

Abstract
Blood vessels are required for a tumor to grow and functional lymphatic vessels are required for it to disseminate to lymph nodes. In an attempt to eradicate both the primary tumor and its lymphatic metastasis, we targeted both blood and lymphatic vessels using two different vascular endothelial growth factor receptor (VEGFR)-2 and -3 tyrosine kinase inhibitors (TKIs), cediranib and vandetanib. We found that while both cediranib and vandetanib slowed the growth rate of primary tumors and reduced blood vessel density, neither agent was able to prevent lymphatic metastasis when administered after tumor cells had seeded the lymph node. However, when administered during tumor growth, cediranib reduced the diameters of the draining lymphatic vessels, the number of tumor cells arriving in the draining lymph node and the incidence of lymphatic metastasis. On the other hand, vandetanib had minimal effect on any of these parameters, suggesting that vandetanib did not effectively block VEGFR-3 on lymphatic endothelial cells in our animal model. Collectively, these data indicate that the response of lymphatic vessels to a TKI can determine the incidence of lymphatic metastasis, independent of TKI's effect on blood vessels.
Keywords: lymphangiogenesis, lymphatic metastasis, tyrosine kinase inhibitors, intravital microscopy
Introduction
Dissection of the molecular, cellular and physical mechanisms of lymphatic metastasis is beginning to yield strategies to prevent lymphatic metastasis (1-7). Specifically, blocking VEGFR-3 signaling with monoclonal antibodies has been shown to prevent lymphatic metastasis, but not control lymphatic metastasis after cancer cells have seeded the lymph node (8-10). To reveal which step of lymphatic metastasis is inhibited by blocking VEGFR-3 signaling, we recently monitored each step in the process of lymphatic metastasis using quantitative intravital microscopy (11) and found that blocking VEGFR-3 signaling reduces the delivery of cells to the lymph node.
These studies lead to a critical question: Can the blockade of both lymphangiogenic and angiogenic signaling pathways prevent formation of lymphatic metastasis after cancer cells have seeded the draining lymph node? This is a timely question in light of the vast pipeline of tyrosine kinase inhibitors of the VEGF receptor family currently undergoing clinical trials (12). In this first use of tyrosine kinase inhibitors (TKIs) that block both VEGFR-2 and -3 signaling to prevent and treat lymphatic metastasis, we used two orally available TKIs, cediranib and vandetanib (AstraZeneca, Macclesfield, UK) (13, 14), in our ear model of lymphatic metastasis that permits imaging of each step in the metastatic process (11).
Materials and Methods
Cell lines
VEGF-C overexpressing (T241-VEGF-C-GFP) and mock-transduced (T241-GFP) T241 murine fibrosarcoma cell lines that constitutively express green fluorescent protein (GFP) under the EF1α promoter were established previously and cultured as reported in 10% FBS/DMEM (11). Human dermal blood microvascular endothelial cells (BECs) and human dermal lymphatic microvascular endothelial cells (LECs) were purchased from Cambrex Corp (East Rutherford, NJ) and were grown in endothelial basal medium 2 containg EGM-2 MV SingleQuots. LECs and BECs were grown on 10 μg/ml fibronectin-coated plates.
