

Abstract
Plasma protein binding of antimicrobial agents is considered to be a key characteristic of antibiotics as it affects both their pharmacokinetics and pharmacodynamics. However, up to the present, no standard methods for measuring protein binding or for quantification of the influence of protein binding on antimicrobial activity exist. This short-coming has previously led to conflicting results on antibacterial activity of highly protein-bound antibiotics. The present review, therefore, set out to summarize (1) methods for quantification of protein binding, (2) microbiological growth media used for determination of the impact of protein binding on antimicrobial activity of antibiotics, and (3) different pharmacodynamic in vitro studies that are used in this context. The advantages and disadvantages of a wide range of different approaches are discussed and compared. The urgent call for international standardization by microbiological societies and laboratories may be considered as a logical consequence of the presented data.
Key words: antimicrobials, pharmacodynamics, protein binding
INTRODUCTION
The importance of protein binding (PB) on the pharmacokinetics (PK) of antibiotics is well-documented and accepted (1,2). In contrast, no general consensus has been reached, whether PB also pharmacodynamically impacts antimicrobial activity by reducing the free, i.e., nonprotein-bound, fraction of an antibiotic (3,4). The unfavorable effect of PB on bacterial killing is generally accepted for β-lactams (5,6), whereas doubts about the usefulness of extrapolation of findings from β-lactams to other antimicrobial classes, such as fluoroquinolones, persist (3,4). There is currently no standardization of in vitro pharmacodynamic (PD) models, which account for the impact of PB of antimicrobials (7). Instead, the use of different investigational media containing variable amounts of protein has previously led to diverging results (8,9). There is thus a need to review and standardize currently used methodologies which investigate the impact of protein binding in in vitro PD models.
In order to evaluate the influence of PB on the PD of an antibiotic, it is necessary to measure the free antibiotic fraction precisely. For this purpose, different approaches are described in the literature. The present review, therefore, starts with a short overview on the most commonly used methods for the determination of PB. The implications of PB on the antimicrobial efficacy of an antibiotic shall be outlined and the current position of PB in in vitro PD models of antibiotics will be discussed. As extensive reviews on impact of PB on PK of drugs are available, the present work will not go into details on this topic (3,10–14).
PRINCIPLES OF PLASMA PROTEIN BINDING OF ANTIBIOTICS
Since over 30 years, the important influence of PB on the antibacterial activity of β-lactam antibiotics in vitro is known and was proven in early studies by Kunin or Merriken (15,16). The relevance of PB on PKs and the microbiological efficacy of an antibiotic were considered clinically important in case of highly bound agents (17). It is well-established that only the protein unbound fraction of a drug in plasma can readily penetrate into and equilibrate with the extravascular space (3). Sufficient tissue penetration of drugs is, in turn, a prerequisite for successful treatment of the majority of bacterial infections, which occur in the interstitial fluid (ISF) rather than in the blood (18). PB also affects drug clearance from the body. For renaly eliminated antibiotics excreted by glomerular filtration, high PB is associated with a decreased elimination rate, since only the free drug is filtered (19). On the contrary, if a drug is eliminated by tubular secretion or undergoes hepatic metabolism, plasma protein binding (PPB) may promote drug elimination by retaining the drug in the bloodstream for delivery to the excretory system.
Mathematical models have been developed to characterize drug–protein interactions and to estimate PB parameters (2). In brief, the fraction of drug bound to plasma proteins is a function of unbound drug concentration (CU), protein concentration (P), the number of binding sites (n), and the equilibrium association constant (KA), which describes the affinity of the drug–protein association. High KA values (105 to 107 L/mol) are associated with high degrees of binding (90% to 99.9%) (2):
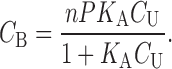 |
1 |
Nowadays, computer-based models are often applied for estimating binding parameters (2). Mathematical models have also been previously used to describe the impact of PBB on the pharmacodynamics of antimicrobials. In this context, data obtained from a number of in vitro experiments can be used to estimate the impact of PPB on antimicrobial killing for various antibiotics and bacteria (20).
Certain antibiotics, such as aminoglycosides and β-lactams (1,21–23), have been shown to display saturable, concentration-dependent, serum protein binding within the concentration range that can result from a normal therapeutic dose (1). As the concentration of a drug in plasma increases, binding sites on proteins are increasingly saturated, resulting in higher percentages of unbound drug in plasma, such is the case for ceftriaxone (22). This may allow for more rapid excretion and may increase free tissue concentrations.
Numerous physiological conditions such as age (e.g., in neonates), body temperature, plasma pH, or a variety of diseases like uremia, hepatitis, hypoalbuminemia, acute viral hepatitis, cirrhosis, nephrotic syndrome, and epilepsy may lead to significant decreases in PB (1,2,24). Furthermore, drug–drug interactions can lead to changes of unbound fractions of protein-bound drugs (25). The mechanism may be either competitive, meaning that drugs bind to the same site, or noncompetitive, with one drug causing a conformational change in the protein molecule, which, in turn, inhibits the binding of the other drug (19). Drugs interacting for binding sites on plasma proteins often additionally interact at the level of metabolism and excretion, resulting in a potentiation effect (19). However, although changes in PPB can have an important influence on individual PK parameters, it was shown that changes in PPB will usually not influence the clinical exposure of a patient to the drug (24). As a consequence, no adjustments in dosing regimens is considered necessary except in rare cases when a drug with narrow therapeutic index that is given parenterally or a drug with a narrow therapeutic index that is given orally and has a very rapid PK–PD equilibration time. Likewise, in case drugs with a narrow therapeutic range and a high degree of PB (>80%) are administered to critically ill patients with reduced organ function, monitoring of the free drug may become necessary (25).
METHODS FOR DETERMINING PLASMA PROTEIN BINDING OF ANTIBIOTICS
Different approaches for the determination of PB are described in the literature. In the following, we will give a short overview on the most commonly used methods. Table I provides an overview on the advantages as well as the short-comings of different approaches for measuring the protein-bound fraction of a drug in vitro and in vivo (1,2,26–44). In the following, we will discuss only the two currently most relevant and frequently used methods of determining PPB, equilibrium dialysis (ED) and ultrafiltration (UF). In addition, in vivo microdialysis will be introduced as it represents the only method of measuring the protein unbound fraction of a drug in vivo.
Table I.
