

Abstract
Objective
Allograft Inflammatory Factor-1 (AIF-1) is a calcium binding scaffold protein which is rapidly induced in vascular smooth muscle cells (VSMCs) in response to injury and inflammation. A transgenic mouse in which AIF-1 expression was driven by a VSMC-specific SM22α promoter was generated to establish a direct relationship between AIF-1 expression and intimal hyperplasia.
Methods and Results
Morphological analysis of partially ligated carotid artery demonstrate a significant increase in neointimal area of AIF-1 Tg versus wild-type mice (569±64 um versus 256±49um, P=0.004). Immunohistochemistry using antibody to the proliferation marker Ki-67 show a significantly greater number of proliferating cells in the AIF-1 Tg lesion compared with wild-type arteries (10.6%±1.0 versus 3.6%±.9, P=0.0007). AIF-1 Tg arteries also had a greater number of cells with activated signal transduction kinase p38 (55.4%±7.0 versus 22.6%±5.4, P=0.002) and PAK1 (67.5%±6.7 versus 35.3%±10.2, P=0.02) compared with wild-type. Cultured VSMCs explanted from AIF-1 Tg proliferate (55.5±3.6×103 versus 37.2±2.0×103 cells/mL, P=0.0001) and migrate more rapidly (39.2±3.2 versus 17.1±1.5 VSMCs per HPF, P=0.0003) than wild-type, and have significantly greater levels of activated p38 and PAK1 than did VSMCs from wild-type littermates (P<0.05).
Conclusions
These data indicate that AIF-1 expression results in increased signal transduction, neointimal formation, and VSMC proliferation in injured mouse carotid arteries.
Keywords: allograft inflammatory factor-1, carotid ligation, proliferation, signal transduction
Intimal hyperplasia subsequent to mechanical and immunologic insult remain clinically significant obstacles limiting the success of vascular interventions and solid organ transplantation.1,2 Common to both of these injuries is a localized inflammatory reaction in which injured endothelial and immune cells secrete growth and inflammatory cytokines which elicit activation of normally quiescent medial vascular smooth muscle cells (VSMCs).3,4 As part of the vascular response to injury, VSMCs migrate from the media into the lumen of the vessel where they proliferate and synthesize cytokines which they respond to in an autocrine fashion, sustaining the progression of intimal hyperplasia. Identification of proteins implicated in this process, and of signaling pathways involved in migration and proliferation, is an important approach in development of modalities to combat this syndrome.
Allograft Inflammatory Factor-1 (AIF-1) is a 143-aa calcium binding protein. AIF-1 has signatures of a cytoplasmic scaffold signaling protein. It contains several PDZ interaction domains, which are important in mediating interactions of multiprotein complexes, and a QXXER motif, which has been shown to mediate Gβγ interactions.5,6 High levels of AIF-1 are constitutively expressed in inflammatory tissue and glial cells. Data from several groups advocate an important role for AIF-1 in inflammatory processes, primarily involving macrophages and inflammatory cells.7-9 AIF-1 expression is evolutionarily conserved and is expressed in the allograft response of such phylogenetically distant species as Carp and marine sponges.10 In humans, the AIF-1 gene maps to the Major Histocompatibility Complex (MHC) class III region on chromosome 6p21.3, a region which is densely populated with genes involved in the inflammatory response, including complement cascade proteins, tumor necrosis factor (TNF)α and β, and NF-κB.11 Taken together, this suggests an important role for this protein in the inflammatory response, however the mechanism of AIF-1 activity remains unclear.
Our laboratory has focused on the role of AIF-1 in VSMC pathophysiology. Although constitutively expressed in lymphoid tissue, AIF-1 is not expressed in unstimulated VSMCs, but is rapidly induced in response to injury and inflammatory cytokines.12 AIF-1 expression in cardiac allografts is associated with rejection and development of clinical coronary artery vasculopathy (CAV).13 In quiescent VSMCs, AIF-1 interacts with actin, but translocates to leading edge lamellipodia in stimulated PDGF-VSMCs.14 Over expression of AIF-1 in VSMCs results in increased proliferation and cell cycle protein expression, increased migration, and activation of the small GTPase Rac1.14-16
Although these data suggest that AIF-1 plays an important role in regulation of VSMC activation, a more direct relationship between AIF-1 expression and development of intimal hyperplasia in vivo has not been established. To this end, we generated a transgenic mouse in which AIF-1 expression is driven by a modified SM22α promoter in which expression is restricted to large and medium sized arteries,17,18 with the goal to determine whether AIF-1 overexpression enhances intimal hyperplasia in vivo. In this study we report that AIF-1 Tg mice have an enhanced response to ligation injury of the carotid artery, characterized by an increased neointima, enhanced activation of signal transduction proteins PAK1 and p38, and increased proliferation of VSMCs. VSMCs explanted from these mice also display increased proliferation, migration, and an enhanced activation of the same signal transduction kinases in response to stimuli. Collectively, these data point to the contribution of the Rac1/PAK1/p38 signal transduction pathways in development of intimal hyperplasia in ligation-injured murine arteries, and identifies AIF-1 expression as an important link in that pathway and in the VSMC response to injury.