Characterization of gene expression by RT-PCR
Total RNA was extracted from cultured cells using TRIzol Reagent (Invitrogen, Carlsbad, CA) and treated with DNase I before hybridization to the oligo(dT) primer. The first strand cDNA was synthesized using SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). PCR reaction was conducted with the following primer pairs using HotStar® Taq Plus PCR kit (Qiagen, Valencia, CA). The sense and anti-sense primers used for VEGF-C: 5′-CAAGGCTTTTGAAGGCAAAG-3′ and 5′-TGCTGAGGTAACCTGTGCTG-3′; VEGFR-1: 5′-CTTTCTCAAGTGCAGAGGGG-3′ and 5′-TCATGTGCACAAGTTTGGGT-3′; VEGFR-2: 5′-GCTTTCGGTAGTGGGATGAA-3′ and 5′-GGAATCCATAGGCGAGATCA-3′; VEGFR-3: 5′-TTGGCATCAATAAAGGCAG-3′ and 5′-CTGCGTGGTGTACACCTTA-3′; PDGFR-α: 5′-ACCTTGCACAATAACGGGAG-3′ and 5′-GAAGCCTTTCTCGTGGACAG-3′; PDGFR-β: 5′-TGCCTCAGCCAAATGTCACC-3′ and 5′-TGCTCACCACCTCGTATTCC-3′; EGFR: 5′-CTGCCAAGGCACAAGTAACA-3′ and 5′-ATTGGGACAGCTTGGATCAC-3′; FGFR1: 5′-GGAAGAGAGACCAGCTGTGATGAC-3′ and 5′-AGATCCGACAGGTCCTTCTCCGTT-3′; c-Kit: 5′-GCATCACCATCAAAAACGTG-3′ and 5′-GATAGTCAGCGTCTCCTGGC-3′; Ret: 5′-TGGCACACCTCTGCTCTATG-3′ and 5′-CTGTTCCCAGGAACTGTGGT-3′; β-Actin: 5′-TGTATGCCTCTGGTCGTACC-3′ and 5′-CAACGTCACACTTCATGATGG-3′. The amplified gene products were resolved on an agarose gel. Mouse embryo mRNA was used for positive controls.
In vitro proliferation assays
The effect of cediranib and vandetanib on net proliferation of T241-GFP, T241-VEGF-C-GFP, BECs and LECs was analyzed using a colorimetric WST-1 based assay (Cell Proliferation Reagent WST-1; Roche Applied Science, Indianapolis, IN). In short, cells were plated at 2000 cells/ml (T241-GPF or T241-VEGF-C-GFP) or 3500 cells/ml (BECs or LECs) and cultured in their growth media as described above without any added stimulation. After 24 hours, different concentrations of cediranib or vandetanib were added. WST-1 assay was performed following the manufacturer's protocol 72 hours later.
Tyrosine Kinase Inhibitor (TKI) dosing
Cediranib (3 mg/kg/day) (14) and vandetanib (50 mg/kg/day) (13) were administered by oral gavage to animals in 1% Tween in PBS for 5 consecutive days followed by two consecutive days with no treatment. This schedule would constitute one week of treatment. 1% Tween in PBS was used as the vehicle control for both compounds.
Animal model
Experiments were performed in nude mice and were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital. To establish tumors we injected 50 μl of tumor cell suspension (containing 5×106 cells) in the tip of the ear. Tumor cell suspension was created from source tumors grown in the flank of 4-6 mice as previously described (11).
Treatment protocols
Prevention Protocol
To test the ability of TKI administration to prevent lymph node metastasis, ear tumors were implanted and a TKI or vehicle was administered during tumor growth. When tumors grew to 40 mm3 in volume, treatment was discontinued and the primary tumor was resected. 28 days after tumor removal (i.e., 42 days after original tumor implantation), microscopic and macroscopic metastases were assessed.
Intervention Protocol
To test the ability of TKI administration to control metastases after tumor cell seeding, ear tumors were implanted and left untreated for 14 days after implantation. On day 14, animals were distributed to ensure equal mean tumor size, primary tumors were resected and a TKI or vehicle was administered for the next 4 weeks. 28 days after tumor removal (i.e., 42 days after original tumor implantation) microscopic and macroscopic metastases were assessed.
Lymphangiography and tumor cell delivery to the lymph node
To examine the effects of TKI administration on tumor cell delivery, ear tumors were implanted and a TKI or vehicle was administered during tumor growth. When the tumors grew to 40 mm3 in volume, functional lymphatic diameter and tumor cell delivery to the draining lymph node were measured as described below.