Methods of Determining Protein Binding
| Advantages | Limitations | References | ||
|---|---|---|---|---|
| Equilibrium dialysis | Separated by a semipermeable membrane, unbound drug diffuses from plasma into protein-free buffer, until equilibrium is reached | Reference method | Long duration | 1,2,26–38,40,42 |
| Simple | Requires use of unphysiological buffer | |||
| Reliable results | Volume shifts | |||
| Membrane adsorption | ||||
| Microdialysis | Dialysate buffer is pumped through an implanted probe, containing a microdialysis membrane. Free drug diffuses from blood into dialysate | In vivo determination of protein binding | Probe insertion and position | 1,2,31–34,43,44 |
| Lack of volume shifts or dilution effects | Changing drug concentration over time | |||
| Can also be used to measure free tissue concentration | Membrane adsorption | |||
| Requires equilibration in vivo | ||||
| For determination of PPB measurement of total antibiotic concentration in vivo is necessary | ||||
| Ultrafiltration | Plasma water and unbound drug is forced through a semipermeable filter, retaining protein–drug complexes | Simple | Ultrafiltrate volumes should be limited to ≤40% of the initial plasma sample because of changes in protein concentration | 2,29,35,36,39,41 |
| Short duration | Leakage of membrane | |||
| Does not require use of unphysiological buffer | Membrane adsorption | |||
| Ultracentrifugation | Dissociation of protein and low-molecular-weight components occurs only by gravitation (centrifugation) | Simple | Long duration | 1,26,28,37 |
| Lack of membrane adsorption, dilution, volume shifts, drug–protein leakage | Concentration gradient from bottom to top can result in false high binding values | |||
| Does not require use of unphysiological buffer | Less suitable for high molecular weight substances | |||
| Chromatography and capillary electrophoresis | Chromatographic methods includes a range of techniques, based on separation of substances (including the bound and unbound fraction of an antibiotic) on the basis of different physical or chemical properties such as molecular size, charge, affinity etc. In capillary electrophoresis, separation of fractions is based on the differential movement of a substance in an electrical field, depending on its degree of ionization. | Accurate methods | Expensive and elaborate | 38 |
| Absence of membrane adsorption, dilution, volume shifts, drug–protein leakage | Poorly sensitive for drugs with low-affinity binding | |||
| Fluorescence spectroscopy | Higher energy photons are used to excite a sample, which then emit lower energy photons. The change in fluorescence at changing ligand/protein concentrations is used to calculate the concentration of bound drug. | Enables direct determination of bound drug concentrations | Poorly sensitive for drugs with low-affinity binding | 1 |
| Elaborate technique | ||||
Equilibrium Dialysis
Although there is no standard method for PB measurements, ED is often regarded as the “reference method” for determining the PB profile of a drug (1,28). It is generally easily practicable, inexpensive, and precise (30). Dialysis cell consist of two reservoirs, separated by a semipermeable dialysis membrane, available in various molecular weight cutoffs (2,29). Plasma and buffer are placed in their respective reservoirs and free drug diffuses from the plasma into the protein-free buffer until equilibrium has been reached. Figure 1 shows a schematic representation of an ED cell. The concentration of free drug (CU) is directly measured in the buffer solution (28). The concentration of bound drug (CB) is obtained by means of Eq. 2:
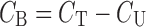 |
2 |
where CT is the total drug concentration measured in the plasma compartment.
Fig. 1.

Schematic representation of an ED cell. Unbound drug diffuses from plasma into protein-free buffer, separated by a semipermeable membrane, until equilibrium is reached
However, there are numerous variables that must be controlled in an ED experiment. Most notably, a temperature of 37°C and a physiological pH should be maintained in order to mimic the in vivo situation (26,30). The time to reach equilibrium should be determined in the course of preliminary experiments by analyzing buffer and plasma concentrations at increasing time periods (2). Equilibrium has been reached when no change is detected in plasma and buffer concentrations within several time points. Long equilibration periods of up to 20 h are considered a disadvantage of ED and require beforehand measurement of chemical stability of the respective drug over time (1,2).
A further limitation of ED is that, due to volume shifts from buffer to the plasma compartment, the final drug concentration in the plasma compartment is lower than the initial plasma concentration (2). Oncotic pressure exerted by plasma protein is the driving force of this phenomenon. Dextran may be added to improve the isotonicity of the buffer or mathematical correction factors may be used to compensate for fluid shifts. Furthermore, a rigid dialyzing system may be used which allows for compensation of oncotic activity by an adequate pressure within the protein-containing compartment (26).
In addition, for drugs displaying concentration-dependent PB, CU at the end of the experiment may differ from the initial CU values. In order to assess the actual value of the initial free drug concentration, the experimental measurements must be corrected mathematically (29). Finally, part of the drug administered to an ED system may be adsorbed to the dialysis membrane, depending on the membrane material, the drug concentration, and the degree of ionization (26). However, nonspecific drug adsorption can be determined and corrected for in the final calculation of the free drug ratio (29). Silanization of container membranes and/or the addition of blank plasma into the buffer container are further possibilities to reduce this error source (27).
Ultrafiltration
A UF unit consists of two reservoirs, separated by a semipermeable filter, which retains plasma proteins and large molecules, while plasma water and low-molecular-weight compounds are collected in the ultrafiltrate. Centrifugal forces usually represent the driving force for the passage of plasma water across the filter membrane. After a short UF period of 10–15 min, CU can be directly determined in the ultrafiltrate. Similar to ED, UF is a simple and reliable procedure for measuring the protein unbound fraction of a drug in plasma. Figure 2 shows a schematic representation of a centrifugational UF system.
Fig. 2.

Schematic representation of a centrifugational UF system. Plasma water and unbound drug is forced through a semipermeable filter (by means of centrifugal forces), retaining protein–drug complexes
One advantage of the UF method is undoubtedly given by the simplicity and short duration of the procedure. UF experiments are performed with small sample volumes of less than 1 mL without the need of employing unphysiological buffer solution. As is the case for ED, samples should be kept at a physiological and constant pH and a constant temperature of 37°C. Adsorption of drug to ultrafiltrate membranes as well as leakage of drug through the filter have been described, but can be compensated for by conducting preliminary UF experiments in a protein-free medium (2,29,36). As the protein concentration in the plasma sample is concentrated, as plasma water is filtered, only a small volume of ultrafiltrate (maximum 40% of the initial plasma sample) should be collected, since the protein concentration in the upper reservoir rises during UF.
Microdialysis
In vivo microdialysis (MD) is most commonly used for determining unbound drug concentrations in the ISF of various tissues (31) but may be used alternatively in order to determine PPB in the blood compartment (34). MD is based on the diffusion of compounds along their concentration gradient from tissue into dialysate (31). Figure 3 shows a schematic representation of a MD probe. For the purpose of defining PPB, a MD probe containing a dialysis membrane is surgically implanted into a blood vessel (2). Dialysate buffer is then pumped through the probe and the unbound drug in plasma diffuses across the membrane into the probe. According to the molecular weight cut-off of the semipermeable membrane, large molecules like proteins will be retained by the membrane (33). Microdialysate samples can then be collected over time for subsequent analysis of CU (34). In order to determine CT, plasma samples must be collected and analyzed separately (2).
Fig. 3.

Schematic representation of a MD probe. Dialysate is pumped through a surgically implanted probe containing a semipermeable membrane. Free drug passively diffuses from tissue into dialysate. Direction of flow as indicated by arrows
As a result of the continuous perfusion of the MD probe, equilibrium between plasma and the recovered dialysate is incomplete, therefore, CU > Cdialysate (33). The factor by which the concentration has to be interrelated is termed relative recovery. The appropriate correction factor is obtained during an in vivo calibration procedure like the retrodialysis procedure (45). The principle of this method relies on the fact that the diffusion process through the semipermeable membrane is quantitatively equal in both directions. Thus, the probe is perfused with a fluid containing a known concentration (Cperfusate) of the drug of interest and its “disappearance rate” through the membrane is determined. The in vivo recovery value is thus calculated as:
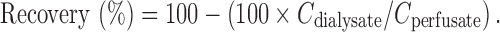 |
3 |
Consecutively, concentrations of the unbound drug in plasma will be calculated by the use of the recovery value:
 |
MD offers the advantages of in vivo measurement of CU and a constant binding equilibrium due to the absence of fluid shifts and concentration effects during the collection of the individual samples. Nevertheless, several disadvantages are referred to the method. Firstly, the dialysis probe must remain inserted in a blood vessel for the duration of the experiment, which may be inconvenient for the individuals (2). Furthermore, as dialysate is collected over time, drug concentrations may change during the collection interval. Finally, only small volumes of dialysate are collected in each sample. This means that relatively sensitive analytical techniques are required to measure drug concentrations in MD experiments (1).
