalizing factor) showed that this increase exceeds the molar amount of remaining run-off product by roughly 5–8 fold, which is close to a 10-fold decrease of the run-off product upon PNA binding to the non-template strand of the long insert. Thus, these truncated products could account for the run-off transcript that was “lost” due to PNA binding. An alternative explanation, that PNA binding somehow inhibited transcription initiation seems to be less likely in our system, because PNA binding to the non-template strand in the case of the short PNA binding site doesn’t detectably affect transcription.
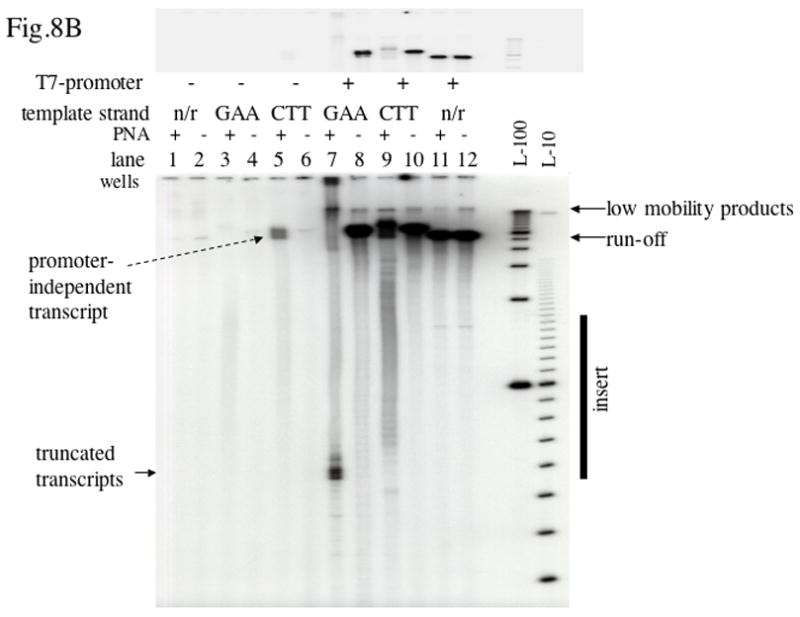
Fig. 8. Promoter-independent transcription.
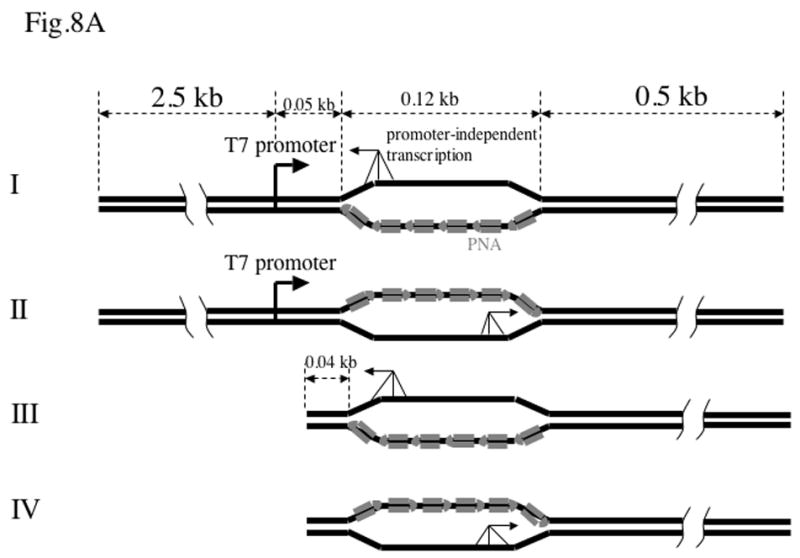
A: Substrates with and without T7 promoter. Only PNA-bound substrates are shown. Promoter-independent transcription (shown by three-legged arrows) in principle could start from any point within single-stranded regions, but it might have some preference for the single-strand/duplex junction. Once it starts, it would continue to the end of the substrate.
