

Abstract
Physiologically based pharmacokinetic (PBPK) models are composed of a series of differential equations and have been implemented in a number of commercial software packages. These models require species-specific and compound-specific input parameters and allow for the prediction of plasma and tissue concentration time profiles after intravenous and oral administration of compounds to animals and humans. PBPK models allow the early integration of a wide variety of preclinical data into a mechanistic quantitative framework. Use of PBPK models allows the experimenter to gain insights into the properties of a compound, helps to guide experimental efforts at the early stages of drug discovery, and enables the prediction of human plasma concentration time profiles with minimal (and in some cases no) animal data. In this review, the application and limitations of PBPK techniques in drug discovery are discussed. Specific reference is made to its utility (1) at the lead development stage for the prioritization of compounds for animal PK studies and (2) at the clinical candidate selection and “first in human” stages for the prediction of human PK.
Key words: absorption, clearance, distribution, PBPK, pharmacokinetics
INTRODUCTION
During the drug discovery process, many compounds are screened for their absorption, distribution, metabolism and excretion (ADME) properties using both in vitro and in vivo resource. To optimize the use of such screening, there has been a growing demand to predict human pharmacokinetics (PK) as early as possible. Accurate predictions of human PK would reduce failures in clinic related to poor ADME properties by enabling the early selection of the best candidates for development and the rejection of those with a low chance of success. Such PK predictions could be used to help choose the first dose for a clinical trial, to test the suitability of the compound for the particular dosage regimen, and even to predict the expected variability in the intended population. A large number of methodologies have been established for PK prediction, including allometric scaling (1–4) and physiologically based pharmacokinetic (PBPK) modeling (5,6). Until recently, the use of PBPK modeling had been limited in drug discovery and development due to the mathematical complexity of the models and the large amounts of in vivo animal tissue concentration data required. However, advances in the prediction of hepatic metabolism (7–10) and tissue distribution (11–17) from in vitro and in silico data have made these models more attractive (18–25). PBPK models provide the opportunity to integrate key input parameters from different sources not only to estimate PK parameters and predict plasma and tissue concentration–time profiles but also to gain mechanistic insight into compound properties.
Several commercial PBPK packages have become available (26) (http://www.simulations-plus.com/; http://www.simcyp.com/; http://www.pk-sim.com/; http://www.cyprotex.com/), and strategies for the application of PBPK in the drug discovery setting have been published and evaluated recently (23,24). This paper describes the PBPK methodology used in drug discovery, together with the applications and limitations of such models with reference to specific case studies within Pfizer.
PBPK MODEL STRUCTURE AND METHODS USED TO GENERATE THE NECESSARY INPUT DATA
PBPK Model Structure
PBPK models are composed of many compartments corresponding to the different tissues of the body, e.g., adipose, bone, brain, gut, heart, kidney, liver, lung, muscle, skin, and spleen, which are connected by the circulating blood system (arterial and venous). A schematic of a PBPK model is shown in Fig. 1. Each compartment is defined by a tissue volume and a tissue blood flow rate which is specific for the species of interest. These physiological parameters are available from several sources in the literature (23,27,28). A tissue can be described as either perfusion-rate-limited or permeability-rate-limited. Perfusion-rate-limited kinetics tends to occur for small lipophilic molecules that have no trouble crossing membranes; in this case, the blood flow to the tissue becomes the limiting process. Permeability-rate-limited kinetics occurs for polar larger molecules that have difficulty penetrating the tissue; here, the permeability across the cell membrane becomes the limiting process. Generic PBPK models used in drug discovery usually assume perfusion-rate-limited kinetics, and in general, the liver and kidney are considered to be the only sites of elimination. The mass balance differential equations used in the model have been described previously (23) and follow the principles shown below.
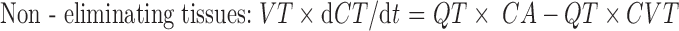 |
1 |
where Q is blood flow (L/h), C is concentration (mg/L), V is volume (L), T is tissues, A is arterial, V is venous, CVT = CT/Kp, where Kp is tissue to plasma partition coefficient of the compound.
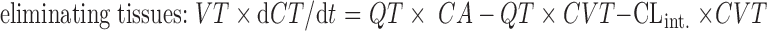 |
2 |
where CLint is the intrinsic clearance of the compound (L/h).
Fig. 1.

A schematic of a PBPK model (Q blood flow, CL int intrinsic clearance)
As well as species-specific physiological parameters, the model requires compound-specific information [i.e., metabolic clearance (CL), tissue to plasma partition coefficients (Kp values), and the rate and extent of absorption] to predict the plasma concentration time profile of the compound in the species of interest (e.g., rat, dog, human) after intravenous (i.v.) or oral administration. The mechanistic model provides a physiological framework facilitating the incorporation of other processes, e.g., active transport processes, when such data are available.
Methods to Predict CL
An important compound-specific parameter required for PBPK modeling is CL. Human CL is often predicted from preclinical species using allometric scaling (1,2,29–34). This can be achieved using preclinical data from multiple species. Such methods assume that differences in CL across species can be predicted by accounting for body weight differences. Corrections for brain weight (BRW) or maximum lifespan (MLP) can also be made. Mahmood and Balian (2) has proposed a rule of exponents to help determine in a prospective manner when and which correction factors should be used. An analysis from Sinha et al. (35) suggested that the most accurate prediction of human oral CL (CL/F) was obtained by scaling unbound CL/F in animal species using the rule of exponents to apply MLP or BRW corrections. For some compounds, the unbound fraction in plasma (fup)-corrected intercept method worked best (34). Some authors have proposed that it is possible to predict human PK from data in a single preclinical species using a fixed exponent method. For instance, Chiou et al. (36) showed that for a set of 54 extensively metabolized compounds, the mean allometric exponent for scaling from rat to human was 0.66. For initial predictions of human PK, we routinely use single species scaling of unbound CL from the rat with a fixed allometric exponent of 0.75.