PROTEIN BINDING IN IN VITRO STUDIES OF ANTIMICROBIAL ACTIVITY
In vitro studies are commonly used to determine the antimicrobial potency of an antibiotic or, alternatively, a combination of antibiotics against distinct pathogens. However, in order to allow for predictions on the efficacy of an antibiotic in humans or animals, in vitro studies must be set out to acknowledge various aspects of in vivo conditions. PB is one of several factors which can substantially influence the PDs of an antibiotic (9,46–48). Similar extents of PB have been shown to differentially affect the antimicrobial activity of highly protein-bound antibiotics (7,9,49,50) and impairment of antimicrobial activity in protein-rich medium may differ between gram-positive and gram-negative pathogens (9). Therefore, the effect of PB on individual pathogens has to be investigated for antibiotics on a case by case base. Table II shows a summary of in vitro studies conducted in the last 13 years (as published on the PubMed U.S. National Library of Medicine database from January 1995 until March 2008), which were aimed at investigating the influence of PB on the activity of different classes of antibiotics (7–9,46–49,51,52). Data depicted in Table II indicate that (1) various media and (2) different microbiological methods are used to investigate the impact of PB and (3) results and interpretations of data are highly variable. Together, these data clearly demonstrate that a standard for the determination of the impact of PB on antimicrobial killing is currently missing.
Table II.
In Vitro Studies on the Influence of Protein Binding on the Antimicrobial Activity of Different Classes of Antibiotics (January 1995–March 2008)
| Antibiotic class | Medium | Determination of PB in test medium (PB %) | In vitro model | Conclusion | Year | Reference |
|---|---|---|---|---|---|---|
| Cephalosporin | MHB+90% HS | Not performed, but cited from literature (cefditoren, 88%) | TKC | Presence of HA (4%) but not HS (90%) significantly reduces bactericidal activity of cefditoren against S. penumoniae, when compared to MHB. This corresponds to the bactericidal activity of the free fraction of cefditoren in MHB | 2007 | 47 |
| MHB+4 g/dL HA | ||||||
| MHB+calculated free fraction of antibiotic | ||||||
| Cephalosporin | Supplemented MHB+4 g/dL HA | Not performed | TKC | Presence of HA (4%) significantly reduces the bactericidal activity of ceftriaxone against S. aureus, when compared to ceftriaxone, cefepime, ceftazidime, and cefotaxime in MHB | 1995 | 46 |
| β-Lactam | MHB+4 g/dL HA | Not performed, but cited from literature (ampicillin, 20%; oxacillin, 60–94%) | TKC, MIC | Presence of HA (4%) significantly reduces the bactericidal activity of ampicillin, oxacillin against S. aureus, when compared to MHB. This corresponds to the bactericidal activity of the free fraction of both antibiotics in MHB | 2004 | 48 |
| MHB+calculated free fraction of antibiotic | ||||||
| Fluoroquinolone | MHB+HA (4–12 g/dL) | Performed in test medium (moxifloxacin, 38%; trovafloxacin, 77%) | TKC | Presence of HA (12%) (which has the same PB capacity for trovafloxacin and moxifloxacin as 100% HS) significantly reduces the bactericidal activity of both fluoroquinolones against S. aureus, but not against P. aeruginosa, when compared to MHB | 2008 | 9 |
| MHB+HS (20–70%) | ||||||
| Fluoroquinolone | MHB+4 g/dL HA | Not performed, but cited from literature (moxifloxacin, 26%) | TKC, MIC | Presence of HA (4%) significantly reduces the bactericidal activity of moxifloxacin against S. aureus, when compared to MHB. This corresponds to the bactericidal activity of the free fraction of moxifloxacin in MHB | 2004 | 48 |
| MHB+calculated free fraction of antibiotic | ||||||
| Fluoroquinolone | MHB+HA (10%, 30%, and 50%) | Not performed | TKC | Presence of HA (10%, 30%, and 50%) does not significantly reduce the bactericidal activity of moxifloxacin, trovafloxacin, and ciprofloxacin against S. aureus, S. penumoniae, and E. coli when compared to MHB | 2000 | 8 |
| Fluoroquinolone | Broth+20% HS | Performed in 100% HS (BAY 12-8039, 26.4–30%) | MIC | Presence of HS (20% and 70%) does not significantly reduce the bactericidal activity of BAY 12-8039 against gram-positive pathogens, when compared to pure broth | 1997 | 52 |
| Broth+70% HS | ||||||
| Iclaprim | MHB+50% HP | Not performed, but cited from literature (iclaprim, 93%) | MIC | Presence of HP (50%) does not significantly reduce the bactericidal activity of iclaprim against MSSA and MRSA, when compared to MHB | 2007 | 51 |
| Fusidic acid | MHB+50% HP | Not performed, but cited from literature (fusidic acid, 97%) | MIC | Presence of HP (50%) significantly reduces the bactericidal activity of fusidic acid against MSSA and MRSA | 2007 | 51 |
| Glycopeptide | MHB+50% HP | Not performed, but cited from literature (teicoplanin, 90%; vancomycin, 55%) | MIC | Presence of HP (50%) does not significantly reduce the bactericidal activity of teicoplanin and vancomycin against MSSA and MRSA | 2007 | 51 |
| Daptomycin | MHB+90% HS | Not performed, but cited from literature (daptomycin, 91.7%) | TKC | Presence of HA (4 g/dL) and HS (90%) significantly delays, but does not significantly reduce the bactericidal activity of daptomycin against E. faecium | 2007 | 49 |
| MHB+4 g/dL HA | ||||||
| MHB+calculated free fraction of antibiotic | ||||||
| Daptomycin | MHB+4 g/dL HA | Not performed, but cited from literature (daptomycin, 91–95%) | TKC, MIC | MIC values of daptomycin against MRSA and VREF increased twofold to eightfold in the presence of albumin (4 g/dL) and decreased twofold in the presence of serum (50%) | 2004 | 7 |
| MHB+50% HS | Presence of HA (4 g/dL) delays, but does not significantly reduce the bactericidal activity of daptomycin against MRSA and VREF | |||||
| MHB+calculated free fraction of antibiotic |
MHB Mueller Hinton broth, HA human albumin, HS human serum, HP human plasma, MSSA methicillin-susceptible S. aureus, MRSA methicillin-resistant S. aureus, VREF vancomycin-resistant E. faecium, MIC minimal inhibitory concentration, TKC time–kill curve
To mimic in vivo conditions as close as possible, an ideal test medium would achieve a level of PB comparable to that of pure serum. On the other hand, the chosen test medium should not influence bacterial growth, when compared to protein-free broth growth media, to allow for quantification of the effect of PB independent from other factors. Table III lists the advantages and limitations of different media for determining antimicrobial activity of protein-bound antibiotics in vitro.
Table III.
