

Abstract
Current extensive genetic research into common complex diseases, especially with the completion of genome-wide association studies, is bringing to light many novel genetic risk loci. These new discoveries, along with previously known genetic risk variants, offer an important opportunity for researchers to improve health care. We describe a method of quick evaluation of these new findings for potential clinical practice by designing a new predictive genetic test, estimating its classification accuracy, and determining the sample size required for the verification of this accuracy. The proposed predictive test is asymptotically more powerful than tests built on any other existing method and can be extended to scenarios where loci are linked or interact. We illustrate the approach for the case of type 2 diabetes. We incorporate recently discovered risk factors into the proposed test and find a potentially better predictive genetic test. The area under the receiver operating characteristic (ROC) curve (AUC) of the proposed test is estimated to be higher (AUC = 0.671) than for the existing test (AUC = 0.580).
Introduction
With the latest improvements and ever-decreasing costs of high-throughput genotyping technologies, large-scale genetic-association studies, and in particular genome-wide association studies, are now being conducted. These studies provide a comprehensive scan of the whole genome and have the potential to identify many more genetic risk variants for common complex diseases. The new findings from these studies, together with previously known genetic and environmental risk factors—whether or not they increase our understanding of the etiology of common complex diseases—offer a potential opportunity for researchers to improve medical care and public health.1
Some previous efforts aimed at combining genetic and environmental findings to predict disease or, more precisely, to develop a predictive genetic test, are discussed elsewhere.2–5 These are important first steps toward the development of successful predictive genetic tests for common complex diseases,6 and such tests have been recognized as comprising the cornerstone of future genomic medicine.7 The hope is that these tests will provide an early discovery of an individual's disease risk so that appropriate prevention strategies can be used to reduce morbidity and mortality. These tests are anticipated to have a large impact on health care and change the form of health care away from treatment toward prevention.8
However, these previous efforts have been limited by the genetic and clinical information available at the time the predictive genetic tests were developed. For instance, the current predictive genetic test for type 2 diabetes is based on three common variants.3 With the genome-wide association studies that have now been conducted for this disease and the novel susceptibility variants identified,9,10 one may want to know how much an existing test could be improved by incorporating into it the newly discovered genetic susceptibility variants.
The identification of risk variants is a progressive process, and so predictive genetic tests will also be subject to change whenever novel genetic susceptibility variants are discovered. This requires a flexible and easily implemented method for the redevelopment of predictive genetic tests with time.6 In this paper, we propose a flexible model to help design a new predictive genetic test. Using information garnered from published genetic-association studies, clinical studies, and even a previously accepted predictive genetic test, this approach provides an estimated classification accuracy of the proposed test and hence an idea of how much improvement over an existing test a proposed new test might achieve.
The clinical performance and applicability of a predictive genetic test rests on four main components: (1) analytic validity, (2) clinical validity, (3) clinical utility, and (4) associated ethical, legal, and social implications.11 Our work here focuses on addressing the clinical validity issue, defined as the ability of a genetic test to detect or predict the associated disorder (phenotype). The assessment of clinical validity is an important step and is the starting point for test building. If the predictive genetic test discriminates well among possible eventual outcomes, we continue to evaluate its clinical utility. If, on the other hand, it has poor accuracy, it is unlikely to have practical value for patient care.12
In this paper, we first derive a general formula to build a predictive genetic test from previous independent association results, extending it to situations where the genetic variants are in linkage disequilibrium or interact with each other. We then indicate how the requisite sample size can be calculated in order to design a study that will have specified power at a given significance level to be sure that this new test attains a desired level of accuracy or has a higher level of accuracy than the existing test. Finally, we illustrate the model with recent association results that have become available for type 2 diabetes.
Material and Methods
Several approaches have been proposed for the evaluation of a predictive genetic test based on multiple disease-susceptibility loci. Among them, the receiver operating characteristic (ROC) curve13 has been recognized as the most suitable measure.14,15 The ROC curve plots a test's true-positive rate (sensitivity) against its false-positive rate (1-specificity) for continuously changing cutoffs over the whole possible range of test results. It evaluates the tests that result from these cutoffs with the entire spectrum of pairs of true-positive rates (TPRs) and false-positive rates (FPRs) and so gives a global description of a test's classification accuracy.16 The ROC curve is also one of the most popular measures used for clinical diagnostic tests and has been widely used in different areas of medicine.17
Among all the techniques available for combining multiple predictors for ROC analysis, the logistic-regression-based approach has been the most commonly used. However, as recent studies have shown, logistic regression will not be optimal if the logistic model does not hold.18 In the statistics literature, the optimality of the likelihood ratio for ROC analysis has long been recognized.19,20 The likelihood ratio—which in this context is defined as the ratio of two density functions of a predictor x, that among cases to that among controls—is useful for the generation of the appropriate (optimal) ROC curve. With Egan's definition,20 the optimal ROC curve results when this likelihood ratio is plotted from its largest value to its smallest value. The optimal ROC curve displays the best possible performance set based on the likelihood ratio in terms of (1) the maximization of the TPR for any fixed value of the FPR, (2) the minimization of the overall misclassification probability, and (3) the minimization of the expected cost. These three ideal properties could be obtained simply by the application of the Neyman-Pearson lemma.20,21 Although the likelihood ratio is ideal for ROC analysis, it was only recently emphasized for combining multiple predictors.21,22 Baker,22 who noted the important role of the likelihood ratio for combining multiple predictors, based his argument regarding its optimality by drawing on cost-effectiveness theory23 rather than on the Neyman-Pearson lemma. Despite this difference, the two arguments are essentially the same, suggesting that for multiple predictors, a test based on the likelihood ratio is optimal for any fixed value of TPR or FPR. This conclusion is elegant, but it has limitations if we are interested in comparing tests with different TPR and FPR pairs or in evaluating the overall performance of the test. For the latter purpose, the ROC curve or its summary indexes are the appropriate criteria to use. Here we choose the area under the ROC curve (AUC), the most popular summary index of the ROC curve, as a measure of a test's performance and prove that the optimal ROC curve has the highest AUC (Appendix A). The AUC measures the probability that test values from a randomly selected pair of diseased and nondiseased individuals are correctly ordered and is thus a convenient global measure for the quantification of classification (diagnostic) accuracy.12 This means that a test based on the optimal ROC curve achieves the highest classification accuracy among all approaches (including a logistic-regression-based approach).
