

Abstract
Protein identification via peptide mass fingerprinting (PMF) remains a key component of high-throughput proteomics experiments in post-genomic science. Candidate protein identifications are made using bioinformatic tools from peptide peak lists obtained via mass spectrometry (MS). These algorithms rely on several search parameters, including the number of potential uncut peptide bonds matching the primary specificity of the hydrolytic enzyme used in the experiment. Typically, up to 1 of these “missed cleavages” are considered by the bioinformatics search tools, usually after digestion of the in silico proteome by trypsin. Using two distinct, non-redundant datasets of peptides identified via PMF and tandem MS, a simple predictive method based on information theory is presented which is able to identify experimentally defined missed cleavages with up to 90% accuracy from amino acid sequence alone. Using this simple protocol, we are able to “mask” candidate protein databases so that confident missed cleavage sites need not be considered for in silico digestion. We show that that this leads to an improvement in database searching, with two different search engines, using the PMF dataset as a test set. In addition, the improved approach is also demonstrated on an independent PMF data set of known proteins which also has corresponding high quality tandem MS data, validating the protein identifications. This approach has wider applicability for proteomics database searching and the program for predicting missed cleavages and masking Fasta-formatted protein sequence databases has been made available via http://ispider.smith.man.acuk/MissedCleave
Keywords: Proteomics, tryptic cleavage, missed cleavage, mass spectrometry, protein identification, scoring systems
Introduction
The accurate analysis of the proteome using mass spectrometry (MS) plays an important role in the understanding of many of the physiological processes that occur in an organism and has become a standard tool used in the identification of proteins. A common approach for protein identification, particularly in high-throughput technologies, is two-dimensional (2D) gel separation followed by enzymatic digestion, matrix-assisted laser desorption/ionization (MALDI)-time-of-flight (TOF) mass measurement and identification by peptide mass fingerprinting (PMF). PMF involves the comparison of experimentally determined peptide masses to molecular weights derived from an in-silico digest of a sequence database. The reliability of a protein match is dependent on the mass accuracy of the data, the validity of the sequence databases searched and the post-translational modification patterns. The task of protein identification is made even more challenging by the occurrence of partial enzymatic protein cleavage, resulting in peptides with internal missed cleavage sites, as proteases frequently fail to digest proteins to their limit peptides.
The endopeptidase trypsin is commonly the enzyme of choice for MS experiments, primarily because of its high cleavage specificity, availability and cost1. As a member of the serine protease family the enzymatic mechanism of trypsin involves the recognition of a target amino acid in a binding pocket and the subsequent cleavage of the C-terminal amide body by a mechanism involving a serine residue on the protease. A negatively charged aspartate residue at the bottom of this deep and narrow binding pocket limits the amino acids to which this enzyme will recognise; only arginine and lysine have the long, basic side chains that are required to form a salt-bridge with this aspartate1.
The most widely used definition of tryptic specificity, implemented in most search tools, is that it cleaves C-terminal to arginine or lysine residues except where these residues are directly followed by a proline residue. The true specificity of any protease, however, is likely to be influenced by other residues in close proximity to the cleavage site in addition to other factors such as local conformation, tertiary structure and experimental conditions2. Indeed, more complex sequence based tryptic cleavage rules have been investigated3,4,5,6 and these rules are summarized in Table 1. Monigatti and Berndt3 investigated the missed cleavage patterns in over 10,000 PMF spectra from two different species, Bacillus subtilitis and human and found rules with some overlap (summarized in Table 1) to Thiede and co-workers4 who discussed a number of missed cleavage patterns in the PMF analysis of 104 protein digests from human Jurkat T cells and Mycobacterium. Yen and co-workers5 investigated missed cleavage patterns in 11,849 high-confidence peptide assignments from the tandem MS of a soluble protein extract from an erythroleukemia K562 cell line and again found overlap with the other two studies (Table 1). In summary the patterns show three principle features: the well documented effect of proline in position P1′, the negative effect of the basic residues lysine and arginine in position P1′ and the negative influence of negatively charged residues such as glutamate and aspartate surrounding the cleavage site.
Table 1. Sequence patterns promoting missed cleavages in trypsin digestions.
| Position relative to cleavage site A | Data Source |
||||||
|---|---|---|---|---|---|---|---|
| P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | |
| [WYF] | [RK] | [^RK] | B | ||||
| [^RK] | [RK] | [WYF] | B | ||||
| [DE] | [RK] | [^RK] | B C | ||||
| [^RK] | [RK] | [DE] | B C | ||||
| [RK] | [^RK] | [DE] | [DE] | D | |||
| [DE] | [DE] | [^RK] | [RK] | D | |||
| [DE] | [^RK] | [RK] | [^RK] | [DE] | D | ||
| [^RK] | [RK] | [RK] | B C | ||||
| [^RK] | [RK] | [H] | B | ||||
| [RK] | [P] | B C | |||||
The sequence surrounding the cleavage site is described as in Schechter and Berger13 as P4-P3-P2-P1-||-P1′-P2′-P3′-P3′, where cleavage occurs between P1 and P1′. Sequence patterns are represented as regular expressions where all letters in the square brackets represent the one letter amino acid codes, and for each set of brackets one of the residues must occur in this sequence position. A ‘^‘ symbol represents that the residue (s) within the brackets are to be excluded.