B: Transcription from substrates shown in A. Lane 7 corresponds to Substrate I, lane 9 corresponds to Substrate II, lane 3 corresponds to Substrate III, lane 5 corresponds to Substrate IV. The designation “n/r” (“no repeats) indicates the vector plasmid pBluescript SK(+). Top panel: a lower exposure of the top area of the same gel containing run-off products is shown.
Qualitatively similar (although more pronounced) effects of PNA binding were obtained for negatively supercoiled DNA (Fig. 5B): in the case of the long inserts in both orientations little or no “run-off” products were observed (lanes 5 and 6). In this case, however, the effect of PNA was also pronounced for the vector plasmid (lanes 9, 10): the “run-off” transcript decreased about four-fold probably due to PNA binding to shorter and mismatched sites in the vector plasmid. (Note that because the substrates in this case were circular, the “run-off” products are not well defined. It is likely that they include a heterogeneous mixture of long products that are not resolved on the gel. For that reason we use the designation “run-off” in quotation marks.)
When PNA is complementary to the non-template strand it is also complementary to the transcript. Thus the PNA remaining in solution could also bind the transcript, which in principle could interfere with further transcription, for example by “pulling out” nascent RNA from the transcription complex. To address this possibility (and other possible effects of free PNA during transcription) we used the fact that although PNA invasion in DNA is inhibited by high salt concentration, the pre-formed DNA-PNA hybrids do not dissociate at high salt [38]. Thus by adding high salt before or after pre-incubation with PNA, we could obtain samples with exactly the same salt and PNA concentrations with and without PNA invasion into DNA. Transcription experiments with these samples (Fig. 6) showed that in our case the effect on transcription was primarily due to preformed DNA-PNA hybrids, rather than PNA hybridization with RNA or other PNA-DNA interactions that might appear during transcription [40].
Fig.6. Effect of the order of PNA and NaCl addition on the transcription inhibition.
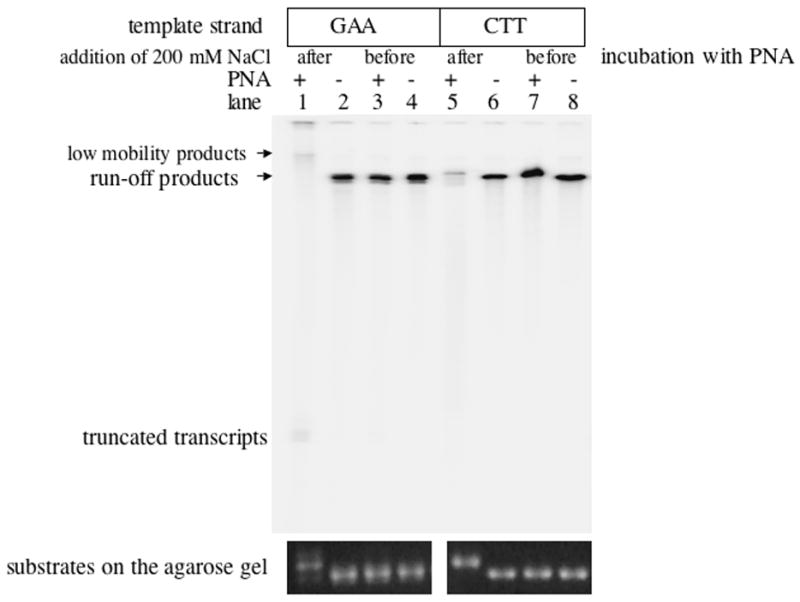
Autoradiograph of the transcription products separated in denaturing acrylamide gel. A pronounced effect was evident only when preincubation with PNA preceded the addition of salt (lanes 1 and 5). Gel-shifts of linear DNA on the agarose gel upon PNA binding (at the bottom of the Figure) confirmed that PNA binds DNA only when 200 mM NaCl were added after preincubation with PNA.