Building a Predictive Genetic Test on the Basis of the Optimal ROC Curve
A General Model for Independent, Noninteracting Loci
Suppose we are interested in constructing a predictive genetic test on the basis of variants at n genetic loci. From previous association studies or other public sources, we obtain the relative risks estimates and population genotype frequency estimates for the ith associated locus, which has possible genotypes. The distribution of these genotypes at the ith locus in the disease (D) population can then be derived from this information, with the formula
| (1) |
The equations displayed in Equation 1 are appropriate for a variety of genetic variants (in particular, single-nucleotide polymorphisms) with known mode of inheritance (Appendix B). The equations can also be modified for the situation where the risk estimates are odds ratios (Appendix B). The information we can use is not limited to that from association studies; it can also come from other genetic studies (Appendix B).
If we assume linkage equilibrium among the n loci, we can calculate the joint probability of the multilocus genotype from the single-locus-genotype frequencies,
| (2) |
where K denotes the total number of multilocus genotypes possible from the n disease-susceptibility loci and its maximal value is . If we further adopt a multiplicative model, which assumes that the joint effect of the n genetic variants is proportional to the product of the individual variants' main effects, then the probabilities of the multilocus genotypes given disease status can be expressed as
| (3) |
where denotes absence of disease and ρ denotes the disease prevalence. Given these probabilities, we can calculate the likelihood ratios (LRs):
| (4) |
We rank the multilocus genotypes in descending order of their LRs, from the highest rank to the lowest rank, and plot the test's TPR (sensitivity) against its FPR (1-specificity) for each possible cutoff between adjoining pairs of multilocus genotypes that might be used in the prediction of disease. This gives us the empirical optimal ROC curve, which simply consists of a set of TPR and FPR pairs:
| (5) |
where is the κ th genotype in the sequence of likelihood ratios. Because the LRs are in descending order and the LRs correspond to the slope of the ROC curve, this indicates that the optimal ROC curve is always concave.
Once the optimal ROC curve has been built, we obtain the explicit expression for the area under the optimal ROC curve by applying the trapezoid rule:
| (6) |
where . This measures the estimated discriminative ability of the test and leads to the highest value of the AUC among all approaches to designing a predictive test. Other statistics we might be interested in, such as predictive values, can be directly obtained from the optimal ROC curve (Appendix C). Proof of the optimality of the predictive values obtained this way follows from the fact that the optimal ROC curve maximizes the TPR for any fixed value of the FPR and the equations in Appendix C.
Genetic Loci in Linkage Disequilibrium or Interacting with Each Other
The above model can be extended to incorporate loci that are in linkage disequilibrium (LD) with each other. In this case, we are interested in the multilocus genotypes formed by these linked loci. Assume we have L linked loci, and for each locus we have alleles. Following a notation similar to that in Gorelick and Laubichler,24 we denote by the coefficients of LD between n loci and by the population allele frequency for the th allele at locus l. Assuming that all possible LD coefficient and population allele-frequency estimates can be obtained from previous studies, we can express the haplotype frequency for haplotype as the summation of all possible products of LD coefficients and allele frequencies whose orders (i.e., the number of loci) add to L,
| (7) |
where is the coefficient of linkage disequilibrium between loci24.
Normally, the coefficients of LD with order higher than two are rarely reported in genetic studies. Denote by the pairwise LD between the alleles and at loci l and . Assuming that for each pair of loci l and all possible pairwise LD coefficient estimates can be obtained from previous studies (in particular, for SNPs), we can approximate the haplotype frequency by only using the pairwise LD () and the population allele frequencies ,
| (8) |
Given the haplotype frequency and the assumption of Hardy-Weinberg equilibrium (HWE), we can derive the distribution for the phased multilocus genotype ,
Provided that the haplotype-relative risk information is available, we can derive the relative risks for the phased multilocus genotype , on the basis of an additive model, as
Although the above equations assume an additive model, any other model (e.g., recessive) can also be adopted according to any prior knowledge of the disease. By treating these L linked loci as comprising one set of genotypes and applying Equation 1, we can incorporate linked loci into the approach.
In a similar manner, we can extend the model to handle interacting loci. In this scenario, we group all possible multilocus genotypes from the interacting loci into a few clusters, each with a different associated disease risk. In the simplest situation, we have just two clusters, a high-risk cluster and a low-risk cluster. At the other extreme, each multilocus genotype itself represents a cluster. Then, by obtaining the relative risks and the distribution of these clusters, and again applying Equation 1, we can incorporate interacting loci into the model.
We illustrate this by using a simple example. Assume there is an interaction between two SNPs (A and B) and the underlying interaction follows a threshold model,25 defined as implying there is a single high risk for all individuals having at least one of the disease-susceptibility alleles at each of the two loci and a common low risk for all other individuals. We denote by rr the relative risk of the high-risk group compared to the low-risk group and by f the population frequency of the high-risk group. Then the distribution of the high- and low-risk groups in cases can be written as
where, assuming Hardy-Weinberg equilibrium and that and are the frequencies of the disease-susceptibility alleles for the two loci, . Although we illustrate the model by using genetic variants, the equations also apply for clinical risk factors and to the situation where there is gene-environment interaction.
Sampling Variability of the Empirical AUC and Comparison of Empirical ROC Curves
The above procedure provides an estimated classification accuracy of the new test in terms of the AUC. The estimated AUC could be subject to large variability if the sample size is small. We introduce here two methods for the calculation of the AUC variance, which enables us to quantify its precision.