Data from a PMF study by Monigatti et al., 2005 3
Data from a statistical study of proteolysis specificity by Keil, 1992 6
Data from a tandem MS study by Yen et al., 2006 5
Although not widely reported, the missed cleavage of tryptic peptide bonds is a common phenomenon. For example, in the PepSeeker database of peptide identifications from tandem MS7, over 40% of the total unique top-ranking tryptic peptides contain one or more missed cleavage. Indeed, several different amino acid sequence patterns associated with missed cleavages in the tryptic digestion of proteins have been reported in previous studies3,4,5,6. However, none have as yet been formally incorporated into PMF tools. Yen and co-workers5 applied a set of simple “missed cleavage” rules in the form of regular expressions to create a restricted search database for tandem MS analysis. Their method improved the discrimination between correct and incorrect peptide assignments for both Mascot8 and SEQUEST9 searches although they observed only a small increase (3%) in true positive identifications5. A slightly different approach has been adopted by Gattiker and co-workers10 where missed cleavage information has been incorporated into their protein digestion tool, PeptideCutter, although this is not yet incorporated in to search tools or scoring systems. Recently, Brown and colleagues demonstrate that an excess of fully digested peptides relative to peptides containing one or more missed cleavages is an excellent marker for true protein identifications11.
Other studies have investigated the effect of the partials setting in PMF searches, for instance Ossipova and co-workers12 found that the minimum number of partials allowed for each search can influence the protein identification results, moreover they found this can vary between different PMFs. Indeed, the ability to locate those potential tryptic cleavage sites that are probably not cleaved during the digestion process is likely to aid in the reliable identification of proteins by PMF.
Here we describe the use of information theory in the generation of probabilities associated with sequence patterns leading to missed cleavages and the subsequent incorporation of this information into two different PMF protein identification tools. By masking the database for high-confidence missed cleavages, we show that this leads affects the number of candidate tryptic peptides per protein, and this leads to improved identifications using a variety of test systems. We believe that the technique offers a generalized approach to limiting proteome database searching by masking putative missed cleavages.
Experimental Section
Using an information theoretic approach we have investigated patterns in the amino acids surrounding potential cleavage sites in proteins digested with trypsin. The goal was to extract the relationship between the cleavage patterns (i.e. missed/cleaved) of a particular tryptic site and the surrounding amino acid sequence, with the ultimate aim of applying this information to PMF tools to improve the protein identification process. A large data set of high quality peptide identifications from tandem MS studies were used to generate missed cleavage rules and these were then tested on a dataset of PMF data from the Manchester Mascot8 server and also to an experimentally obtained PMF data set of known proteins.
Information theory
We have applied an information theoretic approach, to calculate a log-likelihood score, I, estimated from a training set of high confidence peptide identifications from tandem MS (see training data). The score I represents the information supplied about the state S of a putative tryptic site i by each amino acid in a 9 residue window centred about position i. The information score at i for an amino acid type R at position j in the window is shown below in Equation 1.
| Equation 1 |
where FS/Rj is the frequency of a particular residue type R at position j with state S (missed or cleaved) at position i, FR is the general frequency of residue R at position j, FS the frequency of state S and N the total number of amino acids in the dataset. The 9 residue window corresponds to P5-P4-P3-P2-P1-∥-P1′-P2′-P3′-P4′ using the nomenclature of Schechter and Berger13 where cleavage occurs between P1 and P1′. Two information matrices were calculated, one from the observed missed cleavage data (Mm) and a second for the observed actual cleavage data (Mc) for the 20 standard amino acids observed at all nine positions (the eight residues surrounding the cleavage site and the potential tryptic K/R residue). Hence, for every potential tryptic site in a protein two scores SC and SM were calculated by summing appropriate values in Mm and Mc as shown in Equation 2 and Equation 3 below.
| Equation 2 |
| Equation 3 |
Training data
High confidence tandem MS peptide identifications with a Mascot expect score better than 0.05 were used to create the information theory matrices. This data was taken from the PepSeeker database7 and a total of 23,077 top ranking, unique peptide sequences with associated high confidence Mascot identifications. Of these, 9,654 (42%) peptides contained at least one missed cleavage, 4161 (42%) contained one or more internal arginine residues, 5705 (58%) peptides contained one or more internal lysine residues, and 212 (2%) of peptides had both. Here we formally define a missed cleavage as any uncut lysine/arginine peptide bond, including those preceding a proline, although the vast majority of the training data here are non-proline missed cleavages: 8638 (89%) are non-proline.
Test data
The test data for both of the information matrices and the PMF algorithms was a set of PMF queries taken from a local Mascot server8 in Manchester, consisting of 13,179 queries, 9997 of which are unique and have 63,224 associated identified peptides with corresponding peaks in the mass spectrum. The data was provided by a range of laboratories, using a variety of different protocols, ensuring there is no inherent bias towards a single laboratory or protocol in the data.
PMF database searches were re-run using Mascot and an in-house tool Imprint (see Results), using the same search parameters used to obtain the identification by local laboratories. For example the error tolerances, post-translational modifications, enzyme and charge remained the same as the original query. The number of potential partial cleavages was reset, and the taxonomical filter was set to All Entries. For the Mascot searches, the search database remained the same as the original query, whilst all Imprint searches were run against the Swissprot (version 44.5) database due to time constraints. Hence, for consistency, only results of those Imprint searches (5401 in total) for which the protein identified by Mascot had a close homologue (>70% identity) in the Swissprot database are presented.
Benchmarking proteomics search tools is inherently difficult. The correct protein identification for each query is, strictly speaking, unknown; as a result, confidence can be assigned to the protein hits through comparison with the standard score threshold defined by Mascot, at a 95% confidence limit based on the database size. Taking only those top ranking proteins that have scores greater than the Mascot threshold for each particular non-redundant search, there are 12,526 peptides corresponding to 1,423 unique queries in the PMF dataset.