Advantages and Limitations of Different Media for Determining the Antimicrobial Activity of Protein-Bound Antibiotics In Vitro
| Advantages | Limitations | References | ||
|---|---|---|---|---|
| MHB with the calculated protein-free concentration of an antibiotic | MHB is a liquid medium used for antibiotic susceptibility studies | No impairment of bacterial growth | Absence of proteins | 9,47,48,53,54 |
| Good overall comparability of results (widely used) | Does not account for reversible PB | |||
| Does not account for differences of influence of PB for gram-negative/gram-positive bacteria | ||||
| MHB with serum | Human serum is admixed to test medium at different concentrations (ranging from 20% to 100% serum) | Mimicry of in vivo conditions (including pH) | Intrinsic antibacterial effect of serum hampers bacterial growth | 7,50,52,55–65 |
| PB capacity of 100% serum approximates PB capacity of whole blood | Variety of serum content by different investigators | |||
| Relatively high costs and limited availability of human serum | ||||
| MHB with albumin | Albumin is admixed to broth (e.g. MHB) at a concentration of 4 g/dL | Absence of intrinsic antibacterial effect | Absence of serum proteins other than albumin | 7,9,47,49,66,67 |
| Bacterial growth equal to pure MHB | Limited PB capacity, compared to serum | |||
| Relatively high costs of human albumin | ||||
| PB capacity dependent on presence of ions like Ca++ | ||||
MHB Müller Hinton Broth
The most commonly determined PD parameter for bacterial susceptibility to an antibiotic is the minimum inhibitory concentration (MIC). In this context, Mueller Hinton broth (MHB) is a standard broth, which the Clinical and Laboratory Standards Institute recommends for susceptibility testing of the majority of aerobic and facultative anaerobic bacteria (54). MHB is a liquid medium containing beef infusion solids (4 g/L), starch (1.5 g/L), and casein hydrolysate (17.5 g/L) and exhibits a pH of 7.4 ± 0.2 at 37°C. Standard broth, such as MHB, provides optimum conditions required for bacterial growth. In contrast, some protein- or serum-rich media cause impairment of bacterial growth due to the presence of antibacterial-acting substances. Because MHB is widely used for determining bacterial susceptibility, results of in vitro simulations in MHB can be easily compared and interpreted. Although it is an artificial medium, MHB more closely resembles human serum in terms of pH, Na+, K+, and Cl− content and osmolality than other broths (53).
Due to the absence of protein, in vitro PD studies using MHB as a test medium cannot usually account for the influence of PB on antimicrobial activity. Therefore, to investigate the impact of PB of bacterial killing of antibiotics, bacterial susceptibility is often determined in MHB and MICs are consecutively compared to those determined in a protein-rich medium (9,47,48). An alternatively used approach to assess the influence of PPB calculates the protein-free concentration of an antibiotic in serum or growth medium with addition of albumin and simulates antimicrobial activity of the total and the calculated free fraction in MHB (47). However, this approach does not account for reversible PPB and differences of influence of PPB for gram-positive/gram-negative bacteria, i.e., it may overestimate the influence of PPB.
As serum can be considered to represent PB in vivo, serum-containing medium is frequently used for incorporating PB into in vitro studies of antimicrobial activity. Although animal serum has previously been used for this purpose (68), serum proteins from animal species have been shown to exhibit different binding characteristics compared to human serum (65). Therefore, only human serum might be considered appropriate for mimicking in vivo PB in men. Human serum for admixture may be obtained from volunteers on site or are acquired by purchase. In the serum-manufacturing process, whole blood is collected aseptically and allowed to clot. In the case of purchased serum, the serum of hundreds of individuals is then pooled, meaning that variability between lots can be widely excluded.
Using 100% serum for antimicrobial testing would best mimic in vivo conditions, including pH and protein concentration. However, human serum on the one hand is less optimal for bacterial growth than microbiological standard media and on the other hand contains substances that possess antibacterial activity. Growth rates of Staphylococcus aureus, for instance, have been shown to be reduced in media containing human serum (61). Furthermore, when bacterial strains are exposed to antibiotics in active serum, even subinhibitory antibiotic concentrations can exert a bactericidal effect due to the synergistic effect of antibiotics and serum on bacterial growth (7,57,59). There have been attempts to acclimate test strains to growth in human serum (61). Indeed, during these experiments, the tested organisms adapted to conditions in serum, despite the presence of antibacterial molecules. However, they again lost their serum resistance after several passages in microbiological media, precluding their use as reference strains.
A wide variety of serum concentrations in test medium, ranging from 20% to 100% serum, have been used by different investigators in studies examining changes in antimicrobial activity in relation to PB (50,52,55,56,58,60,62–64). In order to minimize the growth-inhibiting effects of serum on bacterial growth, most studies have limited the human serum concentration in test media to 70%. Nevertheless, no cut-off value for the impairment of bacterial growth by the addition of serum could be defined so far, as serum may inhibit bacterial growth even at concentrations of <50% (61).
Alternatively to using serum, broth containing human albumin as a protein supplement is utilized (7,48). In this approach, concentrations of albumin usually around 5% are used, which widely excludes factors hampering bacterial growth (9). Similar to serum, due to interspecies differences in binding affinities for antibiotics, human albumin appears most representative for human in vivo conditions (66). An albumin concentration of 4 g/dL is widely used, as this is equivalent to the albumin concentration in human serum (7,46,47,49,62,67). However, a major limitation of this approach is that, due to the possibility of binding to serum proteins beside albumin, this approach does not necessarily provide the same PB capacity as human serum (9). In this context, a recently conducted study tried to develop a more appropriate test medium with regard to PB capacity by up-titrating the concentration of albumin until a PB level equal to that in pure serum was achieved (9). MHB containing 12% albumin (corresponding to 114 g/L albumin) was shown to have the same PB capacity for two fluoroquinolones as pure serum, but did not, at the same time, impair bacterial growth of S. aureus and Pseudomonas aeruginosa over a period of 24 h when compared with pure MHB.
Besides serum and albumin, other body fluids have previously been used for testing antimicrobial activity with regard to impairment of antimicrobial activity, such as cerebrospinal fluid (69), prostatic extract (70), urine (70,71), and pus (8). The presence of (50%) pus in broth did not result in a significant reduction of bactericidal activity of trovafloxacin, moxifloxacin, and ciprofloxacin against S. aureus, both under aerobic and anaerobic conditions.
In addition to different media, two main PD approaches for determining the effect of PPB on bacterial killing in vitro can be distinguished: those based on the MIC and those based on a kill curve approach (72). The MIC is obtained by making twofold dilutions of the test antibiotic in a liquid culture medium containing or lacking proteins, inoculating it with 5 × 105 cfu/mL microorganisms, and incubating it at 35–37°C for 18–24 h. The smallest amount of antibiotic that inhibits visible growth of the microorganism represents the MIC. The MIC90 is defined as the antibiotic concentration at which 90% of the tested pathogens do not show visible growth over a period of 18–24 h. Determination of the minimum bactericidal concentration (MBC) can additionally be determined for antibiotic substances which induce bacterial death (bactericidal antibiotics). For this purpose, the same procedure used to determine the MIC is repeated. After 18–24 h of incubation, plates in which no visible growth is detectable are used to make subcultures in an antibiotic-free medium. These are reincubated for an additional 18–24 h. The MBC is defined as the concentration of an antibiotic that either totally eradicated bacteria or results in a >99.9% decrease in the initial inoculum (i.e., a 3 − log10 reduction in colony forming units per milliliter).