Asymptotically, the variance of the estimated AUC depends only on the ROC curve itself and the numbers of cases and controls:26
| (9) |
where and are the sample sizes of the disease and nondisease samples and and are given by
| (10) |
This provides a simple variance estimate when all the associated genetic variants used for the test are based on one study. If the genetic variants come from different studies, we cannot use the above equations because and are not defined. In this case, we can adopt a bootstrap approach to estimate the variance. Suppose the n genetic variants for the test are from U independent studies. For the ith associated variant in the uth study , we apply Equation 1 to compute its genotype frequency in the case sample and repeat this step for all genetic variants in the uth study. Further, by using Equations 2 and 3 to combine these genetic variants, we can obtain the probabilities of the multilocus genotypes given disease status. Given the total number of diseased and nondiseased individuals in the uth study, we can derive the observed numbers of all possible multilocus genotypes given disease status. On the basis of these observed numbers, we draw a bootstrap sample and use the sample to calculate the genotype frequencies, given disease status, for each of the variants. We repeat this procedure for all the other U-1 studies and then apply Equations 2–6 to construct the optimal ROC curve and compute the AUC estimate. By drawing a large number of bootstrap samples (e.g., 1000), we can obtain the bootstrap variance for the AUC estimate, denoted . Although we illustrate the bootstrap approach for genetic variants, the same approach also applies for clinical risk factors and to the situation where the genetic loci are in LD or interacting with each other.
With the variance estimates, we can easily determine the significance of the difference between two AUC estimates, and , for two different predictive tests. If the associated variants on which the two tests are based come from different studies, the variance of the AUC difference is equivalent to the sum of the variances of the two AUC estimates,
where and are the variances of and . On that basis, we construct an appropriate test statistic that under the null hypothesis and in large samples follows a standard normal distribution:
| (11) |
If some or all of the associated variants on which the two tests are based come from the same studies, then we need to take the covariance of and into account. We can adopt the same bootstrap approach here, but now forming two optimal ROC curves from each bootstrap sample. The bootstrap variance of the AUC difference can be expressed as
where , , and are the bootstrap variances of and and the bootstrap covariance between and .
Sample-Size Calculation
If the classification accuracy estimate (i.e., the AUC) of the new test appears to be superior to existing tests, or if it reaches a desired accuracy level, it might be worth further developing for clinical use. However, the clinical validity of the test, i.e., its classification and/or prediction accuracy, should be comprehensively evaluated before considering the test for clinical use.11 For that purpose, a new replication study is necessary.27 Such a study serves the purpose of verifying the test's estimated classification accuracy, which has been so far estimated on the basis of assumptions and information from published genetic studies. To conduct the study, we set up a hypothesis of interest and design the study with the requisite sample size to test that hypothesis. If we assume is the AUC that measures the classification accuracy of a previous test, or is the minimum desired level of classification accuracy, we are interested in knowing whether the performance of the new test is superior to . Our null hypothesis for this purpose is , with the alternative , where A is the AUC for the new test. An appropriate sample size can then be determined that will ensure, with specified power at an appropriate significance level, that the new test exceeds the minimal acceptable AUC value . For this, we adopt the general approach based on asymptotic theory.28 Assume α and are the specified type I error and power we require for our test; the required sample size for the test can then be expressed as
| (12) |
where and are the sample sizes required for the disease and nondisease samples, respectively, , and Φ is the standard normal cumulative distribution function.
Application to Type 2 Diabetes
With the numerous genetic and clinical studies conducted so far, our understanding of the causes of type 2 diabetes has greatly improved. Thus, now might be the right time to begin applying the more recent findings into clinical use, developing for type 2 diabetes a predictive test that combines all possible genetic variants and environmental factors. In particular, we use here novel susceptibility loci that have been identified in recent genome-wide association studies conducted for type 2 diabetes.9,10
Searching for a successful predictive genetic test for type 2 diabetes has already been initiated. Recently, Weedon et al.3 used three common variants, rs5219 (Glu23Lys) of KCNJ11, rs1801282 (Pro12Ala) of PPARG, and rs7903146 of TCF7L2, to predict the risk of type 2 diabetes. We start with their study to test how consistent the result from our approach is with their findings and then investigate how much we might be able to improve their test by utilizing the newer findings from genome-wide studies.
From the study by Weedon et al.,3 we obtained the allele frequencies among cases and controls for the three variants. We computed the genotype frequencies among cases and controls assuming Hardy-Weinberg equilibrium for both the case and the control populations. Applying Equation 3, we computed the multilocus genotype probabilities given disease status, as detailed in Table 1. From that, we could construct the optimal ROC curve (Figure 1). The area under the optimal ROC curve is estimated to be 0.580 with Equation 6. The estimated standard error of the AUC is 0.0076 and 0.0075 with Equation 9 and the bootstrap approach, respectively. If we chose 0.0076 as the estimate of the standard error, the corresponding 95% confidence interval (CI) for the AUC is [0.565, 0.595]. The area under the optimal ROC curve has exactly the same value as the one obtained from the logistic regression performed in the original paper.3 This is not surprising, because only three loci are involved in the test and there is no evidence of interaction among these loci,3 so that the ROC curve from linear logistic regression should well approximate the optimal ROC curve. Also, from Figure 4 in the original paper, the logistic-regression-based ROC curve is a concave curve that corresponds closely to the optimal ROC curve.
Table 1.