A further experimental test set was produced from 13 known, well characterised proteins with additional tandem MS peptide identifications. Five of these protein samples were prepared using standard methods for silver and coomassie staining techniques and in-gel digestion and extraction. Gel pieces were reduced and, alkylated with iodoacetamide, prior to overnight trypsin digestion and peptides extraction with washes of 20 mM ammonium bicarbonate and 5% formic acid in 50% Acetonitrile. The remaining eight samples were standards, dissolved in buffer, and processed in a similar way. MALDI-TOF analysis was performed on a Voyager DE STR mass spectrometer (Applied Biosystems), and Tandem mass spectrometry was performed on a Q-TOF Micro mass spectrometer (Waters) attached to a Cap-LC nano-chromatography system (Waters). The resultant data was searched against Swissprot using MASCOT for both PMF and tandem MS searches, although the true identities were known a priori.
Results
Missed cleavage patterns from Information theory
The effect on potential missed cleavage can be represented for each of the 20 standard amino acid types, plotting the information scores from the two matrices MC and MM surrounding a generalized tryptic site, shown in Figure 1. Interestingly there are patterns in Figure 1 that are shared with the patterns from other studies summarized in Table 1. For instance the negatively charged amino acids aspartate and glutamate favour missed cleavages in all of these studies, particularly at positions P3, P4, P1′ and P2′. These two acidic residues may well form salt bridges with the basic residues arginine and lysine, competing with the complementary aspartate at the base of trypsin's P1 pocket, which is part of the enzyme's recognition system, thereby inhibiting their interaction with the enzyme. Figure 1 also suggests that in addition to lysine and arginine, which appear to compete with themselves for cleavage by trypsin, glycine, methionine and serine have a weak but notable association with missed cleavages. Indeed, the plots represent a more holistic treatment of the relative effects each residue type has on the likelihood of cleavage by trypsin at a local candidate site, over and above the simple regular expressions of Table 1.
Figure 1.
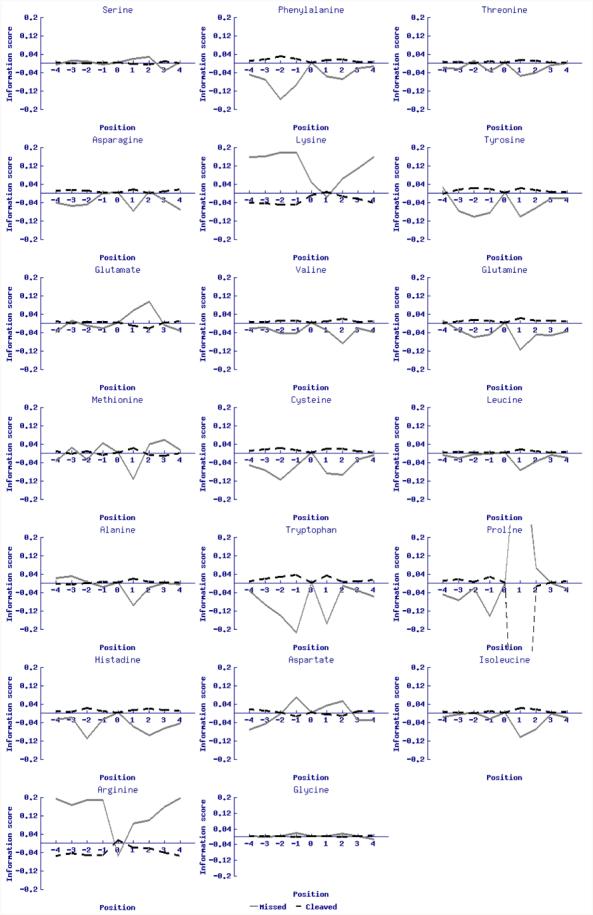
Information theory results for the missed cleavages (solid light grey line) and actual cleavages (dashed dark grey line) in all amino acids.
Predicting cleavage states from SM and SC
The score difference SM-SC was used to predict cleavage states for all putative sites from the PMF test data set, and the results are shown in Figure 2. The predictions are based on a threshold, where a score of SM-SC >threshold is predicted to be a missed cleavage and all others as a cleaved site. In this study, two thresholds of 0.25 and 0.50 were compared. As shown in Figure 2, a 0.25 threshold results in the maximum correct identifications for both missed cleavages and actual cleavages. However, the Matthews Correlation Coefficient (MCC) 14 for this threshold is 0.007 and there is a moderately high percentage (12%) of actual cleavages falsely predicted to be missed. As shown in Figure 2, the 0.5 threshold minimizes the number of actual cleavage sites falsely predicted as missed cleavage sites (4%) whilst still predicting 85% of missed cleavages correctly and had an improved MCC of 0.18 compared to a 0.25 threshold. As described above the matrices were trained on high quality tandem MS data and the data in Figure 2 was generated from testing with high confidence PMF data. Similar results were seen when applied to the original training data (data not shown).
Figure 2.