Methodologies available for predicting hepatic CL (CLH) from in vitro systems (e.g., hepatocytes and microsomes) have been described in detail by Houston (7) and have been validated extensively in the rat (37–39). With advances in the storage and availability of human tissue, these approaches have been used successfully to predict human CLH in a number of cases (8–10). When scaling an in vitro CL to an in vivo CL value, the first step is to obtain an intrinsic CL (CLint) value from microsomal or hepatocyte data. In the discovery setting, such data are normally obtained from substrate depletion assays. CLint values are normalized for cell or microsomal protein concentration to obtain units of microliters per minutes per 106cells or microliters per minute per milligram of protein and subsequently corrected for any non-specific binding to give an unbound CLint [CLint(u)]. Binding to microsomes can be measured in vitro using techniques such as equilibrium dialysis and ultrafiltration or can be predicted using a number of in silico methods (40–43). In vitro CLint(u) can then be scaled to in vivo CLint(u) using formal scaling procedures accounting for the microsomal recovery or hepatocellularity and liver weight as described by Houston (7). Values for microsomal recovery and hepatocellularity are available in the literature for both rat and human (8,44–46). Finally, the in vivo CLint(u) value is used together with blood binding data and liver blood flow within a liver model, e.g., the well-stirred liver model to predict CLH. The main liver models used are the well-stirred model, the parallel-tube model, and the dispersion model. The different attributes of these models have been compared extensively in the literature (37,38).
The prediction of human renal CL (CLR) is normally made using in vivo preclinical data, as no in vitro model is available to scale CLR. In this context, allometric scaling from one or multiple species are usually used. If a compound is cleared by glomerular filtration only and is not reabsorbed or subjected to active uptake processes, CLR can be predicted from the glomerular filtration rate and plasma protein binding.
Methods to Predict Distribution
The development of mechanistic tissue composition equations (11–17) for the prediction of in vivo Kp values, and hence distribution in rat and human, have revolutionized and greatly extended the applicability of the PBPK approach to early drug discovery by reducing the need for in vivo experiments.
The first set of tissue composition equations was developed by Poulin and coworkers (11–13) and later modified by Berezhkovskiy (14). A detailed explanation of the mechanistic basis for these equations has been described (11–14). Briefly, these equations assume that the drug distributes homogenously into the tissue and plasma by passive diffusion where two processes are accounted for: (1) non-specific binding to lipids estimated from drug lipophilicity data (LogP and LogD) and (2) specific reversible binding to common proteins present in plasma and tissue estimated from fup. The main compound-specific input parameters are the pKa, LogD, and fup. The species-specific tissue composition parameters have been reported by Poulin and Theil (13).
Rodgers and co workers (15–17) extended and improved upon these tissue composition equations by incorporating ionization/charge considerations. A detailed explanation of the mechanistic basis for these equations has been described (15–17). Briefly, these equations account for four main processes: (1) partitioning of unionized drug into neutral lipids and neutral phospholipids, (2) dissolution of ionized and unionized drug in tissue water, (3) electrostatic interactions between ionized drug and acidic phospholipids for strong ionized bases, and (4) interactions with extracellular protein for neutrals, weak bases, and acids. The main compound-specific input parameters are the LogP, pKa, blood to plasma partitioning, and unbound fraction in plasma. The species-specific tissue composition parameters have been reported in Rodgers and Rowland (17).
Furthermore, Vss (volume of distribution at steady state) can be calculated using Eq. 3 as described previously (47).
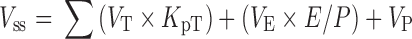 |
3 |
where E is erythrocyte and E/P is the erythrocyte/plasma ratio, which was estimated from B/P and the hematocrit content in blood (Ht).
Methods to Predict Absorption
Various simulation packages are available for the prediction of the rate and extent of oral absorption in human and preclinical species, e.g., GastroPlus™, SimCYP Ltd, PKSIM®, and ChloePK®. The models underlying these packages are based on the CAT model described by Yu and Amidon (48). The GastroPlus™ “advanced compartmental absorption transit” model (ACAT) (49), the SimCYP© “advanced, dissolution, absorption and metabolism” model (ADAM) (50), and the PKSIM® absorption model (51,52) have been described in detail in the literature. In brief, these absorption models are physiologically based transit models consisting of several compartments corresponding to different segments of the digestive tract, which describes the release, dissolution, degradation, metabolism, uptake, and absorption of a compound as it transits through the different segments of the digestive tract. The simulation software uses a variety of in vitro and in silico input data such as solubility, permeability, particle size, LogP, pKa, and dose together with a series of differential equations to model the kinetics associated with each of these processes.
Prediction Accuracy
Prediction accuracy is normally assessed in terms of the fold error from the observed value. In our work, we have considered less than twofold error to be an accurate prediction, i.e., if something is predicted to be within twofold error of the observed value, then the error is not greater than twofold. However, for some parameters and compounds, better prediction accuracy may be required. This is discussed later in the manuscript.
APPLICATIONS AND LIMITATIONS OF PBPK METHODOLOGY WITHIN PROJECTS AT PFIZER
PBPK Modeling in Lead Development (Prioritization for Animal PK Studies)
One potential application of PBPK modeling approaches is to predict animal PK before conducting studies with the aim of prioritizing compounds for animal studies and reducing the number of animal experiments needed. To do this, it is necessary to predict the absorption (if dosing orally), distribution, and CL of the compounds in question. Unlike later stages where information from preclinical species can be used to aid the predictions, in drug discovery, the prediction of these processes relies on the use of physicochemical properties, in vitro data, and increasingly in silico data. While there has been a great deal of progress in recent years in predicting absorption and distribution of compounds, the most difficult process to predict early in the life of a project is the CL. The prediction of these different processes will be discussed below in the context of animal PK at the lead development stage.
PBPK methods can be used within the discovery environment effectively to predict absorption and may help reduce the number of animals used. For example, Fig. 2 shows the accurate simulation of oral plasma concentration time profiles for a number of diverse (LogD, pH 7.4, 1.2–3.8; fup 0.008–0.52; PAMPA permeability 4–32 × 10−6 cm/s), soluble analogues from a discovery program following doses of 1 or 2 mg/kg to the rat. The only inputs into the GastroPlusTM ACAT model were in vitro solubility (as measured in phosphate buffer) and permeability inputs (routinely available at the discovery stage) combined with known i.v. disposition. In vitro flux in MDCK-MDR1 cell lines indicated that these analogues were not substrates for P-glycoprotein. It was assumed that gut metabolism was not limiting oral absorption. Despite this assumption, confidence gained by accurate prediction of oral PK for several compounds in the series reduced the need for further studies by allowing the scientist to rely totally on simulation.
Fig. 2.

Prediction of absorption in rat for a series of analogues using GastroPlus. Solid line, prediction; open square, observed data; dotted line, lower limit of quantification (if applicable to scale)
In addition to this, we have shown that this approach is valid for compounds from across our project portfolio. This retrospective analysis of oral PK profiles in rat allowed accurate prediction (within twofold) of Cmax (maximum concentration), Tmax (time of maximum concentration), and area under the plasma concentration time curve (AUC) in 60, 90, and 46% cases, given intravenous disposition, in vitro solubility in a physiologically relevant media (e.g., FeSSIF, fed state simulated intestinal fluid), and permeability estimates. The apparently poorer AUC prediction accuracy was skewed by poor performance at high doses (only 20% of the predictions were within twofold error when the oral dose was >20 mg/kg). The success of these retrospective analyses built confidence within the project team that these techniques could be employed prospectively to both reduce animal use and the costs associated with routine pharmacokinetic profiling, while also increasing the speed with which compound selection could proceed, given routinely available in vitro data.