Calculation of an Optimal ROC Curve: Three SNPs
| Allele Freq.a (Case;Control) |
Genotype Freq.b (Case;Population) |
Multilocus Genotype Freq.c (Case;Control) |
Likelihood Ratiod | Rank Ordere | ||||
|---|---|---|---|---|---|---|---|---|
| rs5219 | rs1801282 | rs7903146 | rs5219 | rs1801282 | rs7903146 | rs5219x rs1801282x rs7903146 | ||
| 0.384;0.354 | 0.099;0.123 | 0.384;0.3 | 0.147;0.126 | 0.010;0.015 | 0.147;0.093 | 0.0002;0.0002 | 1.236 | 7 |
| 0.473;0.423 | 0.0007;0.0006 | 1.086 | 11 | |||||
| 0.379;0.484 | 0.0005;0.0006 | 0.954 | 15 | |||||
| 0.178;0.214 | 0.147;0.093 | 0.0039;0.0024 | 1.590 | 3 | ||||
| 0.473;0.423 | 0.0124;0.0089 | 1.394 | 6 | |||||
| 0.379;0.484 | 0.0100;0.0082 | 1.223 | 9 | |||||
| 0.812;0.771 | 0.147;0.093 | 0.0177;0.0086 | 2.052 | 1 | ||||
| 0.473;0.423 | 0.0566;0.0316 | 1.794 | 2 | |||||
| 0.379;0.484 | 0.0454;0.0289 | 1.570 | 4 | |||||
| 0.473;0.458 | 0.010;0.015 | 0.147;0.093 | 0.0007;0.0008 | 0.855 | 16 | |||
| 0.473;0.423 | 0.0022;0.0029 | 0.753 | 19 | |||||
| 0.379;0.484 | 0.0018;0.0027 | 0.663 | 23 | |||||
| 0.178;0.214 | 0.147;0.093 | 0.0124;0.0114 | 1.094 | 10 | ||||
| 0.473;0.423 | 0.0399;0.0415 | 0.962 | 13 | |||||
| 0.379;0.484 | 0.0320;0.0378 | 0.846 | 17 | |||||
| 0.812;0.771 | 0.147;0.093 | 0.0566;0.0404 | 1.402 | 5 | ||||
| 0.473;0.423 | 0.1817;0.1476 | 1.231 | 8 | |||||
| 0.379;0.484 | 0.1457;0.1349 | 1.080 | 12 | |||||
| 0.379;0.415 | 0.010;0.015 | 0.147;0.093 | 0.0005;0.0009 | 0.590 | 24 | |||
| 0.473;0.423 | 0.0018;0.0034 | 0.521 | 26 | |||||
| 0.379;0.484 | 0.0014;0.0031 | 0.459 | 27 | |||||
| 0.178;0.214 | 0.147;0.093 | 0.0100;0.0133 | 0.753 | 20 | ||||
| 0.473;0.423 | 0.0320;0.0483 | 0.663 | 22 | |||||
| 0.379;0.484 | 0.0257;0.0440 | 0.584 | 25 | |||||
| 0.812;0.771 | 0.147;0.093 | 0.0454;0.0473 | 0.960 | 14 | ||||
| 0.473;0.423 | 0.1457;0.1725 | 0.845 | 18 | |||||
| 0.379;0.484 | 0.1169;0.1572 | 0.743 | 21 | |||||
Details of using the optimal ROC-curve approach for the three SNPs used by Weedon et al.3
Allele-frequency estimates obtained from Weedon et al.3
Genotype frequencies are computed assuming Hardy-Weinberg equilibrium separately for the case and the control populations.
Multilocus genotype probabilities given disease status calculated with Equation 3.
Likelihood ratios calculated with Equation 4.
Rank order of the LRs.
Figure 1.
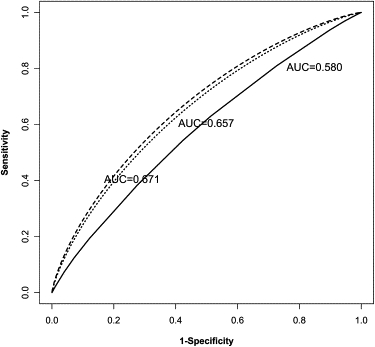
ROC Curves for Type 2 Diabetes
The three lines in the plot from bottom to top correspond to the ROC curves of three type 2 diabetes predictive tests: the rebuilt existing predictive genetic test based on three SNPs, the new predictive test combing the previously associated SNPs, four environmental factors, and four novel risk SNPs from the confirmatory stage of the genome-wide association study,9 and the improved new predictive test with five additional novel risk SNPs discovered in the second genome-wide association study of type 2 diabetes.10 The estimated AUC values of these three tests are 0.580, 0.657, and 0.671, respectively.
We can improve the existing genetic test for type 2 diabetes in at least two ways. Clinical studies have shown that diet, physical activity, cigarette smoking, and alcohol consumption affect the risk of type 2 diabetes.29 These environmental factors could potentially increase the accuracy of the test and, perhaps more importantly, there is the possibility that they interact with the genetic variants to cause the disease.5,9 With regard to genetic variants, a two-stage genome-wide association study has now been completed for type 2 diabetes.9 This study confirmed the association with rs7903146 in the TCF7L2 gene and, in addition, seven SNPs were discovered representing four novel disease-susceptibility loci. We combined the information from these new loci, four important environmental factors, and the three variants used in the previous genetic test to create a new predictive genetic test. To avoid overestimating the test's discriminative ability, we chose four SNPs from the seven new SNPs to represent the four novel loci, removing the remaining three SNPs, which are in linkage disequilibrium with the selected loci. The information for these four novel loci, as well as for rs7903146, comes from the confirmatory stage (stage 2)9 of the study because estimates from that stage are more reliable (i.e., from a well-designed, large-scale association study). Partial details of the calculation are given in Table 2, and we find that the estimated AUC for the new test is 0.657 (Figure 1). In principle, we could also incorporate the three removed SNPs into the tests if the haplotype risk estimates were available, and then the estimated AUC would be even higher.
Table 2.