Materials and Methods
Generation of AIF-1 Transgenic Mouse and Ligation Injury
The G/C-modified SM22α promoter construct subcloned into pGL described by Regan et al17 was a generous gift of Dr Gary Owens, University of Virginia, Charlottesville, Va.. The AIF-1 proteincoding region, preceded by a Kozak consensus sequence previously described,15 was subcloned distal of SM22αG/Cdel into an EcoR1 site of pGL. The SM22αC/Gdel-AIF-1 transgene was liberated from the vector and injected into donor eggs from B6,SJL mice by the University of Pennsylvania Department of Genetics Core Facility. Founder mice were identified by standard polymerase chain reaction (PCR) and Southern blot analysis of tail DNA using primers specific for SM22α and HA-tagged AIF-1 sequence. Mice were not backcrosssed to B6. Age-matched (16 to 20 weeks of age) wild-type and heterozygous transgenic littermates were identified by PCR of tail DNA using primers specific for SM22α and HA-tagged AIF-1 sequence. Male and female mice were used for these studies. The Shear-stress ligation model of injury has been described.19 Mice were anesthetized by injection of ketamine and xylazine. The left common carotid artery was dissected and ligated near the bifurcation for 28 days. Severity of hyperplasia is strain dependent; the SJL and C57 lineages develop robust and moderate intimal hyperplasia, respectively.20 After 28 days, mice were euthanized and tissue prepared for immunohistochemistry and morphological analysis, n=8 animals for each group.
Immunohistochemistry and Morphology
Digitized images were measured and averaged from at least 5 representative 5-μm-thick stained tissue sections at least 75 to 100 μm apart panning between 200 and 800 micrometers per carotid artery were examined using Image Pro Plus (Media Cybernetics). Eight mice were used for morphology, and 3 for IHC. The circumference of the lumen, the area encircled internal elastic lamina (IEL), and the external elastic lamina (EEL) were quantitated. The medial area was calculated by subtracting the area defined by the IEL from the area defined by the EEL, and intimal area calculated as the difference between the area inside the IEL and the luminal area. Tissue fixation, processing, AIF-1 antibody, and immunohistochemical staining was performed as described.13 Ki-67 antibody was from Laboratory Vision Inc, and anti-phospho p38 and PAK1 were from Cell Signaling Inc.
VSMC Culture, Proliferation, and Migration
Abdominal aorta from wild-type and transgenic mice were removed, endothelial layer scraped off, and VSMCs isolated as described.21 Cell culture, proliferation, and migration assays were performed as described14,15; please see Supplemental Materials and Methods (available online at http://atvb.ahajournals.org).
RT-PCR and Western blotting
RNA isolation, PCR, and Western blotting was performed as previously described10,14; please see supplemental materials.
Statistics
Results are expressed as mean±SE. Differences between groups were evaluated with the use of ANOVA to evaluate differences between individual mean values or by t tests where appropriate. Differences were considered significant at a level of P<0.05.
Results
Generation of AIF-1/SM22α Mice
A cause and effect relationship between AIF-1 expression and intimal hyperplasia has not been reported. Because of AIF-1 constitutive expression in immune cells and the demonstrated role that these cells play in vascular injury and remodeling, it was necessary to restrict AIF-1 expression to vascular smooth muscle cells. A transgenic mouse was constructed where AIF-1 expression was driven by the modified SM22α promoter in which AIF-1 expression is restricted to large and medium sized arterial VSMCs (Figure 1A). This promoter has been modified such that expression is maintained even in the context of injury in which VSMC dedifferentiate and has been successfully used in several studies.17,18 The AIF-1 cDNA was tagged with the HA sequence at the 3′ end to distinguish it from the endogenous AIF-1 gene or mRNA. Founder mice were identified for presence of the transgene by PCR using primers specific for SM22α and HA-tagged AIF-1 sequence (Figure 1B), and tissue-specific transgene expression was determined by semiquantitative RT-PCR (Figure 1C). Expression of HA-tagged AIF-1 protein in these arteries was validated by immunoblot using HA antibody, which recognizes only transgene expression (Figure 1D), and by immunohistochemistry using AIF-1 antibody (Figure 1F), and confirms the efficacy of this promoter in driving AIF-1 expression in a tissue-specific manner.
Figure 1.