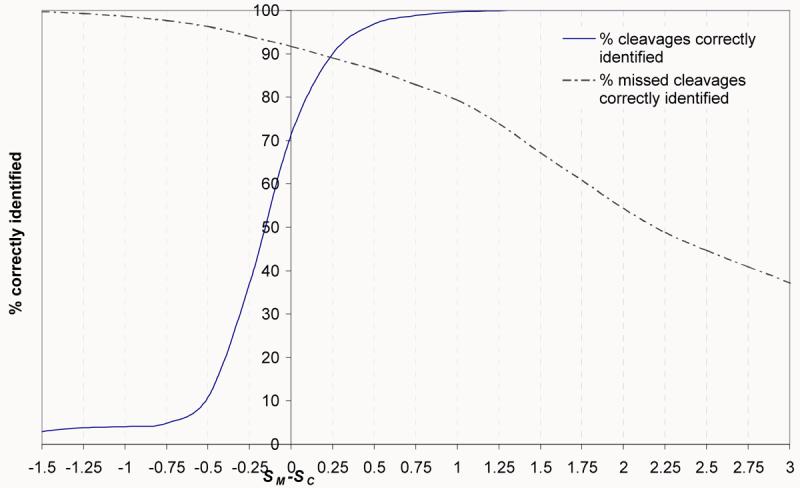
The percentage of cleavages in a high quality PMF dataset correctly identified following calculation of the information scores, SM and SC, for all cleaved peptides and all peptides containing a missed cleavage. The distribution of the difference in the two scores (SM-SC) can be used to determine a threshold in the predictions.
Application of Information Theory to test the PMF data
We evaluated the information theoretic matrices on an independent, non-redundant test set of PMF experiments, reporting only those proteins which had scores greater than the standard Mascot threshold at 95% confidence. This PMF dataset contained only around 7% of the same peptides present in the tandem MS training dataset, based on identical amino acids, and was hence deemed to be a suitable test. The matrices were applied to the observed missed cleavage sites (3,430) and the N- and C-terminal cleavage sites (K/R) of all the peptides observed in the PMF dataset (23,842). If a threshold of SM-SC>0 was applied then 74% of cleavages were correctly identified as missed or cleaved with a sensitivity of 0.71 and specificity of 0.92. If a threshold of SM-SC >0.25 is applied however, 90% of cleavages were correctly identified as missed or cleaved with a sensitivity of 0.90 and specificity of 0.89. Finally, if a threshold of SM-SC >0.5 was applied, 96% of cleavages were correctly identified as missed or cleaved with a sensitivity of 0.97 and specificity of 0.86. These results demonstrate the predictive power in considering local sequence patterns surrounding a putative tryptic cleavage site to make predictions. The next step was to integrate this information into known PMF tools.
Applying the rules from Information theory to different PMF tools
The missed cleavage information was incorporated into PMF tools with the aim of improving the protein identification ability of the software, either by increasing the number of true positives (correct protein identifications) and/or by improving the score for the nominally correct protein identification. Two PMF tools were used in this study, Mascot8 and Imprint. Mascot is widely regarded as an industry standard, and uses a measure of absolute probability based on MOWSE15, the precise details of which are not published. Imprint (unpublished) is an in-house PMF tool, which, for this particular analysis, uses the Piums16 scoring algorithm. This was chosen as it is well documented and because it uses the number of theoretical proteolytic peptides in calculating the score and this value will be changed if putative cleavages are assigned as “uncleavable” by our algorithm. This is a key point, since by forcing some tryptic bonds to be “uncleavable”, without any other effect on the score we would universally be degrading search capability. However, since the masking also changes the number of available peptides per protein, there should also be a competing counter-effect which rewards matches to proteins where we more accurately estimate the chances of a random match (because there are fewer available peptides in masked proteins).
The Piums16 score (σ) for a particular protein j is given in Equation 4, where L is the total number of peaks in the experimental peak list (x), r is the number of common peaks between x and the theoretical peak list for the protein (z(j)), P is the probability for at least one match between x and z(j) and Q is (1-P).
| Equation 4 |
In our case, the array of theoretical peaks z(j) is clearly affected using our algorithm, and will therefore alter the values of P and Q in the above equation. In order to test whether the results derived from the information theory approach could improve current protein identification strategies, they had to be incorporated into the identification algorithms. Since Mascot is proprietary software the approach adopted in this study was to use the two matrices, MM and MC, derived from information theory, to alter the sequence database that was searched by both tools. To do this two additional amino acids, J and O, were introduced. SM and SC were calculated for each Lysine residue (K) in each of the search databases and if SM-SC >threshold then K was replaced by J (i.e. it is likely to be a missed cleavage). Similarly for Arginine (R), R was replaced by O when SM-SC >threshold. In both search algorithms the amino acids J and O were assigned the same monoisotopic and average masses as K and R respectively, but were not cleaved during the in silico digestion of the search database. For the rest of this manuscript the original database will be referred to as the normal database (DBN) and the database with missed cleavage sites replaced by J and O as the missed database (DBM).
The effect of the algorithm was examined on a candidate Fasta-formatted protein database. As expected the removal of some tryptic cleavage sites (by the replacement of K/R with J/O respectively) affected the distribution of both the number of peptides per protein and the length of these peptides. As shown in Figure 3 the replacement of potential tryptic sites (K/R) by amino acids J and O where the cleavage site is predicted to be missed in the Swissprot database leads to a decrease in the number of peptides per protein (Figure 3) and an increase in peptide length (Figure 3B). As mentioned previously, since the Piums scoring function (Equation 4) is based upon the probability P calculated from the in silico peptide mass frequency distribution and the number of peptides in the theoretical peak list z(j), this has implications for the overall Piums score, which is hoped will lead to improved identification scores and estimated significance values.
Figure 3.

The effect of the replacement of predicted missed cleavage residues by J/O in DBM compared to the original database DBN. (A) The effect on the number of peptides per protein in Swissprot and (B) the effect on the length of these peptides.