In time–kill curves (TKC), the change of bacterial counts, measured in colony forming units per milliliter, represents the monitored PD parameter. Colony forming units per milliliter are plotted against time in the presence of different antibiotic concentrations (48,72,73). For the determination of the impact of PPB, bacterial counts at different time points are commonly obtained as follows: Culture tubes containing the medium and desired concentration of protein and antibiotic are kept in a water bath at 37°C to allow for PB to take place. Subsequently, tubes are inoculated with strains of a microorganism at a standardized inoculum of 5 × 105 cfu/mL. At predefined time points, samples are removed and serially diluted with 0.9% sodium chloride and plated onto agar plates, which are incubated for 24 h at 37°C. The colonies are then counted and backextrapolated to the original volume to determine colony forming units per milliliter. For a more realistic simulation of in vivo PK, changing antibiotic concentrations in culture tubes for dynamic pharmacokinetic/pharmacodynamic studies may be applied (74,75). This can either be provided by continuous adjustment or by means of a dynamic circulation model.
Table IV provides a comparison on the advantages and short-comings of the MIC and the TKC approach. The MIC of an antibiotic for different bacterial strains can be easily determined in different investigational media. One major advantage of MIC is that it provides a fast way of screening for the presence of an influence of PB on bacterial killing. However, in this approach, antibiotic concentrations are not increased continuously, but in twofold dilution steps. In addition, MIC does only detect visible growth, i.e., increase of bacterial counts to approximately 1 × 107 cfu/mL after 18–24 h of incubation, but cannot distinguish between less pronounced growth and bacterial killing. Therefore, the MIC does not account for distinct differences in bacterial killing between protein-free and protein-containing media and may be considered inappropriate for the investigation of antibiotics displaying moderate PB (of less than 50%). TKC, on the other hand, provides information about the interaction between bacteria and anti-infectives as a function of concentration and time (48,73) and allow for the exact determination of colony forming units per milliliter. The main disadvantage of the TKC approach lies in the more elaborate procedure compared to the MIC method.
Table IV.
Advantages and Limitations of the MIC and the TKC approach
| Advantages | Limitations | References | |
|---|---|---|---|
| MIC | Easy to perform | Static approach, which does not account for dynamic pharmacokinetics | 61,72 |
| Currently the most widely used pharmacodynamic parameter for pharmacokinetic/pharmacodynamic modeling of antibiotic | Implies subjective assessment of turbidity changes | ||
| MBC can be determined | Only twofold dilution steps, small changes might be overseen | ||
| Permits easy analyzing of effects of drug combinations on bacterial growth | No information on “killing-kinetics” | ||
| For visible growth colony forming units per milliliter of 107 necessary, i.e., less pronounced growth is overseen | |||
| TKC | Microbial killing and growth is observed as a function of both time at a given antibiotic concentration, i.e., “killing-kinetics” is depicted | Elaborate technique | 48,72,73 |
| Curves in the presence (kill curves) and absence (growth curves) of antibiotic can be compared | Commonly only static approach, which does not account for dynamic pharmacokinetics | ||
| Antibiotic concentrations can either be held constant or changed to mimic an in vivo concentration profile | |||
| Exact determination of colony forming units per milliliter |
The MIC is obtained by making twofold dilutions of the test antibiotic in a liquid culture medium, inoculating it with 5 × 105 cfu/mL of bacteria, and incubating it at 35–37°C for 18–24 h. The smallest amount of antibiotic that inhibits visible growth of the microorganism represents the MIC. The TKC is obtained by plotting bacterial growth against time in the presence of chosen antibiotic concentrations. Bacterial counts are obtained as by predefined time points, samples are removed from the culture tubes, serially diluted, and plated onto agar plates, which are incubated for 24 h at 37°C. The colonies are then counted and backextrapolated to the original volume to determine colony forming units per milliliter
MIC Minimal inhibitory concentration, TKC Time Kill Curve
Some authors have previously presented results which disagree with the theory that only the free concentration of a drug exhibits antimicrobial activity. In an in vitro experiment on the antimicrobial activity of daptomycin against Enterococcus faecium, total drug concentrations in MHB containing physiological albumin concentration (4 g/dL) were sufficient to achieve bactericidal activity, whereas no bactericidal activity was determined for the unbound fraction of daptomycin in MHB (49). In another paper, it has been shown that whereas the time to achieve bactericidal activity of daptomycin was delayed in the presence of albumin, the extent of overall kill was generally unchanged, when compared to results in MHB (7). The lack of effect of albumin despite the high PB of above 90% was ascribed to the hypothesized reversibility in the PB of daptomycin (47). Analogous effects were found for dicloxacillin where PB reduced bacterial killing during the first 6 h but not after 24 h (76). Attempts to interpret cefditoren activity against Streptococcus pneumoniae on the basis of the calculated free drug fraction were shown to result in underestimation of antimicrobial activity (47). However, there are several possible methodological pitfalls of in vitro simulation models used to investigate the impact of PB on antimicrobial killing (9). For most experiments which failed to detect decreased bactericidal activity of highly bound antibiotics in protein-rich medium, PB had not been determined in the particular test medium but PB values were estimated from literature. Furthermore, the albumin concentration of 4 g/dL applied in most of the experiments may not achieve PB levels comparable to serum in case of highly protein-bound antibiotics. Although pure serum and MHB containing 4% albumin were shown to contain identical amounts of albumin, the PB capacity for fluoroquinolones in 4% albumin broth was considerably lower than that of pure serum (8.6% in 4% albumin versus 38% in pure serum and 36.8% in 4% albumin versus 77.1% in pure serum for moxifloxacin and trovafloxacin, respectively). This finding was explained by binding of fluoroquinolones to serum proteins other than albumin or by differences in the binding potency of albumin within these environments (9). Thus, it cannot be excluded that methodological errors lead to those results that neglect the impact of PB on antimicrobial activity.
When determining PB, inappropriately performed methods may easily lead to overestimation or underestimation of the extent of protein binding in a given medium. Likewise, differences between the medium used for the determination of PBB and the microbiological medium used for the determination of antimicrobial action may lead to differences in the actual percentage of bound antibiotic and, consecutively, misinterpretation of data. Therefore, every effort should be made to use identical media for the determination of PB and bacterial killing, ideally both representing in vivo conditions as close as possible.
CONCLUSION
Currently, no standard method for the investigation of the impact of PB on antimicrobial killing of antibiotics exists. This might explain somewhat contradictory results obtained from different studies. In this context, international microbiological societies are called upon to develop appropriate standards. However, it may not be possible to find standard mediums and test conditions which are appropriate for all antibiotics. In this case, it is recommended to carefully choose both test medium and PD model on a case by case base. An individual, multiple-step approach, depending on the antibiotic under investigation as well as on the bacterial strains tested, may be most appropriate. Measurement of PB in the finally chosen medium as well as determination of bacterial growth compared to standard growth media is considered mandatory.