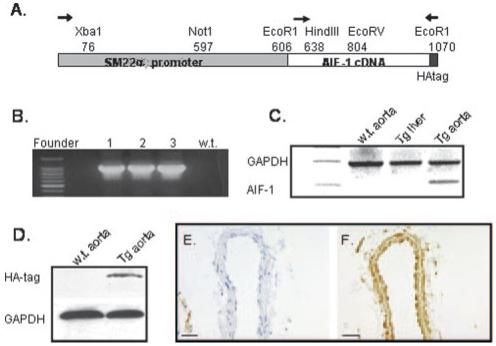
A, Schematic of SM22αG/C promoter-driven AIF-1 expression. Arrows indicate primers used for diagnostic PCR or RT-PCR of mRNA. B, Diagnostic PCR for AIF-1 transgene in wild-type and 3 founder mice. C, Diagnostic RT-PCR for AIF-1 mRNA expression in tissues of transgenic mice. D, Immunoblot of HA-tagged AIF-1 protein in carotid artery using HA antibody. Immunohistochemical detection of AIF-1 protein in naïve wildtype (E) and transgenic (F) carotid artery. Magnification is 400×, bar=20 μm.
Enhanced Intimal Hyperplasia in Response to Injury in AIF-1 Transgenic Mice
To ascertain the effects of AIF-1 expression on the vascular response to injury, the left common carotid artery of wildtype and AIF-1SM22αG/C AIF-1 transgenic littermates were ligated near the carotid bifurcation. After 28 days, arteries were recovered, sectioned, and stained for morphological analysis of vascular compartments. Representative micrographs are depicted in Figure 2A through 2D. No difference in morphology was noted in uninjured arteries (Figure 2A and 2B). However, morphological analysis demonstrate a significant increase in neointimal area of ligation-injured AIF-1 Tg (569.6±64 μm) versus wild-type mice (256.1±49 μm; P=0.003). This corresponds to a significant difference in the neointimal/medial ratio in AIF-1 Tg (0.66±0.048) and wildtype mice (0.37±0.052; P<0.002). The lumen of AIF-1 Tg arteries (308±87 μm) were significantly smaller than wildtype arteries (667±61.2 μm; please see supplemental Figure I). No statistical differences were noted in the overall diameter or medial area of these arteries. In angioplasty injured rat and swine arteries, AIF-1 expression is rapid, peaking 7 to 14 days after injury, and declining to basal levels by 28 days after injury.12 The relative increase in expression of AIF-1 in transgenic carotid arteries compared with wild-type carotid arteries after injury was examined by immunohistochemistry. Figure 2E and 2F shows that little to no AIF-1 is detectible in wild-type arteries, but abundant AIF-1 expression is detected in transgenic arteries at 28 days after injury, demonstrating AIF-1 overexpression is maintained even in dedifferentiated intimal SMCs. Together, these experiments suggest that AIF-1 expression contributes to the vascular response to injury by promoting neointimal formation, rather than vessel remodeling or medial hypertrophy.
Figure 2.
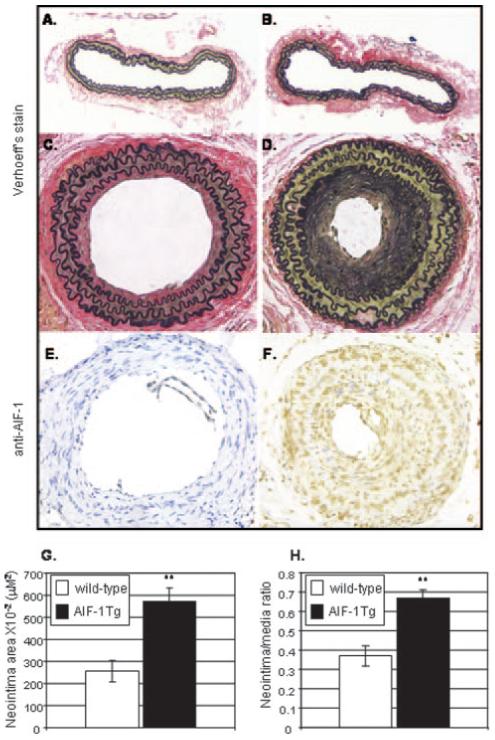
Carotid arteries from AIF-1 transgenic mice have an enhanced response to injury. A-D, Representative micrographs of ligation-injured murine carotid arteries. E and F, Immunohistochemical detection of AIF-1 protein in wild-type (E) and AIF-1 Tg (F) carotid artery 28 days after ligation, using anti-AIF-1 antibody. G and H, Morphological analysis demonstrates a significant increase in neointimal area and neointimal/medial ratio of AIF-1 Tg vs wild-type mice. Magnification is 200×, bar=20 μm.