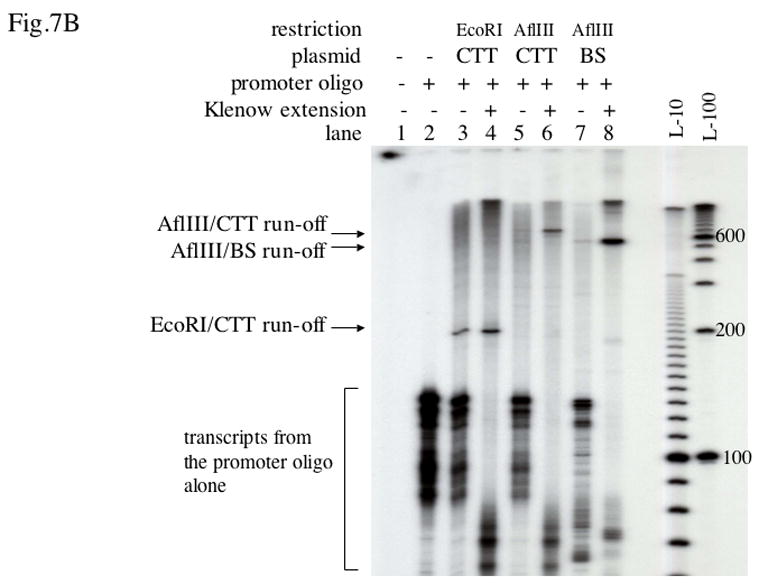
To address the question of whether the single-stranded state of the template strand alone could explain the inhibitory effect of PNA binding to the non-template strand, we used single-stranded circular DNA corresponding to the T7-template strand of pBluescript plasmid with and without the (CTT)39 insert. A complementary oligonucleotide containing the T7 promoter sequence (“promoter oligo”) was hybridized upstream of the (CTT)39 insert covering the entire region from the promoter to the start of the insert. To obtain a defined run-off product, another oligonucleotide containing a restriction site (“restriction oligo”) was hybridized downstream from the insert, and then the circle was linearized by the corresponding restriction enzyme (Fig. 7A). This single-stranded template then could be converted into the double-stranded form by filling with the Klenow enzyme using the promoter oligo as a primer.
Fig.7. Transcription from partially single-stranded DNA substrates.
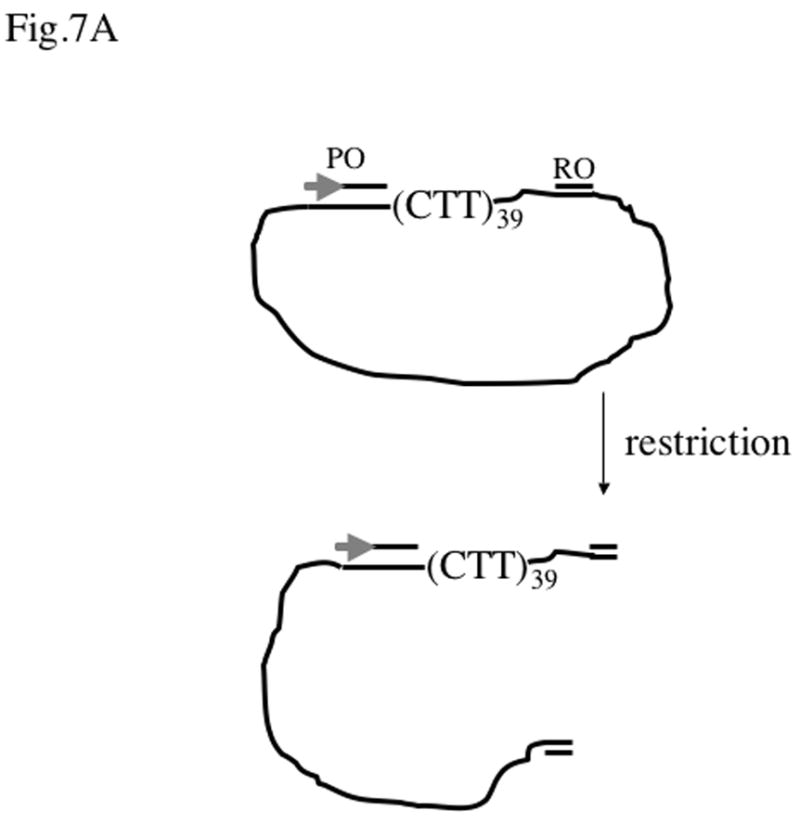
A: The substrate used for transcription. Single-stranded circular DNA corresponding to the template strand of pBlue-iCTT39 plasmid was annealed to the promoter oligo (PO) and to the restriction oligo (RO) that contains the corresponding restriction site that allows linearization of the template. The first 17 nucleotides of the promoter oligo for pBlue-iCTT plasmid correspond to the top strand of the T7 promoter (symbolized by the gray arrow), the rest of the oligo (54 nucleotides) covers the area from the promoter to the CTT insert. A similar promoter oligo was used for the vector plasmid pBluescriptSK(+), except that it covered 48 nucleotides of the downstream area instead of 54. The distance from the CTT-insert to the EcoRI cleavage site was 21 nt, thus the expected run-off in this case is around 0.19 kb. The expected run-off for the pBlue-iCTT39 plasmid and the AflIII oligo is 0.63 kb. The expected run-of for the pBluescript SK(+) plasmid and the AflIII oligo is 0.51 kb.