There has been an improvement in the ability to predict the distribution of candidate agents from physicochemical properties and in vitro data. Methodologies proposed by both Rodgers and Rowland and Poulin and Theil have been outlined in the previous section and can be employed to prioritize candidates for progression to pre-clinical in vivo studies. A representative selection of the literature in this area, in addition to the experience in our own laboratories, is summarized in Table I.
Table I.
V ss Prediction Accuracy
| Authors | Year | Species | Prediction methodology | Prediction accuracy | Comments | |||
|---|---|---|---|---|---|---|---|---|
| Mean fold error | Percentage within twofold | |||||||
| K p/K pu | V dss | K p/K pu | V dss | |||||
| Original methodology manuscripts | ||||||||
| Poulin and Theil (11,12) | 2000/2001 | Mix | / | 1.26 | 70 | Non-adipose/adipose tissues (Note: combined rat, rabbit, mouse and human predictions) | ||
| Mix | / | 1.17 | 100 | |||||
| Rodgers and Rowland (15) | 2005 | Rat | / | 63 | Modified in accordance with Berezhkovskiy (14). Moderate–strong bases | |||
| Rodgers and Rowland (16) | 2006 | Rat | / | 63 | Modified in accordance with Berezhkovskiy (14). Weak bases, neutrals, and zwitterions | |||
| Retrospective analyses employing original methodologies to diverse datasets | ||||||||
| Poulin and Theil (13) | 2002a | Rat/Human | Poulin and Theil | 1.06 | 80 | Significant under-prediction of cationic-amphiphilic agents | ||
| Parrott et al. (20) | 2005 | Rat | Poulin and Theil | 1.9 | 62 | Over-prediction for high LogD/low fup acids; Under-prediction for basic compounds | ||
| Jones et al. (23) | 2006 | Human | Poulin and Theil | 2.3 | 41 | V dss or Vz/F | ||
| Germani et al. (53) | 2007 | Rat | Poulin and Theil | 3.2 | 30 | |||
| De Buck et al. (24,54) | 2007 | Human | Rodgers and Rowland | 1.1 | 85 | |||
| Poulin and Theil | 2.1 | 32 | ||||||
| Rat | Rodgers and Rowland | 1.17 (a) | 83 (a) | (a) basic compounds, (b) neutral compounds | ||||
| Poulin and Theil | 1.6 (a), 1.5 (b) | 60 (a), 42 (b) | ||||||
| Rodgers and Rowland (17) | 2007 | Rat | Rodgers and Rowland | 0.94 | 78 | |||
| Human | 0.86 | 64 | ||||||
| Pfizer | ongoing evaluation | Rat | Rodgers and Rowland | 2.1 | 60 | |||
| Human | 1.7 | 75 | ||||||
| Rat | Poulin and Theil | 1.8 | 60 | |||||
| Human | 2.1 | 45 | ||||||
A number of different authors (12,17,20,54) have reported the encouraging performance of these mechanistic equations, each having demonstrated accurate prediction of rat Vss (within twofold of observed) for greater than 60% of candidates. Poorer prediction accuracy has been reported by Germani et al. (53); however, this work focused primarily on the equations proposed by Poulin and Theil. Our own experiences confirm the utility of these approaches with accurate prediction of rat Vss being achieved in 60% of cases for both Poulin and Theil and Rodgers and Rowland approaches. One advantage to the prediction of Vss using these mechanistic tissue composition equations is the generation of Kp values which can be used within PBPK models, potentially resulting in better predictions of plasma concentration time profiles particularly in terms of shape when concentration versus time profiles are multi-exponential.
In our laboratories, we have investigated the distribution of a small number of candidates (n = 6) into rat heart, muscle, and lung tissues following infusion to steady state. We note the broad agreement (67% within twofold error of the observed value) between predicted and observed distribution into muscle and heart tissue. In addition, while the overall prediction of distribution into lung tissue was quantitatively less accurate (33% within twofold), it is interesting to note that our findings do not mirror the tendency to under-predict distribution into the lung tissue that has been demonstrated previously (15,54).
Perhaps the most difficult PK parameter to predict is CL; however, again, this is an essential input parameter for PBPK modeling. When simulations are performed before the compound is dosed to animals, in vivo data are, by definition, not available. Therefore, this parameter needs to be predicted from in vitro systems. In an analysis of recent in vivo rat PK studies at Pfizer Sandwich, it was found that when rat liver microsomal data were scaled to an in vivo blood CL (CLb), accounting for microsomal, plasma protein binding, and for the blood/plasma ratio, there was an average fivefold under-prediction of actual CLb. The predicted CL was within twofold of the actual value for only five of 37 compounds (13%; Fig. 3a). This trend for fivefold under-prediction was also seen when the dataset was expanded to 89 compounds [where the blood/plasma ratio was assumed to be 1 for the cases where it had not been measured; this assumed value is close to the mean blood/plasma ratio (0.98) of the 37 measured values available].
Fig. 3.

Rat in vivo CL prediction accuracy from rat in vitro systems. a Predicted (from rat microsomal CLint) versus observed in vivo blood clearance (CLb) for a series of 37 compounds for which in vivo intravenous rat PK data were determined at Pfizer, Sandwich. The solid line is the line of unity. b Predicted (from rat microsomal CLint) versus in vivo CLint for a series of 23 compounds. The solid line is the line of unity. c Comparison of rat liver microsomal data and hepatocyte date for 37 compounds. The solid line is the line of unity. d Predicted (from rat hepatocyte CLint) versus in vivo CLint for a series of 24 compounds. The blue- and pink-colored compounds are the same compounds in the rat liver microsomal plot (b). There is much less distinction between blue and pink compounds when hepatocyte data are used
Although the scaling shown in Fig. 3a used an in silico estimate of microsomal fraction unbound (fumic) (43), the fivefold under-prediction was also seen for a smaller subset of 20 compounds where the fumic had been experimentally determined by equilibrium dialysis. The under-prediction seems to be due to an underestimation of CLint from the in vitro incubation. Possible reasons for this observed under-prediction include metabolism by hepatic enzymes other than P450, metabolism of the compound in extrahepatic tissues, CL by non-metabolic mechanisms, e.g., hepatic uptake, and the enzyme activity in the in vitro system possibly not being representative of the activity in vivo.