AIF-1 Expression Increases VSMC Proliferation In Vivo and Ex Vivo
To test the hypothesis that an increase in the proliferative state of arterial VSMCs might be responsible for the dramatic increase in neointimal hyperplasia in ligation-injured AIF-1 Tg carotid arteries, 2 different but complementary experiments were performed. First, serial sections of ligation-injured carotid arteries were immunostained with Ki-67, a marker of cell proliferation. Figure 3 shows a significant increase in Ki-67-stained nuclei in AIF-1 transgenic carotid compared with wild-type carotid (10.6±1.0 versus 3.6±.9, P=0.0007). These cells were CD45 (pan-leukocyte) negative (please see supplemental Figure II). VSMC cultures were established from explants from aorta of AIF-1 Tg and wild-type mice, and the relative increase in expression of AIF-1 cultured VSMCs as compared with wild-type cultured cells after stimulation was examined by Western blot. As expected, wild-type VSMCs demonstrate AIF-1 at barely detectible levels in unstimulated cells, but increase with serum-stimulation (Figure 4A). In contrast, in unstimulated transgenic VSMCs, AIF-1 is expressed at levels which approximate those of stimulated wild-type VSMCs. As expected, immunoblot using anti-HA tag antibody detects protein only in transgenic-derived VSMCs.
Figure 3.
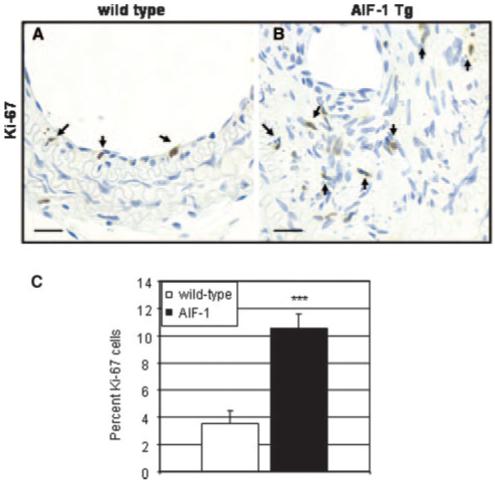
Injured arteries from AIF-1 transgenic mice contain more proliferating cells than do wild-type arteries. Wild-type (A) and AIF-1 Tg (B), immunostained with anti-Ki-67. C, Quantitation of proliferating cells in ligation-injured arteries. Positive staining from media and neointima was counted from at least 4 stained tissue sections from 3 different mice. Values are expressed as percentage of cells. Magnification is 600×, bar=20 μm.
Figure 4.

VSMCs from AIF-1 transgenic mice proliferate more rapidly than wild-type VSMCs. A, Numbers on the y axis indicate cells per well. Shown are mean data from 3 independent experiments. B, Representative blot showing relative expression of endogenous and transgenic AIF-1 in wild-type and Transgenic mice, and AIF-1 detected by AIF-1 antibody or HA tag antibody by Western blot.
To validate differences in proliferative capacity, equal numbers of VSMCs explanted from wild-type or AIF-1 transgenic mice were grown in DMEM supplemented with 1% FCS or 15% FCS growth medium. Figure 4A shows that AIF-1 overexpression in Tg VSMCs is well within physiological levels, as basal levels in Tg VSMCs approximate stimulated levels in wild-type VSMCs. Figure 4B indicates that AIF-1 Tg VSMCs proliferate more rapidly than wildtype VSMCs (55.5±3.6×103 versus 37.2±2.0×103 cells/mL), and even when cultured in reduced serum growth factors, primary VSMCs from AIF-1 transgenic mice prolif-erate significantly more rapidly (24.1×103 versus 14.0×103 cells/mL) than wild-type VSMCs (P=0.001 for each condition). Expression of the cell cycle protein cyclin D1 is also increased in Tg compared with wild-type VSMCs (supplemental Figure III). These experiments indicate that one mechanism whereby AIF-1 expression contributes to increased neointimal hyperplasia in response to injury is by increasing VSMC proliferation.
AIF-1 Expression Influences Signal Transduction
In cultured VSMCs, AIF-1 expression can activate Rac1, and Rac1 is required for AIF-1 activity.15,22 To test the hypotheses that AIF-1 expression might regulate Rac1-mediated signal transduction pathways in vivo, we again pursued complementary in vivo and ex vivo approaches. First, serial sections of ligation-injured carotid arteries were immunostained with anti-phospho p38, a signaling kinase implicated as an important integrator of inflammatory signals,22 and anti-phospho PAK1, a signaling kinase immediately distal to Rac1.23 Immunohistochemical analysis of PAK1 phosphorylation in tissue sections is a useful surrogate to allow cellular localization of AIF-1 mediated Rac1 activation (please see supplemental Figure IV). Figure 5 shows a significantly greater number of phospho PAK1-positive stained cells in ligationinjured AIF-1 Tg carotid arteries compared with wild-type arteries (67.5%±6.7 versus 35.3%±10.2, P=0.02). Perhaps more interesting is the significantly greater number of phospho-PAK-positive cells in naïve AIF-1 Tg arteries compared with wild-type arteries (60.0%±10.5 versus 23.3%±8.0, P=0.002), suggesting a basal level of activated PAK1 in uninjured arteries. There was no significant difference in activated p38 in naïve arteries, but ligated arteries from AIF-1 Tg mice contain significantly greater amounts of activated p38 (55.4%±7.0 versus 22.6%±5.4, P=0.002). These data are the first which indicate that PAK1 and p38 are activated in ligation-injured murine arteries.