B: Transcription products from the substrates described in A with and without Klenow extension, separated in a denaturing acrylamide gel. CTT stands for pBlue-iCTT39 plasmid, BS stands for pBluescript SK(+) plasmid. The positions of corresponding run-off products were consistent with expectation within the gel resolution. Lane 1 corresponds to transcription in the absence of DNA substrates, which did not produce any transcript in our conditions, although in principle such reaction is possible [52]. Lane 2 shows transcripts from the promoter oligo alone, which were also present in the rest of the lanes because of excess of the unbound promoter oligo. Although the exact nature of these products is not known, this kind of reaction was described before and it did not interfere with our analysis. When the samples were treated with Klenow before transcription, these products became shorter most likely because of partial digestion of free promoter oligo by Klenow.
The amount of the run-off product from the 500–600 nt long single-stranded templates was much smaller than the product from the same template treated with Klenow enzyme (Fig. 7B, lanes 5, 6, 7, 8). This could be either because the processivity of T7 RNAP was somewhat lower in the absence of the non-template strand, or because of the presence of secondary structures in the template strand. However, for the shorter single-stranded region of about 120 nt containing (CTT)39, which was about the same as the one that formed when PNA was bound to the non-template strand of double-stranded DNA, the difference was roughly two-fold (lanes 3, 4). Thus, we concluded that although transcription could be less efficient on single-stranded substrates, this doesn’t completely explain the inhibitory effect of PNA binding to the non-template strand (see Discussion).
Note that in this system the single-stranded promoter oligo alone induced some promoter-independent T7 transcription, generating additional transcription products (Fig. 7B, lane 2). The phenomenon of promoter-independent induction of T7 (and other phage RNAPs) transcription from single-stranded or partially single-stranded substrates has been described for many systems ([41–44], see also below). The additional products migrated below our bands of interest in the gel, so they did not affect the interpretation of our results.