Calculation of an Optimal ROC Curve: Adding Four SNPs—One Shown—and Four Environmental Factors—One Shown—to the Calculations in Table 1
| Information for Environmental Risk Factorsa (Percentage;Relative Risk) |
Allele Freq.b (Case;Control) |
Genotype Freq.c (Case;Control) |
Frequencies of Environmentald and Genetic Risk Factors (Case;Population) |
Environmental and Genetic Risk Factors Combination Frequencies (Case;Control) |
Likelihood Ratio | Rank Percentagee | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dietary Score | … | rs5219 | … | rs7903146 | … | Dietary Score | … | rs5219 | … | rs7903146 | … | Dietary Score x … x rs5219x … | ||
| 0.150;1.000 | … | 0.384;0.354 | … | 0.163; 0.084 | … | 0.197;0.150 | … | 0.147;0.126 | … | 0.163;0.089 | … | 8.238e-09;1.450e-09 | 5.680 | 0.20% |
| … | : | … | … | … | : | … | … | … | … | … | … | … | … | |
| 0.270;0.860 | … | 0.384;0.354 | … | 0.163; 0.084 | … | 0.305;0.270 | … | 0.147;0.126 | … | 0.163;0.089 | … | 1.275e-08;2.767e-09 | 4.608 | 0.50% |
| … | : | … | … | … | : | … | … | … | … | … | … | … | … | |
| 0.170;0.770 | … | 0.384;0.354 | … | 0.163; 0.084 | … | 0.172;0.170 | … | 0.147;0.126 | … | 0.163;0.089 | … | 7.189e-09;1.806e-09 | 3.982 | 0.90% |
| … | : | … | … | … | : | … | … | … | … | … | … | … | … | |
| 0.260;0.670 | … | 0.384;0.354 | … | 0.163; 0.084 | … | 0.229;0.260 | … | 0.147;0.126 | … | 0.163;0.089 | … | 9.568e-09;2.869e-09 | 3.335 | 1.75% |
| … | : | … | … | … | : | … | … | … | … | … | … | … | … | |
| 0.150;0.490 | … | 0.384;0.354 | … | 0.163; 0.084 | … | 0.097;0.150 | … | 0.147;0.126 | … | 0.163;0.089 | … | 4.037e-09;1.767e-09 | 2.285 | 5..86% |
| … | : | … | … | … | : | … | … | … | … | … | … | … | … | |
Details of using the optimal ROC-curve approach to combine the SNPs in Table 1, four environmental factors, and four novel risk SNPs from the first genome-wide association study9 by assuming (five combinations only, out of a total of combinations).
Distribution and relative risk of the environmental risk factors obtained from Hu et al.29
Allele frequency estimates obtained from Weedon et al.3
Genotype frequency estimates for cases and controls obtained from the confirmatory stage (stage 2) of the first genome-wide association study.9
Distribution of the environmental factors in cases calculated by applying Equation 1.
100•(rank of LR)/(total number of combinations).
Since this paper was first written, another genome-wide association study has also been completed, and an additional five novel disease risk loci have been discovered10 for type 2 diabetes. We therefore obtained the estimates for these five disease risk loci from the second stage of this genome-wide association study and incorporated them also into the new predictive genetic test; partial details of the calculation are given in Table 3. The estimated AUC for the new test is now increased to 0.671 (Figure 1) with an estimated standard error of 0.0071, and the corresponding 95% CI is [0.657, 0.685]. In the near future, more and more disease risk variants will no doubt be discovered,10 and the relation between these variants (e.g., gene-gene interaction) will become clearer. Thus, for type 2 diabetes, or any other disease, our approach could be adopted to progressively incorporate newly discovered variants, and eventually their interaction effects, gradually improving the classification accuracy of a predictive test.
Table 3.
Calculation of an Optimal ROC Curve: Adding Five SNPs—One Shown—to the Calculations in Table 2
| Environmental and Genetic Risk Factors Combination Frequencies from Previous Testa (Case;Control) |
Genotype Freq.b (Case;Control) |
Genotype Freq. (Case;Population) |
Environmental and Genetic Risk Factors Combination Frequencies (Case;Control) |
Likelihood Ratio | Rank Percentagec | ||
|---|---|---|---|---|---|---|---|
| Dietary Score x … x rs5219x … | rs4402960 | … | rs4402960 | … | Dietary Score x … x rs4402960 x … | ||
| 8.238e-09;1.450e-09 | 0.103;0.115 | … | 0.103;0.114 | … | 1.118e-11;1.160e-12 | 9.640 | 0.02% |
| … | … | … | … | … | … | … | … |
| 1.275e-08;2.767e-09 | 0.103;0.115 | … | 0.103;0.114 | … | 1.731e-11;2.300e-12 | 7.523 | 0.05% |
| … | … | … | … | … | … | … | … |
| 7.189e-09;1.806e-09 | 0.103;0.115 | … | 0.103;0.114 | … | 9.756e-12;1.534e-12 | 6.359 | 0.10% |
| … | … | … | … | … | … | … | … |
| 9.568e-09;2.869e-09 | 0.103;0.115 | … | 0.103;0.114 | … | 1.298e-11;2.492e-12 | 5.210 | 0.21% |
| … | … | … | … | … | … | … | … |
| 4.037e-09;1.767e-09 | 0.103;0.115 | … | 0.103;0.114 | … | 5.478e-12;1.589e-12 | 3.447 | 0.89% |
| … | … | … | … | … | … | … | … |
Details of using the optimal ROC-curve approach to further improve the new predictive genetic test in Table 2 by incorporating five additional novel risk SNPs discovered in the second genome-wide association study of type 2 diabetes10 (five combinations only, out of a total of combinations).
Multilocus genotype probabilities given disease status obtained from the new predictive genetic test in Table 2.
Genotype frequency estimates obtained from the confirmatory stage (stage 2) of the second genome-wide association study.10
100•(rank of LR)/(total number of combinations).
Compared to the existing genetic test (AUC = 0.580), the proposed test has substantially higher estimated classification accuracy (AUC = 0.671). To test the difference in these two AUC values, we calculated
where and are the AUC estimates for the proposed test and existing test, respectively, and is the bootstrap variance of , taking the covariance into account. The corresponding p value for the test is . Although this result is exceedingly significant (owing to being based on an asymptotic result and the large samples involved), the AUC estimates and this test made certain assumptions (e.g., a multiplicative model). Therefore, we should design a study to test whether this accuracy can in fact be achieved. The requisite sample size can be calculated to ensure that the new test has a higher level of accuracy than the existing test. By using Equation 10, we can calculate the variances: and . Assuming a type I error rate (α) of 0.05, power of 0.95, and equal numbers of cases and controls , we compute the necessary sample size using the general formula (Equation 12),
i.e., 191 cases and 191 controls. Note that this particular sample size also applies for testing our hypothesis with any choices of type I error rate and power that satisfy (e.g., and ).