Although lacking scientific rigor, empirically multiplying the in vitro CLint by 10 gives an average fold error of 2.1 with no systematic bias (70% of compounds are predicted to be within twofold of the actual CL).
When a plot of in vitro versus in vivo CLint is made (Fig. 3b) for the same set of compounds (limited to those with CLb < 60 ml min−1 kg−1 and assuming hepatic blood flow to be 70 ml min−1 kg−1), there appear to be two groups of compounds identified (those with <10 and those with >10 fold error). Those shown in diamonds show a reasonable correlation with the in vivo CLint (R2 > 0.7), although there is approximately on average a fivefold under-prediction of CLint and no cases where in vitro CLint was greater than in vivo CLint. The squares represent a subset of compounds with extremely poor predictions of CLint from the rat liver microsomal data (all compounds had greater than tenfold error and the average fold error for this subset was 30).
When data for rat hepatocytes and rat liver microsomes (for 37 of the compounds where data in both systems was available) are compared, the scaled CLint(u) from hepatocytes showed a tendency towards higher values than the rat liver microsomal data (Fig. 3c). However, compared to in vivo CLint(u), there was still an average fold error of 5.5-fold between in vitro (from hepatocytes) and in vivo estimates (n = 24), although for five compounds, the in vitro estimate was higher than that seen in vivo (Fig. 3d). In general, the compounds poorly predicted in rat liver microsomes (average fold error >10) were predicted better using hepatocytes (compare Fig. 3b, d). In vitro estimates of CLint(u) from hepatocytes were used to calculate CL (using the well-stirred model) for 38 compounds. The average fold error for these compounds was 4.4-fold, which, although better than that seen with rat liver microsomes, is still not good enough to be used in PBPK simulations. Five compounds had in vitro CL predictions greater than the actual in vivo CL, and for 31% of compounds, the predicted CL was within twofold of actual value.
Table II summarizes a number of publications which have similarly endeavored to correlate the CLint observed from in vitro rat liver microsome or rat hepatocyte incubations with in vivo CLint. While not intending to be a full and complete review of literature in this area, it is clear that there is a divergence of opinion as to the quantitative nature of the predictive power of the in vitro systems. For example, Houston and Carlile (37) reported successful prediction (70% within twofold) of rat CL from hepatocyte data, while a tendency to under-predict in vivo CL was observed using microsome data. Similarly, Ito and Houston (38) demonstrated an ability to predict rat CL from both microsome and hepatocyte data using either the well-stirred, parallel-tube, or dispersion models of the liver. While the quantitative prediction was more accurate using hepatocytes (accurate prediction in greater than 60% of cases), both methods offered a reasonable prediction of in vivo CLint centered around unity. Clarke and Jeffrey (61) looked at the ability of rat liver microsomal CLint to predict the in vivo CL of 1163 compounds across 48 discovery projects. Despite the limitations of the analysis [(in vitro CLint was compared to in vivo CL (mixture of CLb and CLp) and microsomal binding was not accounted for], this analysis does show that across a broad scope of chemical space, screening data can be used to roughly sort compounds into bins. While this can be of value for initial compound selection, a more defined estimate of CL is required for accurate PBPK simulation.
Table II.
CL Prediction Accuracy
| Authors | Year | Species | Liver model | CLint prediction accuracy | Comments | ||
|---|---|---|---|---|---|---|---|
| Mean fold Error | Percentage within twofold | Predictive tendency | |||||
| Microsomes | |||||||
| Houston and Carlile (37) | 1997 | Rat | Well stirred | 40 | Under-prediction | ||
| Obach (10) | 1999 | Human | Well stirred and parallel tube | WS: 2.3 (a), 4.4 (b), 2.3 (c), PT: 2.3 (a), 4.2 (b), 2.1 (c) | WS: 55 (a), 24 (b), 52 (c) PT: 59 (a), 28 (b), 55 (c) | Under-prediction unless all binding terms are omitted. | Comparison of CL, not CLint, presented. (a) no binding (b) incorporating fub only (c) incorporating both fub and fumic. |
| Ito and Houston (38) | 2004 | Rat | Well stirred, parallel tube, and dispersion | 2.1 (WS), 1.3 (PT), 1.4 (D) | 33 (WS), 39 (PT), 35 (D) | Dispersed around unity | |
| Ito and Houston (55) | 2005 | Human | Well Stirred | 6.17 | 15 | Under-prediction unless fup is omitted | |
| De Buck et al. (54) | 2007 | Rat | Well stirred and parallel tube | WS:1.2 (a), 13.5 (b), 2.5 (c) PT: 1.0 (a), 12 (b), 2.2 (c) | WS:86 (a), 17 (b), 61 (c) PT: 92 (a), 22 (b), 64 (c) | Under-prediction unless all binding terms are omitted | Comparison of CL, not CLint presented. (a) no binding (b) incorporating fub only (c) incorporating both fub and fumic. |
| Naritomi et al. (56) | 2001 | Human | Well stirred, parallel tube, and dispersion | 4 (WS), 3 (PT), 3.2 (D) | 25 (WS), 38 (PT), 50 (D) | Under-prediction | Predictions improved in original manuscript by integration of cross species scaling factor |
| Hepatocytes | |||||||
| Houston and Carlile (37) | 1997 | Rat | Well stirred | 70% | Dispersed around unity | ||
| Parrott et al. (20) | 2005 | Rat | Well stirred | 1.8 | 59 | Dispersed around unity assuming fup = fuinc | Assumes fuhep with diluted serum = fup |
| Germani et al. (53) | 2007 | Rat | Well stirred | 2.3 | 58 | Dispersed around unity assuming fup = fuinc | Comparison of CL, not CLint, presented. Assumes fuhep = fup. blood/plasma ratio assumed = 1 |
| Ito and Houston (38) | 2004 | Rat | Well stirred, parallel tube, and dispersion | 1.4 (WS), 1.3 (PT), 1.1 (D) | 63 (WS), 60 (PT), 66 (D) | Dispersed around unity | |
| Brown et al. (57) | 2007 | Human | Well stirred | 30 | Under-prediction | ||
| McGinnity et al. (58) | 2004 | Human | Well Stirred | 55 | Dispersed around unity assuming fup = fuinc | Assumes fup = fuhep blood/plasma ratio = 1. No acidic compounds | |
| Shibata et al. (59) | 2000 | Rat | Dispersion | 67 | Dispersed around unity assuming fup = fu serum inc | Comparison of CL, not CLint presented. Blood/plasma ratio assumed = 1 | |
| Riley et al. (60) | 2005 | Human | Well stirred | +FCS:86 (a), 3.1 (b), 42 (c) -FCS:13 (a),16 (b), 5.2 (c) | UNDER-PREDICTION unless binding terms are omitted. | Combined dataset from Lave et al. (9), Lau et al. (63), Shibata et al. (59), Naritomi et al. (56). (a) incorporating fub only (b) Assumes fub = fuhep (c) incorporating both fu blood and fu mic. ±FCS indicates the presence/absence of fetal serum albumin | |
| De Buck et al. (24) | 2007 | Human | Well stirred | 2.4 (a) 1.1 (b) | 53 (a) 74 (b) | Under-prediction unless all binding terms are omitted. | NB. Mix of microsome and hepatocyte data presented. (a) Incorporating both fub and fuhep. (b) Assumes fub = fuhep |
WS well stirred, PT parallel tube, D dispersion
Other authors have reported experiences similar to our own laboratory. Under-prediction from rat microsome data has been reported (54), which can only be corrected by the omission of both binding terms. Similar observations have been highlighted for hepatocyte data (20,53) where reasonable quantitative estimates were obtained by assuming that fuhep (fraction unbound in hepatocytes) and fub are equivalent. While in many cases offering accurate predictions and being similar to our own observations, the assumption that fumic is equivalent to fub has been disputed (62). Omission of either term sits uncomfortably with the mechanistically driven approach to predicting the behavior of candidates in vivo.