Promoter-independent PNA induced transcription
As we already mentioned, PNA binding to the template strands in plasmids containing the long insert essentially eliminated normal run-off transcription products. However, we observed an increase of products migrating slower than the normal run-off products (Fig. 5A, lane 5 and Fig. 6, lane 1). These slow-migrating minor products were also present in reactions with control plasmid, most likely due to traces of circular DNA in the samples producing longer transcripts. However, in the case of the (GAA)39 in the template strand the amount of these products appeared to increase upon PNA addition. We hypothesized that there was another source of longer transcripts, due to promoter-independent initiation from the strand displaced by PNA from the duplex. Note that E. coli RNAP promoter-independent transcription from a region opened by PNA was previously demonstrated [9]. As shown in Fig. 8A, promoter-independent transcription in the case of PNA bound to the template strand (Substrate I) would produce a transcript about four times longer than the normal transcript from the T7 promoter. (We will use the designations “template” and “non-template” relative to the normal transcription from T7 promoter). In contrast, when PNA is bound to the non-template strand (Substrate II), the promoter-independent transcript would be about the size of the normal, or shorter, transcript; thus it would be less pronounced and difficult to separate from the remaining normal transcript. These predictions are consistent with the results in Figs. 5A and 6. To check this hypothesis more directly, we removed the promoter from our templates by restriction digestion followed by gel purification. As it is shown in Fig. 8A, in this case the construct with the PNA bound to the non-template strand (Substrate IV) is expected to produce the longest promoter-independent transcripts, with a length close to that of the normal T7 transcript; for the opposite orientation (Substrate III), the transcripts would be much shorter and, because the radioactive labeling is proportional to the length of the transcript, they would produce much lower signal. Furthermore, the promoter-independent transcripts are (most likely) heterogeneous and consequently, the shorter transcripts would be distributed over a greater distance in the gel because of the greater separation of the shorter products during gel electrophoresis. Thus these shorter promoter-independent transcripts might be difficult to detect. Results in shown in Fig. 8B confirm this prediction: the PNA-induced promoter-independent transcript was pronounced only for Substrate IV (lane 5). Quantitation showed that the yield of promoter-independent transcript in this case was about 30-fold less than the yield of the normal transcript.
DISCUSSION
PNA binding and effects on T7 RNAP transcription of DNA containing Friedreich’s ataxia trinucleotide sequence repeats (GAA/CTT)n were investigated. We showed that PNA binds to these repeats, even if binding causes generation of energetically unfavorable positive superhelical stress, in a manner similar to that previously reported for polyA/polyT repeats [38]. In the case of the short PNA binding insert (GAA/CTT)3, PNA binding caused partial transcription inhibition only when PNA was bound to the template strand, but not to the non-template strand. This observation is consistent with the results previously reported for other short homopurine/homopyrimidine sequences [4].
Unexpectedly, for the long insert (GAA/CTT)39, most of the full-length transcription product disappeared, not only when PNA was bound to the template strand, but also when it was bound to the non-template strand (i.e. the template became single-stranded); although in the later case, there were no well-defined predominant truncated products, meaning that truncation occurred with similar probability at any point within the (CTT)39 template strand. Was this effect simply due to a lower processivity of T7 RNAP on the single-stranded template? On the one hand, the non-template strand facilitates transcription elongation by displacing nascent RNA from the template [45], and it allows RNAP to pass gaps in the template strand [46]. On the other hand, it was shown that transcription of a 30 nt DNA template region immediately downstream of the double-stranded T7 promoter had the same efficiency whether this sequence was double- or single-stranded [47]. However, the situation might be different for longer substrates and it could depend upon the template nucleotide sequence. Our results with the single-stranded template prepared from phage DNA showed that for long (500–600 nt) single-stranded templates the yield of run-off transcript was much lower than that for the double-stranded template of the same length. However, for the templates with a shorter single-stranded region (120 nt) the difference between single- and double stranded templates was about twofold, while the inhibition of transcription by PNA bound to the non-template strand (forming a single-stranded region about 120 nt in the template strand within the double-stranded plasmid) was about ten-fold. Thus, other interactions might be involved; for example, a complex of PNA with the non-template strand could create steric hindrances for RNAP moving along the template strand, or positively charged lysine residues from PNA bound to the non-template strand could interact with the template strand, impeding RNAP movement and facilitating transcription termination. In addition, the increased probability of transcription termination might extend into the region downstream of the PNA-binding insert, possibly due to the formation of a persistent RNA-DNA hybrid during transcription of the single-stranded template strand within the insert, and that could interfere with further transcription elongation [48,49].