Figure 5.

Activation of signal transduction proteins by AIF-1. A, Immunohistochemical (see supplemental Figure IV) staining from media and neointima was counted from at least 4 stained tissue sections from 3 different mice. B, Representative immunoblot of activated PAK1 or p38 MAP kinase in VSMCs. C, Combined densiometry values and means from at least 3 experiments using 3 transgenic or wild-type groups of VSMCs.
To validate this in a more controlled environment, equal numbers of pooled VSMCs explanted from abdominal aorta from 3 different wild-type or AIF-1 transgenic mice were serum-starved for 48 hours, then stimulated with 10% fetal calf serum for 7 or 20 minutes. Activation of p38 and PAK1 was determined by Western analysis with phospho-specific antibody. Figure 5 shows that whereas p38 was activated in response to serum for both cell populations, p38 phosphorylation was significantly increased in the AIF-1-expressing VSMCs compared with wild-type VSMCs (P<0.05 for all time points). One notable difference between the 2 cell populations is that whereas PAK1 activation is responsive to serum in wild-type VSMCs, in AIF-1 VSMCs, PAK1 activation appears to be maximal in unstimulated cells, with no difference in phosphorylation between stimulated and unstimulated samples. Together, these data indicate that AIF-1 expression enhances signal transduction cascades in both cultured VSMCs and in vivo, and may be another mechanism whereby AIF-1 increases neointimal hyperplasia in response to injury.
AIF-1 Expression Increases Migration
Because PAK activation is associated with cellular motility, we investigated differences in migration between AIF-1 Tg and wild-type VSMCs. For these experiments, VSMCs were seeded into Boyden chambers, and differences in chemotaxis were quantitated by counting cells which migrated in response to PDGF. Figure 6 demonstrates a significant difference in migration between AIF-1 Tg compared with wildtype VSMCs (39.2±3.2 versus 17.1±1.5 VSMCs per HPF, P=0.0003). This indicates that AIF-1 Tg VSMCs migrate at an increased rate compared with wild-type VSMCs.
Figure 6.
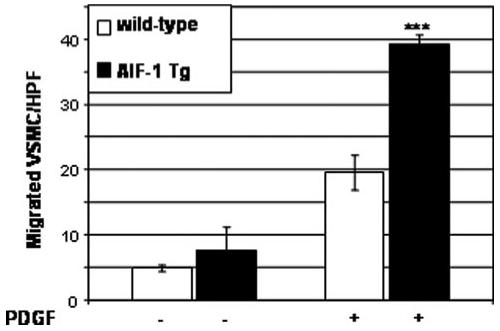
AIF-1 Tg VSMCs migrate at an increased rate compared with wild-type VSMCs. Migrated VSMCs were quantitated by counting 4 high powered fields per membrane. Values are means from experiments performed in triplicate.
Discussion
We have previously shown that AIF-1 expression is a marker of arterial injury and activated VSMCs and is expressed in injured, but not normal, human arteries.12,13 The presence of AIF-1 and the amount of its expression in endomyocardial biopsies from transplanted human hearts correlates with the presence and severity of rejection, and persistent AIF-1 expression is predictive of clinical transplant vasculopathy.13 Although interesting, these data were correlative, and a cause-and-effect relationship between AIF-1 and development of intimal hyperplasia has not been shown. The vascular response to injury is a complex process initiated and propagated by immune cells which create an inflammatory environment resulting in activation of medial VSMCs. AIF-1 expression is considered to be a marker of macrophage activation, though less is known concerning its inflammation-inducible expression in nonimmune cells. Because AIF-1 is constitutively expressed in monocyte/macrophages and T lymphocytes, it was crucial to generate a transgenic mouse in which AIF-1 overexpression was specific to vascular smooth muscle. We used the modified SM22α promoter in which the G/C rich repressor has been removed to allow expression of SM22α even when VSMCs dedifferentiate in response to injury.17,18 Consequently, it was essential to use this promoter to determine the function of AIF-1 in the vascular response to injury. This promoter has been used in several studies, and the constitutive, aortic expression of AIF-1 confirms its efficacy in directing AIF-1 expression in a tissue-restricted fashion. In this study, morphological analysis demonstrate a significant increase in neointimal area and media/intima ratio of ligation-injured AIF-1 Tg versus wild-type mice. The luminal area of AIF-1 Tg mice was also significantly reduced compared with wild-type mice, but the outer elastic lamina was not, indicating that there was no negative remodeling, and the reduction in lumen was attributable to intimal hypertrophy. No difference in morphology between AIF-1 Tg and wild-type uninjured vessels was noted. If AIF-1 does act as a noncatalytic scaffold, it is conceivable that without the background of injury, inflammation-induced signaling proteins are not expressed, or available in an activated form, which may account for the lack of intimal or medial hyperplasia in uninjured Tg mice.