The reasons for the under-predictions from our own data are not completely clear, but as can be seen from Table III, the in vitro CLint values obtained in our in vitro assay are consistent with (or greater than) literature values for a number of well-studied compounds. Thus, the differences may well reflect that the current chemical space being explored by the discovery projects represented in this analysis is populated with compounds that are cleared by non-CYP-mediated mechanism or in extrahepatic tissues.
Table III.
Comparison of Rat Liver Microsome CLint for a Series of Literature Compounds in the Rat Liver Microsome Assay at Pfizer and in the Literature [Ito and Houston (38)]
| Compounds | RLM CLint (μl min−1 mg−1) | |
|---|---|---|
| Literature | Pfizer | |
| Alprenolol | 80 | >646 |
| Diltiazem | 250 | 604 |
| Felodipine | 500 | 2900 |
| Nicardipine | 6,700 | >4,636 |
| Omeprazole | 330 | 178 |
| Diclofenac | 110 | 188 |
| Ketoconazole | 42 | 137 |
| Metoprolol | 44 | 23 |
| Phenytoin | 35 | 38 |
The observed discrepancy in the rat between in vitro and in vivo CLint is a major stumbling block that is currently preventing us from fully implementing PBPK methodology prior to animal studies early in discovery projects. Although as illustrated empirical scaling factors can be used to correct for this deficiency to some extent, an explanation for the systematic under-prediction of CLint in rat from in vitro systems would be a big step forward enabling routine use of the PBPK techniques to prioritize compounds for the first time in animal studies.
PBPK Modeling for the Prediction of Human PK After Intravenous and Oral Administration
In recent years, there have been a number of publications from the pharmaceutical industry describing the use of PBPK methods for the prediction of human PK (19,22–25). Clearly, successful prediction of human PK requires the same description of compound PK as has previously been discussed in preclinical species, namely, distribution, CL, and, following oral administration, absorption. The approaches used to predict these processes in human are largely similar to those methods employed preclinically. A representative summary of the published literature in the distribution and CL areas are shown in Tables I and II, respectively.
Jones et al. (23) proposed and validated a strategy for the use of PBPK for human PK simulations. Essentially, the PBPK simulation is initially performed in animals using animal PBPK models, animal in vitro data (plasma binding and blood/plasma ratio), and compound-specific physicochemical data. The animal simulation is compared with the in vivo data. Providing the simulation in animals is reasonable; the human simulation is performed using a human PBPK model, human in vitro data, and compound-specific physicochemical data. If the simulation in animals is inaccurate, this would indicate a violation of the assumptions of the model; in this case, further experiments may be performed to understand the mismatch, e.g., is it due to non-hepatic elimination processes, permeability-limited tissue distribution, or involvement of active transport processes.
This iterative simulation approach was validated by Jones et al. (23) using a diverse set of 19 orally administered compounds reaching clinical development between 1998 and 2002. For those compounds where good predictions of animal PK were achieved, the accuracy in human in terms of the percentage of compounds with an average fold error of less than twofold was 92%, 67%, and 100% for AUC, Cmax, and Tmax, respectively. The authors illustrated the importance of achieving a good understanding in animals prior to the simulation in human, as the prediction accuracy was reduced (76%, 47%, and 94% for AUC, Cmax, and Tmax, respectively) when the compounds that were judged as having poor predictions in animals were included in the human analysis. In all cases, the authors observed that the PBPK approach was superior to standard allometric scaling methods. The authors state that in general, the compounds that were cleared by hepatic metabolism or renal excretion and whose absorption and distribution were governed by passive processes achieved good predictions using PBPK methods. Significant mispredictions were achieved when other elimination processes or active processes were involved.
Brightman et al. (22) used a PBPK model that had been parameterized using an optimization process (training set 69 intravenously administered compounds taken from the literature) to simulate the human plasma concentration time profiles of a test set of 18 intravenously administered compounds taken from the literature. The prediction accuracy reported for CL was 50% and 61% within two and threefold error of the observed values, respectively.
DeBuck et al. (24) applied the prediction strategy proposed by Jones et al. (23) to 26 compounds reaching clinical development at Johnson and Johnson. Both intravenous and oral administration data were available. For this set of mainly strongly ionized bases, the authors observed that the tissue composition equations developed by Rodgers and Rowland (15) were superior to the Poulin and Theil (13) equations for Vss estimation (84% versus 32% within twofold error of the observed value). This is to be expected for this class of compounds, as these equations incorporate the electrostatic interactions with the acidic phospholipids in the tissues. Using these tissue composition equations together with the ACAT model in GastroPlusTM, the authors achieved a prediction accuracy of 84%, 74%, 65%, and 81% within twofold error of the observed values for Vss, AUC, Cmax, and bioavailability.
In addition, Peters (25) simulated the human oral PK of nine compounds from the literature to within twofold error of the observed values by optimizing the CL and distribution parameters through intravenous profile fitting.