The finding of a transcription-inhibitory effect of PNA binding to the non-template strand for Friedreich’s ataxia triplet repeats is in contrast to the results reported with DNA oligonucleotides, for which binding to the non-template strand facilitated transcription through these repeats, presumably by interference with intramolecular triplex formation [18]. Therefore, it is unlikely that PNA could be used to directly alleviate the transcription insufficiency in Friedreich’s ataxia as proposed for DNA oligonucleotides[18]. However, additional studies under physiological ionic conditions and with eukaryotic RNAPs are required to completely address this question.
The rationale for use of PNA could be for inducing mutagenesis in the repeats, thus alleviating their deleterious effects at the DNA sequence level [19]. PNA binding could facilitate mutagenesis within or near the binding site by several mechanisms: (i) Single-stranded DNA regions and structural distortions within the target DNA produced by PNA binding could create hot spots for DNA-modifying enzymes and homologous recombination. (ii) Blockage of transcription by PNA binding might induce gratuitous TCR [23]. Both of these mechanisms could be potentially utilized to introduce disruptions or deletions into the expanded trinucleotide sequence. Of course, these approaches might be useful for alleviation of the transcription deficiency only if the PNA is removed from the DNA (probably by excision of the entire PNA-DNA hybrid) rapidly enough that it wouldn’t cause prolonged transcription blockage leading to cellular apoptosis. Other homopurine-hompyrimidine sequences in the genome were also shown to cause genomic instability and possibly contribute to carcinogenesis ([14]; reviewed in [50]). Moreover, several homopurine-homopyrimidine sequences are localized in the promoter regions of genes participating in carcinogenesis [51]. Thus the information obtained for PNA effects on transcription in Friedreich’s ataxia triplet repeats could be also applicable for targeting of DNA sequences participating in carcinogenesis.
Acknowledgments
We thank Sergei M. Mirkin for pYES-TTC57 plasmid and helpful discussions, Sergei A. Kazakov for helpful discussions and Graciela Spivak, C. Allen Smith, and Ann Ganesan for critical reading of the manuscript. This work was supported by grant CA77712 to PCH from the National Cancer Institute, NIH.
References
- 1.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254(5037):1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen PE. PNA Technology. Mol Biotechnol. 2004;26(3):233–248. doi: 10.1385/MB:26:3:233. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen PE, Egholm M. An introduction to peptide nucleic acid. Curr Issues Mol Biol. 1999;1(1–2):89–104. [PubMed] [Google Scholar]
- 4.Nielsen PE, Egholm M, Buchardt O. Sequence-specific transcription arrest by peptide nucleic acid bound to the DNA template strand. Gene. 1994;149(1):139–145. doi: 10.1016/0378-1119(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 5.Bukanov NO, Demidov VV, Nielsen PE, Frank-Kamenetskii MD. PD-loop: a complex of duplex DNA with an oligonucleotide. Proc Natl Acad Sci U S A. 1998;95(10):5516–5520. doi: 10.1073/pnas.95.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belotserkovskii BP, Zarling DA. Peptide nucleic acid (PNA) facilitates multistranded hybrid formation between linear double-stranded DNA targets and RecA protein-coated complementary single-stranded DNA probes. Biochemistry. 2002;41(11):3686–3692. doi: 10.1021/bi012017f. [DOI] [PubMed] [Google Scholar]
- 7.Rogers FA, Vasquez KM, Egholm M, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci U S A. 2002;99(26):16695–16700. doi: 10.1073/pnas.262556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faruqi AF, Egholm M, Glazer PM. Peptide nucleic acid-targeted mutagenesis of a chromosomal gene in mouse cells. Proc Natl Acad Sci U S A. 1998;95(4):1398–1403. doi: 10.1073/pnas.95.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mollegaard NE, Buchardt O, Egholm M, Nielsen PE. Peptide nucleic acid. DNA strand displacement loops as artificial transcription promoters. Proc Natl Acad Sci U S A. 1994;91(9):3892–3895. doi: 10.1073/pnas.91.9.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn H, Demidov VV, Nielsen PE, Frank-Kamenetskii MD. An experimental study of mechanism and specificity of peptide nucleic acid (PNA) binding to duplex DNA. J Mol Biol. 1999;286(5):1337–1345. doi: 10.1006/jmbi.1998.2578. [DOI] [PubMed] [Google Scholar]