Discussion
The importance and benefit of predictive genetic tests have been recognized by both researchers and the public.7,8,30–33 In a recent article, genetic tests have been described as the cornerstone of genomic medicine.7 Although the benefit of predictive genetic testing is obvious and this opportunity is important for the field of medical genetics, work on this topic is still limited. This is partly due to our limited knowledge of genetic causes of common diseases. With the recent intensive research on common diseases, especially with the completion of genome-wide association studies, many novel, apparently causal genes have already been discovered, and variants of these genes, whether themselves causal or not, could be usefully implemented into genetic tests. For that purpose, we have described here a general model to both design and evaluate a predictive genetic test. By taking the information from previous, related association studies, our approach has the ability to estimate the proposed test's approximate discriminative ability. By using this result, if it is encouraging, we can formulate a hypothesis of interest to rigorously evaluate the proposed test, and for this purpose, we have provided the formula for sample-size calculation. Our approach is easy to use. In a simple scenario (e.g., for the rebuilt existing predictive genetic test for type 2 diabetes), it can be implemented with Excel. We have also provided sample R source code (OPMDesign.R) on the website noted in the Web Resources.
The model we have introduced for the design of a predictive genetic test is an illustration of the use of the original optimality theory based on the likelihood ratio.19,20 This theory indicates that a decision rule based on the likelihood ratio is best. We have further shown that a test built on the likelihood ratio can achieve the highest discriminative ability among all approaches. We incorporated these ideal properties in designing a predictive genetic test for type 2 diabetes. Our approach is similar to the approaches introduced by Baker22 and McIntosh et al.21 The differences among the three approaches relate to the calculation of the LR, or its one-to-one function, the risk score. Baker22 approached the LR by directly using the joint distributions of predictors among cases and controls in the data, whereas McIntosh et al.21 estimated the risk by logistic regression. From a large-sample simulation21 and a real data application,22 Baker's approach showed a better performance than did the logistic-regression approach,21 but with an over-fitting issue when the sample size is limited. A model is liable to over-fitting whenever too many parameters are estimated for the sample size that is available.34 Baker approached the LR directly from the joint distributions of the predictors, with the result that the model complexity increases exponentially with the number of predictors, leading to an overly optimistic estimate of the ROC performance. For example, if the test is based on n risk SNPs, this approach requires genotype combination frequencies to be estimated from each of the case and control samples. Our approach approximates the likelihood ratios by utilizing the essential information of each genetic variant from previous genetic studies and assumes a multiplicative model. This not only allows us full use of the information for single genetic variants, which is commonly obtained in genetic studies, but also helps mitigate against over-fitting. For the same example, our approach needs only genotype frequencies to be estimated from each of the case and control samples to compute the frequencies of all genotype combinations, and therefore the model complexity is greatly reduced. Our approach can use genetic variants with known mode of inheritance and is easily extended to scenarios where the genetic variants are in linkage disequilibrium or interact with each other. Incorporating this additional information can make the approach more robust to the over-fitting problem and increase the power of the approach. However, the performance of the approach relies on the assumptions being satisfied and the accuracy of the information from previous genetic studies. Assumption violation and inaccurate information can bias the parameters of interest (e.g., the AUC). For instance, if we violate the assumption of a multiplicative model and linkage equilibrium by incorporating all seven SNPs discovered from the first genome-wide association study into the test, the estimated AUC will increase from 0.671 to 0.678. Although this does not seem to affect the result too much, violation of the assumptions when there is a large number of loci or loci having strong effects (i.e., high relative risks) will cause serious bias. In illustration of this, assume two loci near each risk locus are also used for forming the test. If we assume for simplicity that the two loci are in complete linkage disequilibrium with the risk loci, then the estimated AUC is increased to 0.750. To avoid introducing such a bias, we can either apply the extended approach to incorporate these loci given the required information (i.e., the LD estimates), if available, or we can simply remove these extra loci—this leads to a conservative estimate, and the design will still be valid.
We illustrated the proposed approach for the case of type 2 diabetes. The approach was first examined with as an example an existing predictive genetic test,3 and the result from the optimal ROC curve method was found to be highly consistent with the one originally reported. This results from the equivalence between the logistic-regression-based ROC curve and the optimal ROC curve in such a simple scenario (i.e., few loci, no interactions). To further improve the predictive test, we incorporated further risk factors. With both important environmental factors and novel loci discovered from two recent genome-wide association studies taken into account, the new predictive genetic test (AUC = 0.671) could have a significantly higher classification accuracy (P ) than the existing test (AUC = 0.580). Because the variants involved in the new test are either well studied or confirmed, and the corresponding estimates used came from well-designed, large-scale studies, the estimated AUC value for this new test could be considered to be a reasonable approximation of the actual classification accuracy of the test. Because gene-gene and gene-environmental interactions were not studied in the previous association studies, we are unable to incorporate these effects into the new proposed test. If there are strong interaction effects among the predictors, our estimated AUC value would tend to be conservative. The design to study the test will still be valid because any strong interaction effect would lead to a higher value of the AUC and thus be in favor of the alternative hypothesis.
Unlike the current predictive genetic test, we also incorporated environmental risk factors into the new test—not only because by themselves they can increase information on risk to the disease, but also because they could interact with the genetic variants to cause the disease. Without considering them, we cannot study any gene-environmental effect in any proposed new test. The other advantage of studying them is that in some scenarios we can use them as a method to help disease prevention. For example, we could use the equation in Appendix C to calculate the positive predictive values (PPV) for individuals who carry the multilocus genotype with most risk. The chance of type 2 diabetes would then be predicted to decrease from 83.9% to 29.2% if they adopted a healthy life style (exercise/week > 7 hr and dietary score = 5) rather than having a nonhealthy life style (exercise/week < 0.5 hr and dietary score = 1). Because the predictive genetic test can be conducted at an early age, such as at birth, it would be relatively easy to advise high-risk people to adopt a healthier life style when they are young rather than make them change behavior after the disease has been diagnosed.