Application of improved PMF tools to PMF data
Figure 4 shows the results of this analysis for the Mascot8 and the Imprint PMF searches respectively. Each query was run four times with each PMF tool, the first with DBM and no partial cleavages allowed, the second with DBN and up to one partial allowed, the third with DBM and no partials allowed and the fourth DBM with up to one partial allowed. All the results described here refer to DBM searches with a 0.5 threshold unless otherwise stated and up to one partial permitted, as the presence of up to one partial improved the scores on both databases considerably (data not shown). In Figure 4 the results refer to non-redundant searches of protein databases using Mascot and Imprint. Each of the pie charts are divided into two, the first portion represents queries where the top protein hit from DBN is the same as that from DBM; this was true for 87% of Mascot searches and 86% of Imprint searches. For all searches run with Mascot, 24% show an improved score to the top-hit protein when searched with DBM, 68% have the same score and only 8% have a worse score. The second analysis for Mascot (Figure 4B) is a subset of the results from Figure 4A, but where the Mascot protein score was greater than the threshold defined by Mascot. Again the pie chart is divided into two, one representing queries where the top protein hits are the same following a search of DBN and DBM (97%) and the other where the top protein hits are different. This time the scores are a little different with more results having the same score (74%), 18% show an improvement and 8% have a worse score. Figure 4C shows the same results but for searches run with Imprint, of these 57% have the same score, 35% have an improved score and only 8% have a worse score.
Figure 4.

Results of the PMF searches, N is the score for the top ranking protein from DBN and M is the score for the top ranking protein from DBM. Except for the search database all other parameters for the Mascot search remained the same. Where the two searches resulted in the same protein accession ranked in position 1 this is reported as the ‘same top hit’. (A) Results of a non-redundant set of Mascot8 PMF searches, with the threshold for missed cleavages being SM-SC >0.5. (B) Results of a non-redundant set of Mascot8 PMF searches, with the threshold for missed cleavages being SM-SC >0.5 and the protein score greater than the Mascot defined threshold. (c) Results of the Imprint PMF searches, with the threshold for missed cleavages being SM-SC >0.5.
In common with other informatics assessments of proteomic database searching tools, a strictly objective assessment of these results is difficult as true, definitive protein identifications are uncertain. However, we have trained the missed cleavage prediction algorithm on an independent data set, and see clear signs of improvement in protein identification. Unsurprisingly, searches against both treated and native Fasta databases produces the same top hit in the majority (87%) of cases. However, the larger fraction of improved scores compared to lower scores demonstrates the ability of the approach to provide increased statistical certainty for a greater fraction of DBM searches. A total of 9855 unique Mascot searches were performed, of which a total of 1462 searches had a protein score above the standard Mascot threshold (1423 against DBN and 1437 against DBM). Of the 1462 searches, 1413 had the same protein hit when searching with both DBM and DBN and of these, 29 of the searches with DBM actually increased the Mascot score above the confidence threshold, compared to only 19 searches, which lowered the score below the threshold. In contrast only 49 of the 1462 searches had a different protein hit and of these 16 increased the score above the Mascot confidence threshold when searching DBM and only 6 lowered it below. Interestingly, in this small number of cases (49) where this approach suggested alternative top hits with scores above the 95% significance threshold the DBM searches yield improved scores in the majority of cases. These may very well be the “true” hits since the scores have been increased above the 95% confidence limit, offering increased statistical confidence in their veracity. Finally, we note that a larger number of searches resulted in significant Mascot hits against DBM.
We also analysed in Table 2 the frequency of the patterns associated with the missed cleavages in those unique peptides identified by the Mascot approach on the two databases (DBN and DBM). There are only a relatively modest fraction of missed cleaved peptides identified that contain a J/O, which demonstrates that the method is underestimating the full extent of partial cleavages. The strict threshold (0.5) used to mask the search database ensures that only confident cleavage sites are replaced by J/O and as a result up to one partial is still considered in the search (hence why some statistics in Table 2 have a minimum of two missed cleavages per peptide). However, it is fairly common to see both the partially cleaved peptides and one or both of its “daughter” peptides from limit cleavage in the same spectrum, for instance one or more “daughter” peptides are observed in 23% of the partially cleaved peptides identified by Mascot with DBN. Despite this, the approach is still effective at improving protein identifications with two different scoring systems and this might be impaired if the sensitivity of the method were improved at the expensive of specificity. This improvement is expected to be due to the improved estimate of the number of available tryptic peptides from the in silico digest, since it would not be possible for the algorithms to match the daughter peptides from the “uncleavable” one.
Table 2. Missed cleaved peptide statistics.
| Peptide characteristic A |
Minimum number of missed cleavages |
DBN |
DBM |
|---|---|---|---|
| .[K] | 0 | 5813 | 5918 |
| .[R] | 0 | 5188 | 5175 |
| .[K]*. | 1 | 1538 | 1278 |
| .[R]*. | 1 | 1210 | 953 |
| .[J]*. | 1 | 0 | 484 |
| .[O]*. | 1 | 0 | 336 |
| .[K].[J]*. or [J].[K]*. | 2 | 0 | 80 |
| .[K].[O]*. or [O].[K]*. | 2 | 0 | 27 |
| .[R].[J]*. or [J].[R]*. | 2 | 0 | 56 |
| .[R].[O]*. or [O].[R]*. | 2 | 0 | 35 |
| .[K][P]*. | 1 | 424 | 5 |
| .[R][P]*. | 1 | 309 | 2 |
| .[J][P]*. | 1 | 0 | 403 |
| .[O][P]*. |
1 |
0 |
313 |
| Total number of peptides with at least one missed cleavage |
1 | 2700 | 2848 |
Characteristics of observed missed cleavage sites in unique peptides identified from Mascot8 searches of DBN and DBM (0.5 threshold) with a score greater than the standard Mascot threshold. ‘.’ Represents zero or more amino acids, ‘*’ represents any amino acid residue, specific residues are shown in square brackets.