- 12.Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res. 1995;23(2):217–222. doi: 10.1093/nar/23.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol. 2006;16(3):351–358. doi: 10.1016/j.sbi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability. Mutat Res. 2006;598(1–2):103–119. doi: 10.1016/j.mrfmmm.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 15.Wells RD. DNA triplexes and Friedreich ataxia. Faseb J. 2008 doi: 10.1096/fj.07-097857. [DOI] [PubMed] [Google Scholar]
- 16.Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35(10):3383–3390. doi: 10.1093/nar/gkm271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnett R, Melander C, Puckett JW, et al. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA. TTC repeats in Friedreich’s ataxia. Proc Natl Acad Sci U S A. 2006;103(31):11497–11502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabczyk E, Usdin K. Alleviating transcript insufficiency caused by Friedreich’s ataxia triplet repeats. Nucleic Acids Res. 2000;28(24):4930–4937. doi: 10.1093/nar/28.24.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakamoto N, Larson JE, Iyer RR, Montermini L, Pandolfo M, Wells RD. GGA*TCC-interrupted triplets in long GAA*TTC repeats inhibit the formation of triplex and sticky DNA structures, alleviate transcription inhibition, and reduce genetic instabilities. J Biol Chem. 2001;276(29):27178–27187. doi: 10.1074/jbc.M101852200. [DOI] [PubMed] [Google Scholar]
- 20.Mellon I, Bohr VA, Smith CA, Hanawalt PC. Preferential DNA repair of an active gene in human cells. Proc Natl Acad Sci U S A. 1986;83(23):8878–8882. doi: 10.1073/pnas.83.23.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellon I, Hanawalt PC. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature. 1989;342(6245):95–98. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 22.Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51(2):241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 23.Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266(5193):1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- 24.Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem. 2007;282(44):32433–32441. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- 25.Droge P, Pohl FM. The influence of an alternate template conformation on elongating phage T7 RNA polymerase. Nucleic Acids Res. 1991;19(19):5301–5306. doi: 10.1093/nar/19.19.5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peck LJ, Wang JC. Transcriptional block caused by a negative supercoiling induced structural change in an alternating CG sequence. Cell. 1985;40(1):129–137. doi: 10.1016/0092-8674(85)90316-2. [DOI] [PubMed] [Google Scholar]
- 27.Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28(14):2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ditlevson JV, Tornaletti S, Belotserkovskii BP, et al. Inhibitory effect of a short Z-DNA forming sequence on transcription elongation by T7 RNA polymerase. Nucleic Acids Res. 2008;36(10):3163–3170. doi: 10.1093/nar/gkn136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tornaletti S, Park-Snyder S, Hanawalt PC. G4-forming sequences in the non-transcribed DNA strand pose blocks to T7 RNA polymerase and mammalian RNA polymerase II. J Biol Chem. 2008;283(19):12756–12762. doi: 10.1074/jbc.M705003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grabczyk E, Fishman MC. A long purine-pyrimidine homopolymer acts as a transcriptional diode. J Biol Chem. 1995;270(4):1791–1797. doi: 10.1074/jbc.270.4.1791. [DOI] [PubMed] [Google Scholar]