References
- 1.Oravcova J., Bohs B., Lindner W. Drug–protein binding sites. New trends in analytical and experimental methodology. J. Chromatogr. B Biomed. Appl. 1996;677(1):1–28. doi: 10.1016/0378-4347(95)00425-4. [DOI] [PubMed] [Google Scholar]
- 2.Wright J. D., Boudinot F. D., Ujhelyi M. R. Measurement and analysis of unbound drug concentrations. Clin Pharmacokinet. 1996;30(6):445–462. doi: 10.2165/00003088-199630060-00003. [DOI] [PubMed] [Google Scholar]
- 3.Bergogne-Berezin E. Clinical role of protein binding of quinolones. Clin Pharmacokinet. 2002;41(10):741–750. doi: 10.2165/00003088-200241100-00004. [DOI] [PubMed] [Google Scholar]
- 4.Turnidge J. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Drugs. 1999;58(Suppl 2):29–36. doi: 10.2165/00003495-199958002-00006. [DOI] [PubMed] [Google Scholar]
- 5.Kunin C. M. Clinical pharmacology of the new penicillins. 1. The importance of serum protein binding in determining antimicrobial activity and concentration in serum. Clin. Pharmacol. Ther. 1966;7(2):166–179. doi: 10.1002/cpt196672166. [DOI] [PubMed] [Google Scholar]
- 6.Kunin C. M., Craig W. A., Kornguth M., Monson R. Influence of binding on the pharmacologic activity of antibiotics. Ann. N Y Acad. Sci. 1973;226:214–224. doi: 10.1111/j.1749-6632.1973.tb20483.x. [DOI] [PubMed] [Google Scholar]
- 7.Cha R., Rybak M. J. Influence of protein binding under controlled conditions on the bactericidal activity of daptomycin in an in vitro pharmacodynamic model. J. Antimicrob. Chemother. 2004;54(1):259–262. doi: 10.1093/jac/dkh259. [DOI] [PubMed] [Google Scholar]
- 8.Rubinstein E., Diamantstein L., Yoseph G., et al. The effect of albumin, globulin, pus and dead bacteria in aerobic and anaerobic conditions on the antibacterial activity of moxifloxacin, trovafloxacin and ciprofloxacin against Streptococcus pneumoniae, Staphylococcus aureus and Escherichia coli. Clin. Microbiol. Infect. 2000;6(12):678–681. doi: 10.1046/j.1469-0691.2000.00166.x. [DOI] [PubMed] [Google Scholar]
- 9.Zeitlinger M., Sauermann R., Fille M., Hausdorfer J., Leitner I., Muller M. Plasma protein binding of fluoroquinolones affects antimicrobial activity. J. Antimicrob. Chemother. 2008;61(3):561–567. doi: 10.1093/jac/dkm524. [DOI] [PubMed] [Google Scholar]
- 10.Bergan T., Engeset A., Olszewski W. Does serum protein binding inhibit tissue penetration of antibiotics? Rev. Infect. Dis. 1987;9(4):713–718. doi: 10.1093/clinids/9.4.713. [DOI] [PubMed] [Google Scholar]
- 11.Craig W. A., Ebert S. C. Protein binding and its significance in antibacterial therapy. Infect. Dis. Clin. North Am. 1989;3(3):407–414. [PubMed] [Google Scholar]
- 12.Craig W. A., Welling P. G. Protein binding of antimicrobials: clinical pharmacokinetic and therapeutic implications. Clin Pharmacokinet. 1977;2(4):252–268. doi: 10.2165/00003088-197702040-00002. [DOI] [PubMed] [Google Scholar]
- 13.Peterson L. R., Gerding D. N. Influence of protein binding of antibiotics on serum pharmacokinetics and extravascular penetration: clinically useful concepts. Rev. Infect. Dis. 1980;2(3):340–348. doi: 10.1093/clinids/2.3.340. [DOI] [PubMed] [Google Scholar]
- 14.Scheife R. T. Protein binding: what does it mean? DICP. 1989;23(7–8 Suppl):S27–31. doi: 10.1177/106002808902300706. [DOI] [PubMed] [Google Scholar]
- 15.Craig W. A., Kunin C. M. Significance of serum protein and tissue binding of antimicrobial agents. Annu. Rev. Med. 1976;27:287–300. doi: 10.1146/annurev.me.27.020176.001443. [DOI] [PubMed] [Google Scholar]
- 16.Merrikin D. J., Briant J., Rolinson G. N. Effect of protein binding on antibiotic activity in vivo. J Antimicrob Chemother. 1983;11(3):233–238. doi: 10.1093/jac/11.3.233. [DOI] [PubMed] [Google Scholar]
- 17.Wise R. The clinical relevance of protein binding and tissue concentrations in antimicrobial therapy. Clin. Pharmacokinet. 1986;11(6):470–482. doi: 10.2165/00003088-198611060-00004. [DOI] [PubMed] [Google Scholar]
- 18.Wise R. Protein binding of beta-lactams: the effects on activity and pharmacology particularly tissue penetration. II. Studies in man. J. Antimicrob. Chemother. 1983;12(2):105–118. doi: 10.1093/jac/12.2.105. [DOI] [PubMed] [Google Scholar]
- 19.Lindup W. E., Orme M. C. Clinical pharmacology: plasma protein binding of drugs. Br. Med. J. (Clin Res Ed) 1981;282(6259):212–214. doi: 10.1136/bmj.282.6259.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yano Y., Oguma T., Nagata H., Sasaki S. Application of logistic growth model to pharmacodynamic analysis of in vitro bactericidal kinetics. J. Pharm. Sci. 1998;87(10):1177–1183. doi: 10.1021/js9801337. [DOI] [PubMed] [Google Scholar]
- 21.Burkhardt O., Brunner M., Schmidt S., Grant M., Tang Y., Derendorf H. Penetration of ertapenem into skeletal muscle and subcutaneous adipose tissue in healthy volunteers measured by in vivo microdialysis. J. Antimicrob. Chemother. 2006;58(3):632–636. doi: 10.1093/jac/dkl284. [DOI] [PubMed] [Google Scholar]
- 22.Perry T. R., Schentag J. J. Clinical use of ceftriaxone: a pharmacokinetic–pharmacodynamic perspective on the impact of minimum inhibitory concentration and serum protein binding. Clin. Pharmacokinet. 2001;40(9):685–694. doi: 10.2165/00003088-200140090-00004. [DOI] [PubMed] [Google Scholar]
- 23.Mehrotra R., De Gaudio R., Palazzo M. Antibiotic pharmacokinetic and pharmacodynamic considerations in critical illness. Intensive Care Med. 2004;30(12):2145–2156. doi: 10.1007/s00134-004-2428-9. [DOI] [PubMed] [Google Scholar]
- 24.Benet L. Z., Hoener B. A. Changes in plasma protein binding have little clinical relevance. Clin. Pharmacol. Ther. 2002;71(3):115–121. doi: 10.1067/mcp.2002.121829. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta A. Clinical utility of free drug monitoring. Clin. Chem. Lab. Med. 2002;40(10):986–993. doi: 10.1515/CCLM.2002.172. [DOI] [PubMed] [Google Scholar]
- 26.Kurz H., Trunk H., Weitz B. Evaluation of methods to determine protein-binding of drugs. Equilibrium dialysis, ultrafiltration, ultracentrifugation, gel filtration. Arzneimittelforschung. 1977;27(7):1373–1380. [PubMed] [Google Scholar]
- 27.Lin Z. J., Musiano D., Abbot A., Shum L. In vitro plasma protein binding determination of flunarizine using equilibrium dialysis and liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2005;37(4):757–762. doi: 10.1016/j.jpba.2004.10.050. [DOI] [PubMed] [Google Scholar]
- 28.Scholtan W. [Methods of determination and theoretical principles of the serum protein binding of drugs (author’s transl)] Arzneimittelforschung. 1978;28(7):1037–1047. [PubMed] [Google Scholar]