More than one characteristic may appear in a single peptide sequence hence we report the total number of peptides that contain at least one missed cleavage and not the total number of missed cleavages that occur in the dataset. The shaded boxes show the overall statistics that over 30% of the unique peptides in the dataset contain at least one missed cleavage.
Application of improved PMF tools to known PMF data
One problem associated with the analysis described above is that we cannot be completely confident of the correct protein identification assigned by Mascot or Imprint. To address this, we applied both PMF tools to a dataset of 13 known proteins analysed by both MALDI-MS PMF and tandem MS, using the tandem MS searches as a means to produce high quality peptide identifications and therefore a high quality ‘gold standard’ set. The results for all 13 proteins and both PMF tools and the tandem MS data are shown in Table 3.
Table 3. ‘Masking’ the search database improves the overall performance of two PMF scoring algorithms, Imprint and Mascot8, on a dataset of known proteins.
| Protein name | Species | Accession | DB type B | Imprint A | Mascot A | MS/MS | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| score | significance | Id | error (Da)D | score | Expect | Id | Id C | ||||
| Ubiquitin protein ligase |
Human | O95071 | DBN | 60.87 | 1.7e-04 |
|
0.5 | 67 | 0.03 |
|
 (rank 1) |
| DBM 0.25 | 65.16 | 1.1e-04 |
|
0.5 | 81 | 1.3e-03 |
|
||||
| DBM 0.5 | 61.79 | 2.9e-03 |
|
0.5 | 69 | 0.02 |
|
||||
| Chaperone protein dnaK |
E.Coli | P04475 | DBN | 21.53 | 77.00 |
|
0.1 | 42 | 9.10 |
|
(rank 1) |
| DBM 0.25 | 19.63 | 160.00 |
|
0.1 | 42 | 11.00 |
|
||||
| DBM 0.5 | 21.66 | 19.00 |
|
0.1 | 42 | 11.00 |
|
||||
| Methionine synthase reductase |
Human | Q9UBK8 | DBN | 24.78 | 7.6e-03 |
|
1.0 | 52 | 1.10 |
|
(rank 1) |
| DBM 0.25 | 30.90 | 6.8e-07 |
|
1.0 | 52 | 1.10 |
|
||||
| DBM 0.5 | 24.78 | 8.7e-07 |
|
1.0 | 52 | 1.10 |
|
||||
| Serum albumin precursor |
Bovine | P02769 | DBN | 35.53 | 5.0e-11 |
|
2.0 | 61 | 0.12 |
|
(rank 1) |
| DBM 0.25 | 36.03 | 2.9e-11 |
|
2.0 | 63 | 0.08 |
|
||||
| DBM 0.5 | 35.53 | 5.8e-08 |
|
2.0 | 61 | 0.12 |
|
||||
| Beta lactamase TEM precursor |
E.Coli | P62593 | DBN | 61.84 | 7.9e-12 |
|
2.0 | 86 | 3.7e-04 |
|
(rank 1) |
| DBM 0.25 | 61.84 | 1.4e-16 |
|
2.0 | 86 | 3.7e-04 |
|
||||
| DBM 0.5 | 61.84 | 2.7e-26 |
|
2.0 | 86 | 3.7e-04 |
|
||||
| Alcohol dehydrogenase |
Yeast | P00330 | DBN | 29.51 | 7.9e-04 |
|
2.0 | 61 | 0.10 |
|
(rank 1) |
| DBM 0.25 | 29.90 | 0.13 |
|
2.0 | 61 | 0.10 |
|
||||
| DBM 0.5 | 29.51 | 0.13 |
|
2.0 | 61 | 0.10 |
|
||||
| Beta Casein | Bovine | P02666 | DBN | 13.29 | 46.33 |
|
2.0 | 43 | 8.30 |
|
(rank 2) |
| DBM 0.25 | 14.12 | 3.02 |
|
2.0 | 41 | 8.30 |
|
||||
| DBM 0.5 | 13.54 | 5.39 |
|
2.0 | 43 | 8.30 |
|
||||
| Beta galactosidase |
E.coli | P00722 | DBN | 154.10 | 3.6e-22 |
|
2.0 | 211 | 1.3e-16 |
|
(rank 1) |
| DBM 0.25 | 154.53 | 7.0e-28 |
|
2.0 | 211 | 1.3e-16 |
|
||||
| DBM 0.5 | 154.10 | 7.4e-26 |
|
2.0 | 211 | 1.3e-16 |
|
||||
| Cytochrome C | Horse | P00004 | DBN | 12.74 | 6.3e-04 |
|
2.0 | 50 | 1.70 |
|
(rank 1) |
| DBM 0.25 | 23.29 | 1.5e-10 |
|
2.0 | 54 | 0.66 |
|
||||
| DBM 0.5 | 17.32 | 5.2e-06 |
|
2.0 | 50 | 1.70 |
|
||||
| Glceraldehyde 3 phosphate |
Bovine | P10096 | DBN | 26.05 | 7.38 |
|
2.0 | 61 | 0.14 |
|
(rank 1) |
| DBM 0.25 | 26.21 | 0.85 |
|
2.0 | 61 | 0.13 |
|
||||
| DBM 0.5 | 26.05 | 4.84 |
|
2.0 | 61 | 0.14 |
|
||||
| Myoglobin | Horse | P68082 | DBN | 52.80 | 3.7e-18 |
|
2.0 | 124 | 6.3e-08 |
|
(rank 1) |
| DBM 0.25 | 53.80 | 4.0e-10 |
|
2.0 | 129 | 2.0e-08 |
|
||||
| DBM 0.5 | 52.80 | 1.1e-09 |
|
2.0 | 124 | 6.3e-08 |
|
||||
| Ovalbumin | Chick | P01012 | DBN | 37.66 | 4.9e-05 |
|
1.0 | 79 | 1.9e-03 |
|
(rank 1) |
| DBM 0.25 | 44.03 | 7.3e-07 |
|
1.0 | 80 | 1.8e-03 |
|
||||
| DBM 0.5 | 37.66 | 3.4e-04 |
|
1.0 | 79 | 1.9e-03 |
|
||||
| Phosphorylase B | Rabbit | P00489 | DBN | 61.17 | 4.1e-10 |
|
2.0 | 99 | 1.9e-05 |
|
(rank 1) |
| DBM 0.25 | 67.10 | 4.5e-13 |
|
2.0 | 113 | 7.9e-07 |
|
||||
| DBM 0.5 | 61.46 | 1.2e-07 |
|
2.0 | 100 | 1.6e-05 |
|
||||
In all cases the proteins were searched with the fixed modification Carbamidomethyl (C) and variable modification Oxidation (M), with up to one missed cleavage permitted and an error tolerance of two Daltons (Da) except where stated. Those results highlighted in grey are those where the searches of DBM improved upon the score of the corresponding score for the DBN search.