Proliferation of intimal VSMCs is a major contributor to increased intimal hyperplasia in injured arteries and an important maladaptive response of VSMCs to many stimuli. We previously demonstrated that overexpression of AIF-1 in human and rat VSMC line leads to increased proliferation, aberrant cell cycle protein expression, and shortening of the cell cycle,12,15 and we hypothesized that the increased neointima in injured arteries was attributable to proliferation of VSMCs. In injured arteries, significantly more VSMCs stained positive for expression of the proliferation marker Ki-67 in AIF-1 Tg compared with wild-type arteries suggesting that VSMC proliferation was responsible for increased intimal hyperplasia. This was verified using explanted VSMCs in which AIF-1 Tg cells proliferated 91.7% and 83.3% more rapidly in 15% and 1% FCS, respectively. This correlates with our previous work indicating that overexpression of AIF-1 leads to increased VSMC proliferation.
In a previous study, one outcome of RNA knockdown of AIF-1 in mouse macrophages was an inhibition of atherogenic and inflammation-induced signal transduction,18 which lead us to investigate whether overexpression of AIF-1 in Tg arteries would lead to increased activation of inflammation-initiated signaling pathways. We examined p38 MAPK because this kinase is an integrator of inflammation-inducible signaling, and activation of p38 often results in the production and activation of proinflammatory and proliferative mediators.22 p38 phosphorylation mediates VSMC activation, and use of multiple p38-specific pharmacological inhibitors has shown that sustained activation of p38 is an important contributor to the vascular response to injury.24,25 In ligation-injured but not naïve Tg arteries, p38 phosphorylation was significantly increased compared with wild-type arteries. This was validated ex vivo using VSMCs explanted from these mice, where p38 phosphorylation in serum-starved, and also in serum-stimulated, VSMCs was increased in AIF-1 Tg VSMCs compared with wild-type VSMCs. Because of the recognized impact of p38 activity on vascular pathophysiology, AIF-1 activation of p38 is of consequence and may contribute to the increased VSMC proliferation and increased neointima observed in transgenic mice.
We examined PAK1 phosphorylation because PAK1 is a direct effector of Rac1.23 AIF-1 can activate Rac1 in human VSMCs, and Rac1 activation is required for AIF-1 activity, as point mutations in the AIF-1 calcium binding domain create activation-null mutants in which Rac1 activation, VSMC proliferation, and migration are impaired.14,26 Significantly more PAK1 is phosphorylated in the AIF-1 Tg ligation-injured carotid arteries compared with wild-type. There was an unexpectedly high level of PAK1 activation in naïve AIF-1 transgenic arteries, suggesting constitutive activation of Rac1, which is directly proximal to PAK1 and responsible for its activation.29,30 In cultured VSMCs, AIF-1 Tg VSMCs also demonstrate a similarly high level of PAK1 phosphorylation in serum-starved VSMCs, compared with wild-type VSMCs. One noteworthy difference between the 2 cell populations is that in AIF-1 Tg VSMCs, PAK1 activation appears to be maximal in unstimulated cells, with no increase in activation in serum-stimulated samples, whereas wild-type VSMCs do show an expected FCS-induced increase in phosphorylation. In tracheal SMCs, PAK1 activation is proximal to p38 activation.27 The observation that in AIF-1 Tg VSMCs, PAK1 phosphorylation is maximal in unstimulated VSMCs, but p38 activation does increase with stimulation, implies that p38 may be activated as part of a second pathway, and not necessarily be proximal to PAK1, at least in these cells. Differences in cultured VSMCs and in vivo may also explain why p38 activation does not differ in naïve transgenic and wild-type arteries but do differ in cultured VSMCs. Migration is regulated by the activation of Rac1 and its direct effector, the PAK proteins.29,30 Inhibition of Rac1 and its downstream effector PAK1 attenuates VSMC migration.31 Collectively, data in arteries and explanted VSMCs suggests that the increased proliferation observed in Tg VSMCs is likely a consequence of increased activation of the Rac1/PAK pathway. This also corroborates our previous data demonstrating constitutive expression of AIF-1 enhances Rac1 activation, aberrant expression of cell cycle proteins, and proliferation, even in serum starved VSMCs.12,14,28 On the other hand, the observation that uninjured arteries from transgenic mice do not demonstrate differences in intimal or medial VSMC hyperplasia may illustrate recognized differences between in vivo and cell culture conditions. This would suggest that whereas in serum reduced culture conditions AIF-1 can effect proliferation and signal transduction cascades, and in vivo in naïve arteries AIF-1 can induce signal transduction, other components necessary to promote more distal events as proliferation and intimal hyperplasia in naïve arteries are absent. This may also explain why p38 activation does not differ in naïve transgenic and wild-type arteries.