- 29.Sebille B. Methods of drug protein binding determinations. Fundam. Clin. Pharmacol. 1990;4(Suppl 2):151s–161s. doi: 10.1111/j.1472-8206.1990.tb00073.x. [DOI] [PubMed] [Google Scholar]
- 30.Wan H., Rehngren M. High-throughput screening of protein binding by equilibrium dialysis combined with liquid chromatography and mass spectrometry. J. Chromatogr. A. 2006;1102(1–2):125–134. doi: 10.1016/j.chroma.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 31.Joukhadar C., Muller M. Microdialysis: current applications in clinical pharmacokinetic studies and its potential role in the future. Clin. Pharmacokinet. 2005;44(9):895–913. doi: 10.2165/00003088-200544090-00002. [DOI] [PubMed] [Google Scholar]
- 32.Le Quellec A., Dupin S., Tufenkji A. E., Genissel P., Houin G. Microdialysis: an alternative for in vitro and in vivo protein binding studies. Pharm. Res. 1994;11(6):835–838. doi: 10.1023/A:1018973607051. [DOI] [PubMed] [Google Scholar]
- 33.Stahl M., Bouw R., Jackson A., Pay V. Human microdialysis. Curr. Pharm. Biotechnol. 2002;3(2):165–178. doi: 10.2174/1389201023378373. [DOI] [PubMed] [Google Scholar]
- 34.Verbeeck R. K. Blood microdialysis in pharmacokinetic and drug metabolism studies. Adv. Drug Deliv. Rev. 2000;45(2–3):217–228. doi: 10.1016/S0169-409X(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 35.Heinze A., Holzgrabe U. Determination of the extent of protein binding of antibiotics by means of an automated continuous ultrafiltration method. In.t J. Pharm. 2006;311(1–2):108–112. doi: 10.1016/j.ijpharm.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 36.Whitlam J. B., Brown K. F. Ultrafiltration in serum protein binding determinations. J. Pharm. Sci. 1981;70(2):146–150. doi: 10.1002/jps.2600700208. [DOI] [PubMed] [Google Scholar]
- 37.Matsushita Y., Moriguchi I. Measurement of protein binding by ultracentrifugation. Chem. Pharm. Bull. (Tokyo) 1985;33(7):2948–2955. doi: 10.1248/cpb.33.2948. [DOI] [PubMed] [Google Scholar]
- 38.Hage D. S. Chromatographic and electrophoretic studies of protein binding to chiral solutes. J. Chromatogr. A. 2001;906(1–2):459–481. doi: 10.1016/S0021-9673(00)00957-2. [DOI] [PubMed] [Google Scholar]
- 39.Ackerman B. H., Taylor E. H., Olsen K. M., Abdel-Malak W., Pappas A. A. Vancomycin serum protein binding determination by ultrafiltration. Drug Intell. Clin. Pharm. 1988;22(4):300–303. doi: 10.1177/106002808802200404. [DOI] [PubMed] [Google Scholar]
- 40.Gastearena I., Dios-Vieitez M. C., Terraz M. M., Domingo S., Fos D. Determination of the alpha1-acid glycoprotein binding of azithromycin in vitro by equilibrium dialysis. J. Chemother. 1995;7(Suppl 4):26–28. [PubMed] [Google Scholar]
- 41.Hutchins J. E., Tyczkowska K., Aronson A. L. Determination of ampicillin in serum by using simple ultrafiltration technique and liquid chromatographic analysis. J. Assoc. Off. Anal. Chem. 1986;69(5):757–759. [PubMed] [Google Scholar]
- 42.Teraoka H., Nierhaus K. H. Measurement of the binding of antibiotics to ribosomal particles by means of equilibrium dialysis. Methods Enzymol. 1979;59:862–866. doi: 10.1016/0076-6879(79)59131-9. [DOI] [PubMed] [Google Scholar]
- 43.Tsai T. H., Cheng F. C., Hung L. C., Chen C. F. Determination of unbound ceftriaxone in rat blood by on-line microdialysis and microbore liquid chromatography. Int. J. Pharm. 1999;193(1):21–26. doi: 10.1016/S0378-5173(99)00309-9. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt S., Rock K., Sahre M., et al. The effect of protein binding on the pharmacological activity of highly bound antibiotics. Antimicrob. Agents Chemother. 2008;52(11):3994–4000. doi: 10.1128/AAC.00427-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Traunmuller F., Zeitlinger M., Zeleny P., Muller M., Joukhadar C. Pharmacokinetics of single- and multiple-dose oral clarithromycin in soft tissues determined by microdialysis. Antimicrob. Agents Chemother. 2007;51(9):3185–3189. doi: 10.1128/AAC.00532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer S. M., Kang S. L., Cappelletty D. M., Rybak M. J. Bactericidal killing activities of cefepime, ceftazidime, cefotaxime, and ceftriaxone against Staphylococcus aureus and beta-lactamase-producing strains of Enterobacter aerogenes and Klebsiella pneumoniae in an in vitro infection model. Antimicrob. Agents Chemother. 1995;39(8):1764–1771. doi: 10.1128/aac.39.8.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sevillano D., Gimenez M. J., Alou L., et al. Effects of human albumin and serum on the in vitro bactericidal activity of cefditoren against penicillin-resistant Streptococcus pneumoniae. J. Antimicrob. Chemother. 2007;60(1):156–158. doi: 10.1093/jac/dkm115. [DOI] [PubMed] [Google Scholar]
- 48.Zeitlinger M. A., Sauermann R., Traunmuller F., Georgopoulos A., Muller M., Joukhadar C. Impact of plasma protein binding on antimicrobial activity using time-killing curves. J. Antimicrob. Chemother. 2004;54(5):876–880. doi: 10.1093/jac/dkh443. [DOI] [PubMed] [Google Scholar]
- 49.Cafini F., Aguilar L., Gonzalez N., et al. In vitro effect of the presence of human albumin or human serum on the bactericidal activity of daptomycin against strains with the main resistance phenotypes in Gram-positives. J. Antimicrob. Chemother. 2007;59(6):1185–1189. doi: 10.1093/jac/dkm078. [DOI] [PubMed] [Google Scholar]
- 50.Tsuji B. T., Leonard S. N., Rhomberg P. R., Jones R. N., Rybak M. J. Evaluation of daptomycin, telavancin, teicoplanin, and vancomycin activity in the presence of albumin or serum. Diagn. Microbiol. Infect. Dis. 2008;60(4):441–444. doi: 10.1016/j.diagmicrobio.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 51.Laue H., Valensise T., Seguin A., Hawser S., Lociuro S., Islam K. Effect of human plasma on the antimicrobial activity of iclaprim in vitro. J. Antimicrob. Chemother. 2007;60(6):1388–1390. doi: 10.1093/jac/dkm392. [DOI] [PubMed] [Google Scholar]
- 52.Woodcock J. M., Andrews J. M., Brenwald N. P., Ashby J. P., Wise R. The in-vitro activity of faropenem, a novel oral penem. J. Antimicrob. Chemother. 1997;39(1):35–43. doi: 10.1093/jac/39.1.35. [DOI] [PubMed] [Google Scholar]
- 53.Peterson L. R., Shanholtzer C. J. Tests for bactericidal effects of antimicrobial agents: technical performance and clinical relevance. Clin. Microbiol. Rev. 1992;5(4):420–432. doi: 10.1128/cmr.5.4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.N. C. f. C. L. Standards. Methods for determining bactericidal activity of antimicrobial agents. Document M26-P. In: Stand. NCCL, ed. 7(2):35–76; 1987.