- 31.Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62(1):111–121. doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krasilnikova MM, Kireeva ML, Petrovic V, Knijnikova N, Kashlev M, Mirkin SM. Effects of Friedreich’s ataxia (GAA)n*(TTC)n repeats on RNA synthesis and stability. Nucleic Acids Res. 2007;35(4):1075–1084. doi: 10.1093/nar/gkl1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J Biol Chem. 2001;276(29):27171–27177. doi: 10.1074/jbc.M101879200. [DOI] [PubMed] [Google Scholar]
- 34.Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271(5250):802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- 35.Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27(17):6209–6217. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krasilnikova MM, Mirkin SM. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol. 2004;24(6):2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence specific inhibition of DNA restriction enzyme cleavage by PNA. Nucleic Acids Res. 1993;21(2):197–200. doi: 10.1093/nar/21.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cherny DY, Belotserkovskii BP, Frank-Kamenetskii MD, et al. DNA unwinding upon strand-displacement binding of a thymine-substituted polyamide to double-stranded DNA. Proc Natl Acad Sci U S A. 1993;90(5):1667–1670. doi: 10.1073/pnas.90.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bentin T, Nielsen PE. Enhanced peptide nucleic acid binding to supercoiled DNA: possible implications for DNA “breathing” dynamics. Biochemistry. 1996;35(27):8863–8869. doi: 10.1021/bi960436k. [DOI] [PubMed] [Google Scholar]
- 40.Larsen HJ, Nielsen PE. Transcription-mediated binding of peptide nucleic acid (PNA) to double-stranded DNA: sequence-specific suicide transcription. Nucleic Acids Res. 1996;24(3):458–463. doi: 10.1093/nar/24.3.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krupp G. Unusual promoter-independent transcription reactions with bacteriophage RNA polymerases. Nucleic Acids Res. 1989;17(8):3023–3036. doi: 10.1093/nar/17.8.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharmeen L, Taylor J. Enzymatic synthesis of RNA oligonucleotides. Nucleic Acids Res. 1987;15(16):6705–6711. doi: 10.1093/nar/15.16.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seyhan AA, Vlassov AV, Johnston BH. RNA interference from multimeric shRNAs generated by rolling circle transcription. Oligonucleotides. 2006;16(4):353–363. doi: 10.1089/oli.2006.16.353. [DOI] [PubMed] [Google Scholar]
- 44.Daubendiek SL, Kool ET. Generation of catalytic RNAs by rolling transcription of synthetic DNA nanocircles. Nat Biotechnol. 1997;15(3):273–277. doi: 10.1038/nbt0397-273. [DOI] [PubMed] [Google Scholar]
- 45.Gopal V, Brieba LG, Guajardo R, McAllister WT, Sousa R. Characterization of structural features important for T7 RNAP elongation complex stability reveals competing complex conformations and a role for the non-template strand in RNA displacement. J Mol Biol. 1999;290(2):411–431. doi: 10.1006/jmbi.1999.2836. [DOI] [PubMed] [Google Scholar]
- 46.Zhou W, Reines D, Doetsch PW. T7 RNA polymerase bypass of large gaps on the template strand reveals a critical role of the nontemplate strand in elongation. Cell. 1995;82(4):577–585. doi: 10.1016/0092-8674(95)90030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15(21):8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kireeva ML, Komissarova N, Kashlev M. Overextended RNA:DNA hybrid as a negative regulator of RNA polymerase II processivity. J Mol Biol. 2000;299(2):325–335. doi: 10.1006/jmbi.2000.3755. [DOI] [PubMed] [Google Scholar]
- 49.Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell. 2003;12(3):711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Jain A, Wang G, Vasquez KM. DNA triple helices: Biological consequences and therapeutic potential. Biochimie. 2008 doi: 10.1016/j.biochi.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Q, Gaddis SS, MacLeod MC, et al. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol Carcinog. 2007;46(1):15–23. doi: 10.1002/mc.20261. [DOI] [PubMed] [Google Scholar]
- 52.Biebricher CK, Luce R. Template-free generation of RNA species that replicate with bacteriophage T7 RNA polymerase. Embo J. 1996;15(13):3458–3465. [PMC free article] [PubMed] [Google Scholar]