‘DB type’ is the search database that was used in each case, DBM from a threshold of both 0.25 and 0.5 were used.
Id is ‘’ if the top ranking protein hit (unless otherwise stated) is the correct protein, and ‘’ otherwise.
In the Mascot searches, the ‘error’ is the error tolerance used in the search (in Da).
The MS/MS results from the same samples, identified using Mascot8.
As expected there is an improvement in the scores for eleven and five of the 13 proteins for Imprint and Mascot respectively, using a missed cleavage prediction threshold of 0.25. This threshold, however, also results in a false positive result with Imprint. There is an apparent incorrect assignment for chaperone protein dnaK, assigning a greater score to the incorrect protein, whilst the original database and the larger threshold assign the protein correctly. The larger threshold of 0.5 on the other hand has no false positive results in this dataset and shows an improved score for five and two of the 13 proteins for Imprint and Mascot respectively.
Imprint slightly out performs Mascot when using both DBN and DBM with the 0.5 threshold. We observe a better overall improvement using DBM with Imprint compared to Mascot. This may be explained by the scoring algorithms. Imprint uses an implementation of the Piums16 scoring system and this considers the number of peptides in the theoretical protein, which as shown in Figure 3 is changed following the application of the missed cleavage rules. The precise details of the Mascot scoring algorithm are unknown.
Neither search engine was able to correctly identify the Beta Caesin protein; there were only a small number of search peptides for this protein and interestingly in the tandem MS search with the same protein sample, Beta Caesin was identified but it was ranked second and this was therefore a challenging identification for any algorithm to make given the available data.
Discussion
PMF remains a widely used approach particularly in high-throughput MS-based protein identification studies17. The score of the best protein match is affected by factors such as the sequence coverage, the number of matched peptides from the protein, the number of matched/unmatched experimental peptide masses, the mass accuracy, and the matches to random proteins. Importantly, the number of unmatched or missed peptides in a protein is also a factor, as well as the excess of limit peptides 11. Optimally, all the peptide masses of a mass fingerprint should be attributed to the top-ranked protein, although this is rarely true due to a number of factors, including protein mixtures, post-translational modifications, genetic variations and incomplete digestion with the protease. This latter point, as we have demonstrated, can have an impact not only on the score of the top ranking protein but also on the protein that is identified. Indeed, in our dataset alone over 40% of the peptides had missed cleavage sites (11% of which precede a proline residue). We examined the nature of experimentally observed missed cleavage peptides in our dataset where peptides corresponding to the cleavage were also observed, as shown in Table 4. A notable feature of these peptides that we observed as both missed and cleaved was that their “daughter” peptides, produced when the missed peptide bond was also cleaved, showed marked amino acid preferences adjacent to the scissile bond. When the frequency of amino acids is converted into a propensity using the masked DBM version of Swissprot, proline is surprisingly over-represented at the P1′ position, although trypsin is not expected to cleave a lysine/arginine-proline peptide bond. Indeed, this has been recently highlighted by Mann and co-workers 1. They point out that these observations of N-terminal prolines may well be due to in-source decay or other artefacts of gas phase ion chemistry rather than tryptic cleavage. Nevertheless, given that this observation appears to be common in the mass spectrometer, it should be considered by search engines.
Table 4. Amino acid preferences adjacent to the sissile bond in ‘daughter’ peptides produced when a missed peptide bond is also cleaved.