Some researchers have suggested that it would be less costly and more efficient to conduct genetic tests for only high-risk individuals (e.g., individuals with a family history of disease), instead of for the general population.35,36 We therefore also attempted to investigate a predictive test for high-risk diabetes subjects on the basis of results of the initial stage of the first genome-wide association study.9 We found that the resulting test could reach a high level of classification accuracy (AUC = 0.855). However, this result would be liberal because the controls came from the general population rather than from a subpopulation of high-risk individuals. To conduct a genetic test on a particular subgroup of the population requires that we investigate its performance on that subgroup. If a test appears to be superior to the one already in use for the general population, it might be considered as a candidate for a high-risk population, but it must first be carefully tested on such a population.
Our proposed approach should be a useful tool for designing a predictive genetic test. It would help the investigator explore possible hypotheses and make decisions regarding developing a new genetic test. It should function as an exploratory phase of medical test development at little cost. The performance of the approach depends on our knowledge of disease-associated variants and the accuracy of the estimates found in the published association studies. The estimated test's classification accuracy will reflect the actual test's performance if the variants involved in the test have been well studied and their estimates come from well-planned studies. Otherwise, the estimate could be subject to bias. Any result would only be valid for the same population as that used for the association study that produced the estimates, and not necessarily apply to different populations, for which the risk estimates and population frequency estimates could be different.
Appendix A
Proof that the Optimal ROC Curve Has the Highest AUC
When we are dealing with multiple genetic variants, each method combines multiple predictors differently and assigns its own unique score for each multilocus genotype. Because the ROC curve relies only on the ranks of these scores, not the absolute scores, the ROC curve from each approach can be represented by the unique ranks of the multilocus genotypes. Assuming represents the ranks of multilocus genotypes from the optimal ROC approach and represents those from any other approach, we prove that the AUC from the optimal ROC approach is always as great as, or greater than, that from any other approach .
It is easy to show that the rank can always be obtained from by a series of order switches between pairs of multilocus genotypes. We prove that each order switch from the original ranks of the optimal ROC curve can only decrease, or leave unchanged, the AUC value. Assume and are the distributions of the multilocus genotypes in cases and controls, respectively, from the nth order switch. At the (n+1)th step, we switch the order of the ith and jth multilocus genotypes, and the corresponding distributions are then denoted and . Simply by using the trapezoid rule, we can calculate the difference in the AUCs:
where
Because ,
To satisfy the condition the order of the ith to jth multilocus genotypes must keep their original order as in . This requirement also applies to other pair switches and may limit the possible order changing but is always feasible. Therefore, we obtain
and thus prove that the AUC of the optimal ROC curve is at worst equal to that of any other approach. Because is arbitrary, the AUC of the optimal ROC curve is the highest among all ROC curves.
Appendix B
Risk Estimates Measured as Odds Ratios
If the estimates of risk parameters for the genetic variants are odds-ratio estimates, Equation 1 is no longer valid unless we make the rare disease assumption. Under the common disease scenario, we could still obtain , , assuming there are possible (multilocus) genotypes for the ith genetic variant. For each genotype, we denote its odds ratio estimate and the corresponding frequency , . The probability of disease given genotype, , , can then be obtained from the following equations,
| (13) |
By applying Bayes' rule, we have ,
| (14) |
Genetic Variants Are SNPs
We assume the ith genetic variant is a SNP that has two alleles, A and a. From a previous study, we obtain the genotype frequencies for genotypes and the relative risk , the risk for AA (Aa) divided by that for aa. If the genotype frequencies are not available from the previous study, we can estimate them from the allele frequencies on the assumption of HWE. On the basis of Equation 1, we have the genotype distribution in the disease population,
| (15) |
Further modifying the above equations, we can have a similar expression for a different mode of inheritance. For instance, if the variant A is dominant, we have the following conditional probabilities:
| (16) |
where denote the relative risk of the genotypes with the A allele versus the genotype without the A allele.
Using Information from Previous Genetic-Test Studies
If the genetic variants we are interested in have been studied in an existing genetic test, we could also utilize such information. From the ROC curve of the previous genetic test, we can obtain the entire set of TPR and FPR pairs. Assuming there are K TPR and FPR pairs, the distribution of genotype combinations among cases can be derived as
where the are the ordered TPRs from the left side to the right of the ROC curve.
If we are only interested in some of the genetic variants, we sum the above conditional probabilities over all genetic variants of no interest and thus obtain the distribution for the genetic variants of interest.
Appendix C
Calculating Predictive Values
For given disease prevalence ρ, the predictive values can be simply calculated by Bayes' rule:
Web Resources
The URLs for data presented herein are as follows:
R code, http://darwin.cwru.edu/∼qlu/
Acknowledgments
This work was supported by United States Public Health Service Resource grant (RR03655) from the National Center for Research Resources, Research grant (GM28356) from the National Institute of General Medical Sciences, and Cancer Center Support Grant P30CAD43703 from the National Cancer Institute.