- 55.Balcabao I. P., Alou L., Aguilar L., Gomez-Lus M. L., Gimenez M. J., Prieto J. Influence of the decrease in ciprofloxacin susceptibility and the presence of human serum on the in vitro susceptibility of Streptococcus pneumoniae to five new quinolones. J. Antimicrob. Chemother. 2001;48(6):907–909. doi: 10.1093/jac/48.6.907. [DOI] [PubMed] [Google Scholar]
- 56.Bedenic B. Selection of Klebsiella pneumoniae mutants with high-level cefotaxime resistance during growth in serum containing therapeutic concentrations of cefotaxime. Chemotherapy. 2002;48(1):10–14. doi: 10.1159/000048581. [DOI] [PubMed] [Google Scholar]
- 57.Boswell F. J., Ashby J. P., Andrews J. M., Wise R. Effect of protein binding on the in vitro activity and pharmacodynamics of faropenem. J. Antimicrob. Chemother. 2002;50(4):525–532. doi: 10.1093/jac/dkf155. [DOI] [PubMed] [Google Scholar]
- 58.Edwards J. R. Cefotetan: antibacterial activity against Staphylococcus aureus in the presence of human serum. Chemioterapia. 1988;7(4):271–273. [PubMed] [Google Scholar]
- 59.Gustafsson I., Cars O. The influence of protein binding on the antibacterial activity of faropenem against Haemophilus influenzae. Clin. Microbiol. Infect. 2004;10(10):934–937. doi: 10.1111/j.1469-0691.2004.00977.x. [DOI] [PubMed] [Google Scholar]
- 60.Leuthner K. D., Cheung C. M., Rybak M. J. Comparative activity of the new lipoglycopeptide telavancin in the presence and absence of serum against 50 glycopeptide non-susceptible staphylococci and three vancomycin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2006;58(2):338–343. doi: 10.1093/jac/dkl235. [DOI] [PubMed] [Google Scholar]
- 61.Nix D. E., Matthias K. R., Ferguson E. C. Effect of ertapenem protein binding on killing of bacteria. Antimicrob. Agents Chemother. 2004;48(9):3419–3424. doi: 10.1128/AAC.48.9.3419-3424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Odenholt I., Lowdin E., Cars O. Pharmacodynamic effects of telavancin against methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains in the presence of human albumin or serum and in an in vitro kinetic model. Antimicrob. Agents Chemother. 2007;51(9):3311–3316. doi: 10.1128/AAC.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perl T. M., Pfaller M. A., Houston A., Wenzel R. P. Effect of serum on the in vitro activities of 11 broad-spectrum antibiotics. Antimicrob. Agents Chemother. 1990;34(11):2234–2239. doi: 10.1128/aac.34.11.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitz F. J., Boos M., Mayer S., Verhoef J., Milatovic D., Fluit A. C. In vitro activity of faropenem and 20 other compounds against beta-lactamase-positive and -negative Moraxella catarrhalis and Haemophilus influenzae isolates and the effect of serum on faropenem MICs. J. Antimicrob. Chemother. 2002;49(1):220–223. doi: 10.1093/jac/49.1.220. [DOI] [PubMed] [Google Scholar]
- 65.Wise R. Protein binding of beta-lactams: the effects on activity and pharmacology particularly tissue penetration. I. J. Antimicrob. Chemother. 1983;12(1):1–18. doi: 10.1093/jac/12.1.1. [DOI] [PubMed] [Google Scholar]
- 66.Pistolozzi M., Bertucci C. Species-dependent stereoselective drug binding to albumin: a circular dichroism study. Chirality. 2008;20(3–4):552–558. doi: 10.1002/chir.20521. [DOI] [PubMed] [Google Scholar]
- 67.Tsuji B. T., Rybak M. J. Short-course gentamicin in combination with daptomycin or vancomycin against Staphylococcus aureus in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob. Agents Chemother. 2005;49(7):2735–2745. doi: 10.1128/AAC.49.7.2735-2745.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Louie A., Kaw P., Liu W., Jumbe N., Miller M. H., Drusano G. L. Pharmacodynamics of daptomycin in a murine thigh model of Staphylococcus aureus infection. Antimicrob. Agents Chemother. 2001;45(3):845–851. doi: 10.1128/AAC.45.3.845-851.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lutsar I., Friedland I. R. Pharmacokinetics and pharmacodynamics of cephalosporins in cerebrospinal fluid. Clin. Pharmacokinet. 2000;39(5):335–343. doi: 10.2165/00003088-200039050-00003. [DOI] [PubMed] [Google Scholar]
- 70.Aagaard J., Gasser T., Rhodes P., Madsen P. O. MICs of ciprofloxacin and trimethoprim for Escherichia coli: influence of pH, inoculum size and various body fluids. Infection. 1991;19(Suppl 3):S167–169. doi: 10.1007/BF01643691. [DOI] [PubMed] [Google Scholar]
- 71.Zhanel G. G., Karlowsky J. A., Davidson R. J., Hoban D. J. Influence of human urine on the in vitro activity and postantibiotic effect of ciprofloxacin against Escherichia coli. Chemotherapy. 1991;37(3):218–223. doi: 10.1159/000238857. [DOI] [PubMed] [Google Scholar]
- 72.Mueller M., de la Pena A., Derendorf H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: kill curves versus MIC. Antimicrob. Agents Chemother. 2004;48(2):369–377. doi: 10.1128/AAC.48.2.369-377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stratton C. W., Weeks L. S., Aldridge K. E. Comparison of kill-kinetic studies with agar and broth microdilution methods for determination of antimicrobial activity of selected agents against members of the Bacteroides fragilis group. J. Clin. Microbiol. 1987;25(4):645–649. doi: 10.1128/jcm.25.4.645-649.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sauermann R., Delle-Karth G., Marsik C., et al. Pharmacokinetics and pharmacodynamics of cefpirome in subcutaneous adipose tissue of septic patients. Antimicrob. Agents Chemother. 2005;49(2):650–655. doi: 10.1128/AAC.49.2.650-655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeitlinger M. A., Erovic B. M., Sauermann R., Georgopoulos A., Muller M., Joukhadar C. Plasma concentrations might lead to overestimation of target site activity of piperacillin in patients with sepsis. J. Antimicrob. Chemother. 2005;56(4):703–708. doi: 10.1093/jac/dki284. [DOI] [PubMed] [Google Scholar]
- 76.Dudley M. N., Blaser J., Gilbert D., Zinner S. H. Significance of “extravascular” protein binding for antimicrobial pharmacodynamics in an in vitro capillary model of infection. Antimicrob. Agents Chemother. 1990;34(1):98–101. doi: 10.1128/aac.34.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]