| C' terminal Amino Acid |
Total matches to “daughter” peptidesA |
Frequency of matches to “daughter” peptides |
Frequency in Swissprot |
Normalised B frequency of total matches to “daughter” peptides |
|---|---|---|---|---|
| A | 3 | 2.03 | 3.83 | 0.53 |
| C | 0 | 0 | 0.81 | 0.00 |
| D | 1 | 0.68 | 5.27 | 0.13 |
| E | 8 | 5.41 | 8.07 | 0.67 |
| F | 0 | 0.00 | 2.19 | 0.00 |
| G | 3 | 2.03 | 16.33 | 0.12 |
| H | 0 | 0.00 | 1.43 | 0.00 |
| I | 0 | 0.00 | 2.62 | 0.00 |
| K | 0 | 0.00 | 0.48 | 0.00 |
| L | 0 | 0.00 | 5.67 | 0.00 |
| M | 1 | 0.68 | 1.41 | 0.48 |
| N | 0 | 0.00 | 2.38 | 0.00 |
| P | 118 | 79.73 | 17.35 | 4.60 C |
| Q | 1 | 0.68 | 1.65 | 0.41 |
| R | 1 | 0.68 | 3.6 | 0.19 |
| S | 6 | 4.05 | 5.68 | 0.71 |
| T | 1 | 0.68 | 3.08 | 0.22 |
| V | 2 | 1.35 | 4.61 | 0.29 |
| W | 0 | 0.00 | 0.38 | 0.00 |
| Y | 0 | 0.00 | 1.46 | 0.00 |
All ‘daughter’ peptides, (i.e. the mass of the two peptides either side of a J/O in the top ranking protein hit) of the unique protein hits from the Mascot searches were matched to the original experimental search masses.
The data was normalised against the “daughter” peptides surrounding every J/O in the Swissprot database.
There is very high number of matches for proline residues which may be explained as an artefact of gas phase ion chemistry 1.
In this study we have described the use of information theory to generate rules for characteristic patterns in the residues surrounding a missed cleavage site, some of which have been observed previously. When incorporated into two different PMF tools the patterns show improvements in protein identification over standard PMF searches, particularly in the scores associated with the top ranking protein hits. In over 90% of cases the method either improved or had no effect on the protein scores, in some cases it even changed the top ranking protein. This work was further supported by the application to a dataset of known proteins, for which the method showed an overall improvement in the performance.
This study demonstrates that missed cleavages play an important role in protein identification and can be a limiting factor in the scores in some instances. We believe the method is generically applicable to any search engine which considers either the number of potential peptides in a theoretical digest, or number of missed/absent putative peptides. Indeed, similar approaches have appeared recently in the literature applied to PMF and tandem MS database search protocols, although they do not have the general applicability to the database “masking” approach applied here 3,4,5.
Availability
A web tool is available at http://ispider.smith.man.ac.uk/MissedCleave to predict missed cleavage positions in a single protein sequence with the ability to set a threshold. A perl script is also available for download from the same URL to mask entire Fasta formatted databases with J/O replacing missed cleave positions.
Acknowledgments
The authors would like to acknowledge Julian Selley from the Faculty Bioinformatics Core Facility for his technical assistance, all groups who supplied data to the Mascot server, Josip Lovric for useful discussions and the BBSRC for support via the ISPIDER project (BBSB17204).
Abbreviations
- PMF
Peptide Mass Fingerprinting
- MCC
Matthews Correlation Coefficient
References
- 1.Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics. 2004;3:608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys, Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 3.Monigatti F, Berndt P. Algorithm for accurate similarity measurements of peptide mass fingerprints and its application. J. Am. Soc. Mass Spectrom. 2005;16:13–21. doi: 10.1016/j.jasms.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Thiede B, Lamer S, Mattow J, Siejak F, Dimmler C, Rudel T, Jungblut PR. Analysis of missed cleavage sites, tryptophan oxidation and N-terminal pyroglutamylation after in-gel tryptic digestion. Rapid Commun. Mass Spectrom. 2000;14:496–502. doi: 10.1002/(SICI)1097-0231(20000331)14:6<496::AID-RCM899>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Yen C, Russell S, Mendoza AM, Meyer-Arendt K, Sun S, Cios JJ, Ahn NG, Resing KA. Improving sensitivity in shotgun proteomics using a peptide-centric database with reduced complexity: protease cleavage and SCX elution rules from data mining of MS/MS spectra. Anal. Chem. 2006;78:1071–1084. doi: 10.1021/ac051127f. [DOI] [PubMed] [Google Scholar]
- 6.Keil B. Specificity of Proteolysis. Springer-Verlag; Berlin-Heidelberg-New York: 1992. pp. 66–69. [Google Scholar]
- 7.McLaughlin T, Siepen JA, Selley J, Lynch JA, Lau KW, Yin H, Gaskell SJ, Hubbard SJ. PepSeeker: a database of proteome peptide identifications for investigating fragmentation patterns. Nucleic Acids Res. 2006;34:D649–654. doi: 10.1093/nar/gkj066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 10.Gattiker A, Bienvenut WV, Barioch A, Gasteiger E. FindPept, a tool to identify unmatched masses in peptide mass fingerprinting protein identification. Proteomics. 2002;2:1435–1444. doi: 10.1002/1615-9861(200210)2:10<1435::AID-PROT1435>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Stead DA, Preece A, Brown AJP. Universal metrics for quality assessment of protein identifications by mass spectrometry. Mol. Cell. Proteomics. 2006;5:1205–1211. doi: 10.1074/mcp.M500426-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Ossipova E, Fenyo D, Eriksson J. Optimizing search conditions for the mass fingerprint-based identification of proteins. Proteomics. 2006;6:2079–2085. doi: 10.1002/pmic.200500484. [DOI] [PubMed] [Google Scholar]
- 13.Schechter I, Berger A. On the size of the active site in proteases. I. papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 14.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 15.Pappin DJC, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 16.Samuelsson J, Dalevi D, Levander F, Rognvaldsson T. Modular, scriptable and automated analysis tools for high-throughput peptide mass fingerprinting. Bioinformatics. 2004;20:3628–3635. doi: 10.1093/bioinformatics/bth460. [DOI] [PubMed] [Google Scholar]
- 17.Gavin A-C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dümpelfeld B, Edelmann A, Heurtier M-A. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]