In our company, we have compared PBPK modeling (implemented in GastroPlusTM) to a simple one-compartmental PK model prediction method for the simulation of human PK. We performed intravenous and oral simulations for a set of 20 diverse in-house compounds which were selected based on the availability of intravenous human PK data. Initial validation of the PBPK modeling approach and assumptions was performed in rat and dog as proposed by Jones et al. (23). Human IV PBPK predictions were performed using the most appropriate tissue composition equations for distribution, human liver microsomes (accounting for both microsomal and plasma protein binding) for hepatically metabolized compounds, and allometric scaling from a single species for non-hepatically metabolized drugs. Human oral PBPK predictions were performed as above in combination with the ACAT model to simulate the absorption profile. The one-compartmental PK predictions were performed using the same CL value implemented in the PBPK model, a Vss value estimated by assuming unbound Vss is equal across species, an absorption rate constant equal to rat, fraction absorbed equal to rat, and a bioavailability calculated from the predicted CL and the fraction absorbed value. In agreement with others in the literature, the PBPK predictions were shown to be superior to the one-compartmental PK model predictions in the majority of cases in terms of the predicted PK parameters and also profile shape (Table IV). The PBPK method was able to capture the plasma concentration time profile shape more effectively than the one compartmental model. In order to further understand any mispredictions, simulations of intravenous and oral PK were rerun using the observed rather than predicted CL value as input into the model. These simulations showed that in over 90% of cases, distribution and absorption were being predicted accurately for this set of compounds. These improved predictions are likely due to the mechanistic nature of the models allowing the simulation of several phases to a profile together with species differences and nonlinearities in absorption. However, our results indicate that in humans, as in animals, the most challenging parameter to predict was CL and, consequently, bioavailability (results not shown). When considering hepatic CL only, we saw predictions of CL from microsomes that were within twofold of the observed for 70% of the cases, which is an improvement on our observations in the rat. Our results indicate that in many cases, even a prediction accuracy for CL of twofold is not good enough for accurate PBPK simulations, particularly for high CL compounds where small changes in a predicted CL value can have large effects on bioavailability. Further work is ongoing in our company to improve the estimation of human CL. The prediction of in vivo CL from microsomes have been shown by (10,24,55) to be improved when appropriate binding terms are excluded. In our study, we accounted for both microsomal and blood binding. We relied heavily on microsome data rather than hepatocyte data. Several authors (57,58) have reported under-predictions of CL also using hepatocyte data.`
Table IV.
Prediction Accuracy after Intravenous and Oral Administration to Humans for 19 Compounds
| Prediction method | % within twofold error of the observed value (threefold error of the observed value) | ||||
|---|---|---|---|---|---|
| V ss (i.v.) | Clearance (i.v.) | AUC (p.o.) | C max (p.o.) | Terminal half-life (p.o.) | |
| GastroPlus PBPK | 90 (100) | 80 (85) | 50 (72) | 67 (72) | 61 (83) |
| One-compartmental model | 75 (85) | 80 (85) | 33 (56) | 44 (61) | 50 (61) |
In addition, we have used PBPK modeling to prospectively predict the human PK and design the first in human trials of a number of compounds. Figure 4 shows the predicted versus observed data for compound X over a range of doses. The simulation was initially validated in animals following i.v. and oral administration. Simulations were performed in human using CL values estimated from human liver microsomes and from the dog CL (by single species scaling, exponent of 0.75). The simulated profile captured well the observed data particularly in terms of shape. These predictions were used to design the clinical study enabling informed dose setting and more confident calculations of bulk requirements. The model was refined as clinical data became available and was then used to predict food effects and formulation effects as the doses were increased.
Fig. 4.

Comparison of GastroPlus PBPK simulated profiles with the observed data for compound X. Solid line, GastroPlus PBPK prediction from human liver microsome data; dotted line, GastroPlus PBPK prediction from dog CL data; open squares, observed data
Conclusions
In summary, using examples from our own laboratories as well as literature data, we have shown how PBPK techniques can be used in drug discovery. The limitations of the PBPK approaches and some of the improvements required to further increase the utility of PBPK have also been discussed.
Our current focus is on using PBPK methodologies at the lead development stage for the prioritization of compounds for animal PK studies and at the clinical candidate selection and “first in human” stages for the prediction of human PK. A mixture of in silico, in vitro, and in vivo data is used in the PBPK simulations. As the ability to predict the input parameters for PBPK models improves, the need for in vitro and in vivo data will decrease. Eventually, we may be in a situation where human PK can be accurately predicted using in silico inputs only.
Acknowledgments
The authors thank Sue Cole, Rhys Jones, and Anne Heatherington for their input to this work.
Contributor Information
Hannah M. Jones, Phone: +44-1304-644207, FAX: +44-1304-651817, Email: hannah.jones@pfizer.com
Iain B. Gardner, Email: iain.gardner@pfizer.com
Kenny J. Watson, Email: kenny.j.watson@pfizer.com
References
- 1.Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982;10:201–227. doi: 10.1007/BF01062336. [DOI] [PubMed] [Google Scholar]
- 2.Mahmood I., Balian J. D. Interspecies scaling: Predicting pharmacokinetic parameters of antiepileptic drugs in humans from animals with special emphasis on clearance. J. Pharm. Sci. 1996;85:411–414. doi: 10.1021/js950400y. [DOI] [PubMed] [Google Scholar]
- 3.Obach R. S., Baxter J. G., Liston T. E., Silber B. M., Jones B. C., MacIntyre F., Rance D. J., Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- 4.Lave T., Coassolo P., Reigner B. Prediction of hepatic metabolic clearance based on interspecies allometric scaling techniques and in vitro–in vivo correlations. Clin. Pharmacokinet. 1999;36:211–231. doi: 10.2165/00003088-199936030-00003. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka C., Kawai R., Rowland M. Dose-dependent pharmacokinetics of cyclosporin A in rats: Events in tissues. Drug Metab. Dispos. 2000;28:582–589. [PubMed] [Google Scholar]
- 6.Nestorov I. Whole body pharmacokinetic models. Clin. Pharmacokinet. 2003;42:883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- 7.Houston J. B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994;47:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 8.Iwatsubo T., Hirota N., Ooie T., Suzuki H., Shimada N., Chiba K., Ishizaki T., Green C. E., Tyson C. A., Sugiyama Y. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol. Ther. 1997;73:147–171. doi: 10.1016/S0163-7258(96)00184-2. [DOI] [PubMed] [Google Scholar]