In summary, we have constructed a transgenic mouse in which AIF-1 expression is restricted to arterial VSMCs to determine whether AIF-1 expression influences the vascular response to injury. Several novel findings generated from use of this mouse and supported by ex vivo experiments result from this study: (1) PAK1 and p38 are activated in ligation-injured murine arteries; (2) AIF-1 expression results in increased neointima in ligated murine carotid arteries; (3) AIF-1 expression results in increased proliferation of VSMC in injured arteries and in cultured VSMCs; (4) AIF-1 expression results in increased p38 and PAK1 phosphorylation in injured arteries and cultured VSMCs; and (5) AIF-1 expression results in increased VSMC migration. Although the precise molecular mechanism(s) whereby AIF-1 leads to activation of Rac1 and distal signaling proteins remains to be elucidated, these data provide insight into the involvement of the Rac1/PAK/p38 pathway in development of intimal hyperplasia, and identifies AIF-1 as an important molecular component in that pathway. It also supports the hypothesis that AIF-1 is an inflammation-responsive protein which promotes signal transduction resulting in VSMC activation and neointimal hyperplasia in response to injury.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by grant HL-63810 from the National Heart, Lung, and Blood Institute, and in part from grant 0655403U from the American Heart Association, and 146643428, from the Roche Organ Transplant Research Foundation, to M.V.A.
Footnotes
Disclosures
None.
References
- 1.Welt FGP, Rogers C. Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol. 2002;22:1769–1776. doi: 10.1161/01.atv.0000037100.44766.5b. [DOI] [PubMed] [Google Scholar]
- 2.Ventura HO, Mehra MR, Smart FW. Cardiac allograft vasculopathy: current concepts. Am Heart J. 1995;129:791–798. doi: 10.1016/0002-8703(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 3.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 4.IP J, Fuster V, Badimon L, Badimon J, Taubman M, Chesebro J. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiology. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 5.Hung AY, Sheng M. PDZ domains: structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, DeVivo M, Dingus J, Harry A, Li J, Sui J, Carty DJ, Blank JL, Exton JH, Stoffel RH, et al. A region of adenylyl cyclase 2 critical for regulation by G protein beta gamma subunits. Science. 1995;268:1166–1169. doi: 10.1126/science.7761832. [DOI] [PubMed] [Google Scholar]
- 7.Deininger MH, Meyermann R, Schluesener HJ. The allograft inflammatory factor-1 family of proteins. FEBS Lett. 2002;514:115–121. doi: 10.1016/s0014-5793(02)02430-4. [DOI] [PubMed] [Google Scholar]
- 8.Utans U, Arceci R, Yamashita Y, Russell M. Cloning and characterization of allograft Inflammatory Factor-1: A novel macrophage factor identified in rat cardiac allografts with chronic rejection. J Clin Investigation. 1995;95:2954–2962. doi: 10.1172/JCI118003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schluesener HJ, Seid K, Meyermann R. Effects of autoantigen and dexamethasone treatment on expression of endothelial-monocyte activating polypeptide II and Allograft-Inflammatory Factor-1 by activated macrophages and microglial cells in lesions of experimental autoimmune encephalomyelitis, neuritis, and uveitis. Acta Neuropathol. 1999;97:119–126. doi: 10.1007/s004010050964. [DOI] [PubMed] [Google Scholar]
- 10.Kruse M, Steffen R, Batel R, Muler I, Muller W. Differential expression of allograft inflammatory factor 1 and of glutathione peroxidase during auto-and allograft response in marine sponges. J Cell Science. 1999;112:4305–4313. doi: 10.1242/jcs.112.23.4305. [DOI] [PubMed] [Google Scholar]
- 11.Iris FMJ, Bougueleret L, Prieur S, Caterina D, Primas G, Perrot V, Jurka J, Rodriguez-Tome P, Claverie JM, Dausset J, Cohen D. Dense Alu clustering and a potential new member of the NF-kB family within a 90 kilobase HLA class III segment. Nat Genet. 1993;3:137–145. doi: 10.1038/ng0293-137. [DOI] [PubMed] [Google Scholar]
- 12.Autieri MV, Mu A, Carbone C. Expression of Allograft Inflammatory Factor-1 (AIF1) is a marker of activated human VSMC and arterial injury. Arterioscler Thromb Vasc Biol. 2000;20:1737–1744. doi: 10.1161/01.atv.20.7.1737. [DOI] [PubMed] [Google Scholar]