References
- 1.Christensen K., Murray J.C. What genome-wide association studies can do for medicine. N. Engl. J. Med. 2007;356:1094–1097. doi: 10.1056/NEJMp068126. [DOI] [PubMed] [Google Scholar]
- 2.Yang Q., Khoury M.J., Botto L., Friedman J.M., Flanders W.D. Improving the prediction of complex diseases by testing for multiple disease-susceptibility genes. Am. J. Hum. Genet. 2003;72:636–649. doi: 10.1086/367923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weedon M.N., McCarthy M.I., Hitman G., Walker M., Groves C.J., Zeggini E., Rayner N.W., Shields B., Owen K.R., Hattersley A.T. Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Med. 2006;3:e374. doi: 10.1371/journal.pmed.0030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yiannakouris N., Trichopoulou A., Benetou V., Psaltopoulou T., Ordovas J.M., Trichopoulos D. A direct assessment of genetic contribution to the incidence of coronary infarct in the general population Greek EPIC cohort. Eur. J. Epidemiol. 2006;21:859–867. doi: 10.1007/s10654-006-9070-5. [DOI] [PubMed] [Google Scholar]
- 5.Lyssenko V., Almgren P., Anevski D., Perfekt R., Lahti K., Nissen M., Isomaa B., Forsen B., Homstrom N., Saloranta C. Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes. 2005;54:166–174. doi: 10.2337/diabetes.54.1.166. [DOI] [PubMed] [Google Scholar]
- 6.Janssens A.C., van Duijn C.M. Towards predictive genetic testing of complex diseases. Eur. J. Epidemiol. 2006;21:869–870. doi: 10.1007/s10654-006-9084-z. [DOI] [PubMed] [Google Scholar]
- 7.Epstein C.J. Medical genetics in the genomic medicine of the 21st century. Am. J. Hum. Genet. 2006;79:434–438. doi: 10.1086/507610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans J.P., Skrzynia C., Burke W. The complexities of predictive genetic testing. BMJ. 2001;322:1052–1056. doi: 10.1136/bmj.322.7293.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 10.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bookman E.B., Langehorne A.A., Eckfeldt J.H., Glass K.C., Jarvik G.P., Klag M., Koski G., Motulsky A., Wilfond B., Manolio T.A. Reporting genetic results in research studies: summary and recommendations of an NHLBI working group. Am. J. Med. Genet. A. 2006;140:1033–1040. doi: 10.1002/ajmg.a.31195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zweig M.H., Campbell G. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin. Chem. 1993;39:561–577. [PubMed] [Google Scholar]
- 13.Lusted L.B. Signal detectability and medical decision-making. Science. 1971;171:1217–1219. doi: 10.1126/science.171.3977.1217. [DOI] [PubMed] [Google Scholar]
- 14.Janssens A.C., Pardo M.C., Steyerberg E.W., van Duijn C.M. Revisiting the clinical validity of multiplex genetic testing in complex diseases. Am. J. Hum. Genet. 2004;74:585–588. doi: 10.1086/382052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Q., Khoury M.J., Botto L., Friedman J.M., Flanders W.D. Revisiting the clinical validity of multiplex genetic testing in complex diseases: Reply to Janssens et al. Am. J. Hum. Genet. 2004;74:588–589. doi: 10.1086/382052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClish D.K. Analyzing a portion of the ROC curve. Med. Decis. Making. 1989;9:190–195. doi: 10.1177/0272989X8900900307. [DOI] [PubMed] [Google Scholar]
- 17.Hanley J.A. Receiver operating characteristic (ROC) methodology: The state of the art. Crit. Rev. Diagn. Imaging. 1989;29:307–335. [PubMed] [Google Scholar]
- 18.Pepe M.S., Cai T., Longton G. Combining predictors for classification using the area under the receiver operating characteristic curve. Biometrics. 2006;62:221–229. doi: 10.1111/j.1541-0420.2005.00420.x. [DOI] [PubMed] [Google Scholar]
- 19.Green D.M., Swets J.A. John Wiley and Sons; New York: 1966. Signal Detection Theory and Psychophysics. [Google Scholar]
- 20.Egan J.P. Academic Press; New York: 1975. Signal Detection Theory and ROC Analysis. [Google Scholar]
- 21.McIntosh M.W., Pepe M.S. Combining several screening tests: Optimality of the risk score. Biometrics. 2002;58:657–664. doi: 10.1111/j.0006-341x.2002.00657.x. [DOI] [PubMed] [Google Scholar]
- 22.Baker S.G. Identifying combinations of cancer markers for further study as triggers of early intervention. Biometrics. 2000;56:1082–1087. doi: 10.1111/j.0006-341x.2000.01082.x. [DOI] [PubMed] [Google Scholar]
- 23.Weinstein M.C., Fineberg H.V., Elstein A.S., Frazier N.S., Neuhauser D., Neutra R.R., McNeil B.J. W.B. Saunders; Philadelphia: 1980. Clinical Decision Analysis. [Google Scholar]
- 24.Gorelick R., Laubichler M.D. Decomposing multilocus linkage disequilibrium. Genetics. 2004;166:1581–1583. doi: 10.1534/genetics.166.3.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini J., Donnelly P., Cardon L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005;37:413–417. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- 26.DeLong E.R., DeLong D.M., Clarke-Pearson D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 27.Chanock S.J., Manolio T., Boehnke M., Boerwinkle E., Hunter D.J., Thomas G., Hirschhorn J.N., Abecasis G., Altshuler D., Bailey-Wilson J.E. Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 28.Pepe M.S. Oxford University Press; New York: 2003. The Statistical Evaluation of Medical Tests for Classification and Prediction. [Google Scholar]
- 29.Hu F.B., Manson J.E., Stampfer M.J., Colditz G., Liu S., Solomon C.G., Willett W.C. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N. Engl. J. Med. 2001;345:790–797. doi: 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- 30.Beaudet A.L. 1998 ASHG presidential address. Making genomic medicine a reality. Am. J. Hum. Genet. 1999;64:1–13. doi: 10.1086/302217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell J. The new genetics in clinical practice. BMJ. 1998;316:618–620. doi: 10.1136/bmj.316.7131.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins F.S. Shattuck lecture–medical and societal consequences of the Human Genome Project. N. Engl. J. Med. 1999;341:28–37. doi: 10.1056/NEJM199907013410106. [DOI] [PubMed] [Google Scholar]
- 33.Jones, M. (2000). The genetic report card. New York Times Magazine, June 11, p. 80.
- 34.Hastie T., Tibshirani R., Friedman J.H. Springer; New York: 2001. The Elements of Statistical Learning. [Google Scholar]
- 35.Khoury M.J. Genetics and genomics in practice: The continuum from genetic disease to genetic information in health and disease. Genet. Med. 2003;5:261–268. doi: 10.1097/01.GIM.0000076977.90682.A5. [DOI] [PubMed] [Google Scholar]
- 36.Vineis P., Schulte P., McMichael A.J. Misconceptions about the use of genetic tests in populations. Lancet. 2001;357:709–712. doi: 10.1016/S0140-6736(00)04136-2. [DOI] [PubMed] [Google Scholar]