- 9.Lave T., Dupin S., Schmitt C., Valles B., Ubeaud G., Chou R. C., Jaeck D., Coassolo P. The use of human hepatocytes to select compounds based on their expected hepatic extraction ratios in humans. Pharm. Res. 1997;14:152–155. doi: 10.1023/A:1012036324237. [DOI] [PubMed] [Google Scholar]
- 10.Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 11.Poulin P., Theil F. P. A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J. Pharm. Sci. 2000;89:16–35. doi: 10.1002/(SICI)1520-6017(200001)89:1<16::AID-JPS3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 12.Poulin P., Schoenlein K., Theil F. P. Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J. Pharm. Sci. 2001;90:436–447. doi: 10.1002/1520-6017(200104)90:4<436::AID-JPS1002>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 13.Poulin P., Theil F. P. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J. Pharm. Sci. 2002;91:129–156. doi: 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- 14.Berezhkovskiy L. M. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J. Pharm. Sci. 2004;93(6):1628–40. doi: 10.1002/jps.20073. [DOI] [PubMed] [Google Scholar]
- 15.Rodgers T., Leahy D., Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005;94:1259–1276. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 16.Rodgers T., Rowland M. Physiologically based pharmacokinetic modeling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006;95(6):1238–57. doi: 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- 17.Rodgers T., Rowland M. Mechanistic approaches to volume of distribution predictions: Understanding the processes. Pharm. Res. 2007;24(5):918–33. doi: 10.1007/s11095-006-9210-3. [DOI] [PubMed] [Google Scholar]
- 18.Poulin P., Theil F. P. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J. Pharm. Sci. 2002;91:1358–1370. doi: 10.1002/jps.10128. [DOI] [PubMed] [Google Scholar]
- 19.Theil F. P., Guentert T. W., Haddad S., Poulin P. Utility of physiologically based pharmacokinetic models to drug development and rational drug discovery candidate selection. Toxicol. Lett. 2003;138:29–49. doi: 10.1016/S0378-4274(02)00374-0. [DOI] [PubMed] [Google Scholar]
- 20.Parrott N., Paquereau N., Coassolo P., Lave T. An evaluation of the utility of physiologically based models of pharmacokinetics in early drug discovery. J. Pharm. Sci. 2005;94:2327–2343. doi: 10.1002/jps.20419. [DOI] [PubMed] [Google Scholar]
- 21.Brightman F. A., Leahy D. E., Searle G. E., Thomas S. Application of a generic physiologically-based pharmacokinetic model to the estimation of xenobiotic levels in rat plasma. Drug Metab. Dispos. 2006;34:84–93. doi: 10.1124/dmd.105.004804. [DOI] [PubMed] [Google Scholar]
- 22.Brightman F. A., Leahy D. E., Searle G. E., Thomas S. Application of a generic physiologically-based pharmacokinetic model to the estimation of xenobiotic levels in human plasma. Drug Metab. Dispos. 2006;34:94–101. doi: 10.1124/dmd.105.004838. [DOI] [PubMed] [Google Scholar]
- 23.Jones H. M., Parrott N., Jorga K., Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharmacokinet. 2006;45(5):511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 24.De Buck S. S., Sinha V. K., Fenu L. A., Nijsen M. J., Mackie C. E., Gilissen R. A. H. J. Prediction of human pharmacokinetics using physiologically based modeling: A retrospective analysis of 26 clinically tested drugs. Drug Metab. Dispos. 2007;35(10):1766–80. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 25.Peters S. A. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin. Pharmacokinet. 2008;47(4):261–75. doi: 10.2165/00003088-200847040-00004. [DOI] [PubMed] [Google Scholar]
- 26.Rowland M., Balant L., Peck C. Physiologically based pharmacokinetics in drug development and regulatory science: A workshop report (Georgetown University, Washington, DC, May 29–30, 2002) AAPS PharmSci. 2004;6:E6. doi: 10.1208/ps060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies B., Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993;10:1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 28.Brown R. P., Delp M. D., Lindstedt S. L., Rhomberg L. R., Beliles R. P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health. 1997;13:407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 29.Dedrick R. L. Animal scale-up. J. Pharmacokinet. Biopharm. 1973;1:435–461. doi: 10.1007/BF01059667. [DOI] [PubMed] [Google Scholar]
- 30.Mordenti J. Man versus beast: Pharmacokinetic scaling in mammals. J. Pharm. Sci. 1986;75:1028–1040. doi: 10.1002/jps.2600751104. [DOI] [PubMed] [Google Scholar]
- 31.Lave T., Portmann R., Schenker G., Gianni A., Guenzi A., Girometta M. A., Schmitt M. Interspecies pharmacokinetic comparisons and allometric scaling of napsagatran, a low molecular weight thrombin inhibitor. J. Pharm. Pharmacol. 1999;51:85–91. doi: 10.1211/0022357991772006. [DOI] [PubMed] [Google Scholar]
- 32.Tang H., Mayersohn M. A novel model for prediction of human drug clearance by allometric scaling. Drug Metab. Dispos. 2005;33:1297–1303. doi: 10.1124/dmd.105.004143. [DOI] [PubMed] [Google Scholar]
- 33.Tang H., Mayersohn M. A global examination of allometric scaling for predicting tested drugs. Drug Metab. Dispos. 2007;35:1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 34.Mahmood I. Prediction of human drug clearance from animal data: Application of the rule of exponents and ‘fu corrected intercept method’ (FCIM) J. Pharm. Sci. 2006;95(8):1810–1821. doi: 10.1002/jps.20650. [DOI] [PubMed] [Google Scholar]
- 35.Sinha V. K., De Buck S. S., Fenu L. A., Smit J. W., Nijsen M., Gilissen R. A. H. J., Van Peer A., Lavrijsen K., Mackie C. E. Predicting oral clearance in humans how close can we get with allometry? Clin. Pharmacokinet. 2008;47(1):35–45. doi: 10.2165/00003088-200847010-00004. [DOI] [PubMed] [Google Scholar]
- 36.Chiou W. L., Robbie G., Chung S. M., Wu T. C., Ma C. Correlation of plasma clearance of 54 extensively metabolized drugs between humans and rats: mean allometric coefficient of 0.66. Pharm. Res. 1998;15(9):1474–9. doi: 10.1023/A:1011974226596. [DOI] [PubMed] [Google Scholar]
- 37.Houston J. B., Carlile D. J. Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices. Drug Metab. Rev. 1997;29:891–922. doi: 10.3109/03602539709002237. [DOI] [PubMed] [Google Scholar]
- 38.Ito K., Houston J. B. Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm. Res. 2004;21:785–792. doi: 10.1023/B:PHAM.0000026429.12114.7d. [DOI] [PubMed] [Google Scholar]
- 39.Jones H. M., Houston J. B. Substrate depletion approach for determining in vitro metabolic clearance: Time dependencies in hepatocyte and microsomal incubations. Drug Metab. Dispos. 2004;32:973–982. doi: 10.1124/dmd.104.000125. [DOI] [PubMed] [Google Scholar]
- 40.Austin R. P., Barton P., Cockroft S. L., Wenlock M. C., Riley R. J. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos. 2002;30:1497–1503. doi: 10.1124/dmd.30.12.1497. [DOI] [PubMed] [Google Scholar]
- 41.Austin R. P., Barton P., Mohmed S., Riley R. J. The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab. Dispos. 2005;33:419–425. doi: 10.1124/dmd.104.002436. [DOI] [PubMed] [Google Scholar]
- 42.Hallifax D., Houston J. B. Binding of drugs to hepatic microsomes: Comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos. 2006;34(4):724–26. doi: 10.1124/dmd.105.007658. [DOI] [PubMed] [Google Scholar]
- 43.D. B. Turner, A. Rostami-Hodjegan, G. T. Tucker, and K. Rowland-Yeo. Prediction of non-specific hepatic microsomal binding from readily available physicochemical properties. 9th European ISSX Meeting June 4th–7th, Manchester, UK. (2006).