- 13.Autieri MV, Kelemen SE, Thomas BA, Feller ED, Goldman BI, Eisen HJ. Allograft Inflammatory Factor-1 (AIF-1) expression correlates with cardiac rejection and development of cardiac allograft vasculopathy. Circulation. 2002;106:2218–2223. doi: 10.1161/01.cir.0000035652.71915.00. [DOI] [PubMed] [Google Scholar]
- 14.Autieri MV, Kelemen SE, Wendt KW. AIF-1 is an actin-polymerizing and Rac1-activating protein that promotes vascular smooth muscle cell migration. Circ Res. 2003;92:1107–1114. doi: 10.1161/01.RES.0000074000.03562.CC. [DOI] [PubMed] [Google Scholar]
- 15.Autieri MV, Carbone CM. Over expression of Allograft Inflammatory Factor-1 promotes proliferation of vascular smooth muscle cells by cell cycle deregulation. Arterioscler Thromb Vasc Biol. 2001;21:1421–1426. doi: 10.1161/hq0901.095566. [DOI] [PubMed] [Google Scholar]
- 16.Tian Y, Kelemen SE, Autieri MV. Inhibition of AIF-1 expression by constitutive siRNA expression reduces macrophage migration, proliferation, and signal transduction initiated by atherogenic stimuli. Am J Physiol Cell Physiol. 2006;290:C1083–C1089. doi: 10.1152/ajpcell.00381.2005. [DOI] [PubMed] [Google Scholar]
- 17.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106:1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wamhoff BR, Hoofnagle MH, Burns A, Sinha S, McDonald OG, Owens GK. A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ Res. 2004;95:981–988. doi: 10.1161/01.RES.0000147961.09840.fb. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 20.Harmon KJ, Couper LL, Lindner V. Strain-dependent vascular remodeling phenotypes in inbred mice. Am J Pathol. 2000;156:1741–1748. doi: 10.1016/S0002-9440(10)65045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:85–88. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- 22.Tenhunen O, Rysa J, Ilves M, Soini Y, Ruskoaho H, Leskinen H. Identification of cell cycle regulatory and inflammatory genes as predominant targets of p38 mitogen-activated protein kinase in the heart. Circ Res. 2006;99:485–493. doi: 10.1161/01.RES.0000238387.85144.92. [DOI] [PubMed] [Google Scholar]
- 23.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIMkinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 24.Kavurma MM, Khachigian LM. ERK JNK and p38 MAP kinases differentially regulate proliferation and migration of phenotypically distinct smooth muscle cell subtypes. J Cell Biochem. 2003;89:289–300. doi: 10.1002/jcb.10497. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi N, Matsumori A, Furukawa Y, Ono K, Okada M, Iwasaki A, Miyamoto T, Nakano A, Sasayama S. Role of p38 mitogen-activated protein kinase in neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2000;20:2521–2526. doi: 10.1161/01.atv.20.12.2521. [DOI] [PubMed] [Google Scholar]
- 26.Autieri MV, Chen X. The ability of AIF-1 to activate human vascular smooth muscle cells is lost by mutations in the EF-hand calcium-binding region. Exp Cell Res. 2005;307:204–211. doi: 10.1016/j.yexcr.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Dechert MA, Holder JM, Gerthoffer WT. p21-activated kinase 1 participates in tracheal smooth muscle cell migration by signaling to p38 MAPK. Am J Physiol Cell Physiol. 2001;281:C123–C132. doi: 10.1152/ajpcell.2001.281.1.C123. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Kelemen SE, Autieri MV. AIF-1 Expression Modulates Proliferation of human vascular smooth muscle cells by autocrine expression of G-CSF. Arterioscler Thromb Vasc Biol. 2004;24:1217–1222. doi: 10.1161/01.ATV.0000130024.50058.de. [DOI] [PubMed] [Google Scholar]
- 29.Zhao ZS, Manser E. PAK and other Rho-associated kinases-effectors with surprisingly diverse mechanisms of regulation. Biochem J. 2005;386:201–214. doi: 10.1042/BJ20041638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knaus UG, Wang Y, Reilly AM, Warnock D, Jackson JH. Structural requirements for PAK activation by Rac GTPases. J Biol Chem. 1998;273:21512–21518. doi: 10.1074/jbc.273.34.21512. [DOI] [PubMed] [Google Scholar]
- 31.Weber DS, Taniyama Y, Rocic P, Seshiah PN, Dechert MA, Gerthoffer WT, Griendling KK. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ Res. 2004;94:1219–1226. doi: 10.1161/01.RES.0000126848.54740.4A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.