- 44.Carlile D. J., Zomorodi K., Houston J. B. Scaling factors to relate drug metabolic clearance in hepatic microsomes, isolated hepatocytes, and the intact liver: studies with induced livers involving diazepam. Drug Metab. Dispos. 1997;25:903–911. [PubMed] [Google Scholar]
- 45.Wilson Z. E., Rostami-Hodjegan A., Burn J. L., Tooley A., Boyle J., Ellis S. W., Tucker G. T. Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br. J. Clin. Pharmacol. 2003;56:433–440. doi: 10.1046/j.1365-2125.2003.01881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barter A. E., Beaune P. H., Boobis A. R., Carlile D. J., Edwards R. J., Houston J. B., Lipscomb J. B., Pelkonen O. R., Tucker G. T., Rostami Hodjegan A. Scaling factors for the extrapolation of in vivo microsomal protein and hepatocellularity per gram of liver. Curr. Drug. Metab. 2007;8(1):33–45. doi: 10.2174/138920007779315053. [DOI] [PubMed] [Google Scholar]
- 47.Sawada Y., Hanano M., Sugiyama Y., Harashima H., Iga T. Prediction of the volumes of distribution of basic drugs in humans based on data from animals. J. Pharmacokinet. Biopharm. 1984;12:587–596. doi: 10.1007/BF01059554. [DOI] [PubMed] [Google Scholar]
- 48.Yu L. X., Amidon G. L. A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 1999;186:119–125. doi: 10.1016/S0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 49.Agoram B., Woltosz W. S., Bolger M. B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001;50(Suppl 1):S41–67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 50.D. B. Turner, M. Jamei, G. T. Tucker, and A. Rostami-Hodjegan. Supersaturation properties of poorly soluble weak bases are key factors in determining drug absorption: A simulation study of nifedipine using the ADAM model (Simcyp v7.1). “EUFEPS & COST B25 Conference on Bioavailability and Bioequivalence: Focus on physiological factors and variability. October 1–2, 2007, Athens, Greece. (2007).
- 51.Willmann S., Schmitt W., Keldenich J., Dressman J. B. A physiologic model for simulating gastrointestinal flow and drug absorption in rats. Pharm. Res. 2003;20:1766–1771. doi: 10.1023/B:PHAM.0000003373.72652.c0. [DOI] [PubMed] [Google Scholar]
- 52.Willmann S., Schmitt W., Keldenich J., Lippert J., Dressman J. B. A physiological model for the estimation of the fraction dose absorbed in humans. J. Med. Chem. 2004;47:4022–4031. doi: 10.1021/jm030999b. [DOI] [PubMed] [Google Scholar]
- 53.Germani M., Crivori P., Rocchetti M., Burton P. S., Wilson A. G. E., Smith M. E., Poggesi I. Evaluation of a basic physiologically based pharmacokinetic model for simulating the first-time-in-animal study. Eur. J. Pharm. Sci. 2007;31:190–201. doi: 10.1016/j.ejps.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 54.De Buck S. S., Sinha V. K., Fenu L. A., Gilissen R. A. H. J., Mackie C. E., Nijsen M. J. The prediction of drug metabolism, tissue distribution, and bioavailability of 50 structurally diverse compounds in rat using mechanism-based absorption, distribution, and metabolism prediction tools. Drug Metab. Dispos. 2007;35:649–659. doi: 10.1124/dmd.106.014027. [DOI] [PubMed] [Google Scholar]
- 55.Ito K., Houston J. B. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm. Res. 2005;22:103–112. doi: 10.1007/s11095-004-9015-1. [DOI] [PubMed] [Google Scholar]
- 56.Naritomi Y., Terashita S., Kagayama A., Sugiyama Y. Utility of hepatocytes in predicting drug metabolism: Comparison of hepatic intrinsic clearance in rats and humans in vivo and in vitro. Drug Metab. Dispos. 2003;31:580–588. doi: 10.1124/dmd.31.5.580. [DOI] [PubMed] [Google Scholar]
- 57.Brown H. S., Griffin M., Houston J. B. Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab. Dispos. 2007;35:293–301. doi: 10.1124/dmd.106.011569. [DOI] [PubMed] [Google Scholar]
- 58.McGinnity D. F., Soars M. G., Urbanowicz R. A., Riley R. J. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab. Dispos. 2004;32:1247–1253. doi: 10.1124/dmd.104.000026. [DOI] [PubMed] [Google Scholar]
- 59.Shibata Y., Takahashi H., Ishii Y. A convenient in vitro screening method for predicting in vivo drug metabolic clearance using isolated hepatocytes suspended in serum. Drug Metab. Dispos. 2000;28:1518–1523. [PubMed] [Google Scholar]
- 60.Riley R. J., McGinnity D. F., Austin R. P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos. 2005;33:1304–1311. doi: 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]
- 61.Clarke S. E., Jeffrey P. Utility of metabolic stability screening: Comparison of in vitro and in vivo clearance. Xenobiotica. 2001;31:591–598. doi: 10.1080/00498250110057350. [DOI] [PubMed] [Google Scholar]
- 62.Grime K., Riley R. J. The impact of in vitro binding on in vitro–in vivo extrapolations, projections of metabolic clearance and clinical drug–drug interactions. Curr. Drug Metab. 2006;7:251–264. doi: 10.2174/138920006776359266. [DOI] [PubMed] [Google Scholar]
- 63.Lau Y. Y., Sapidou E., Cu X., White R. E., Cheng K. C. Development of a novel in vitro model to predict hepatic clearance using fresh, cryopreserved and sandwich-cultured hepatocytes. Drug. Metab. Dispos. 2002;30:1446–1454. doi: 10.1124/dmd.30.12.1446. [DOI] [PubMed] [Google Scholar]