Lymphangiography was performed by slow injection in the interstitial tissue of the peripheral ear of 500,000 Da TRITC dextran (Invitrogen, Carlsbad, CA). Ear lymphatics were observed with epifluorescence intravital microscopy (E-IVM) and/or multi-photon intravital microscopy (MP-IVM) (15, 16). Lymphangiography images were captured and lymphatic diameters were measured using Image J software (http://rsb.info.nih.gov/ij/) as previously described (11).
To quantify tumor cell delivery following lymphangiography, the cervical lymph node was exposed and imaged with MP-IVM. Images of all the GFP positive cells detectable in each lymph node were captured as image stacks with a 10 μm interval step. 1-8 image stacks per lymph node and 10-51 slices per field were acquired. The number of cells was counted using ImageJ software in a blinded fashion by two investigators.
Histological analysis
For the metastasis assay, formalin fixed and paraffin embedded cervical lymph nodes were stained with hematoxylin and eosin. Multiple sections spaced 200 μm apart spanning the entire lymph node were examined. For blood vessel evaluation in primary tumors, size-matched tumors were fixed in 4% paraformaldehyde by immersion, embedded in paraffin and immunostained with MECA-32 antibody (1:50, BD Biosciences, San Jose, CA). VEGFR-2 (1:500, Cell Signaling, Danvers, MA), VEGFR-3 (1:100, eBioscience, San Diego, CA) and platelet derived growth factor receptor (PDGFR)-β (1:50 Cell Signaling) staining were carried out on T-241-VEGF-C-GFP ear tumors and developed with diaminobenzidine.
Immunofluorescence
T241-VEGF-C-GFP tumors were fixed in 4% formaldehyde in PBS for 2 hrs and followed by 30% sucrose overnight. Tissues were embedded in OCT, frozen and cut into 20 μm thick sections. Sections were fixed in cold acetone and blocked with 3% BSA+5% normal horse serum+0.1% TritonX-100 in PBS. Primary antibody was incubated overnight followed by secondary antibody incubation for 2 hrs. Primary antibodies: Rat anti-CD31 (BD Biosciences), Hamster anti-CD31 (Chemicon, Temecula, CA), Rabbit anti-LYVE-1 (Upstate Biotechnology, Lake Placid, NY); Rabbit anti-VEGFR2 (Cell Signaling), Rat anti-VEGFR3 (eBioscience); Species appropriate secondary antibodies were labeled with Cy3 or Cy5 (Jackson ImmunoResearch, West Grove, PA).
Statistics
Quantitative data are presented as the mean ± SEM. Paired and unpaired Student's t test, Mann-Whitney U-test, ANOVA with Games-Howell post-hoc test and Fisher's exact test were used for statistical analysis. Values of p ≤ 0.05 were considered statistically significant.
Results and Discussion
In vitro and in vivo validation of TKI targets on T241 tumor cell lines
We hypothesized that blocking both VEGFR-2 and VEGFR-3 can inhibit lymphatic metastasis by acting on both blood and lymphatic vessels. To test this hypothesis, we used two TKIs with similar relative biochemical IC50s for VEGFR-2 and VEGFR-3, cediranib and vandetanib (13, 14). Cediranib has additional in vitro activity (IC50 within 10-100 fold of VEGFR-2) against VEGFR-1, c-Kit and PDGFR-β (14). In contrast, vandetanib has additional in vitro activity against EGFR (13) and RET (17). In light of these IC50 data, RT-PCR was used to characterize the in vitro expression of the multiple targets of cediranib and vandetanib in T241-GFP and T241-VEGF-C-GFP cell lines. Of these targets, including VEGFR-2 and VEGFR-3, only PDGFR-α and PDGFR-β were detectable in these cell lines (Figure 1A). We then immunohistochemically stained for VEGFR-2, VEGFR-3 and PDGFR-β in T241-VEGF-C-GFP tumors and found that these receptors were not detectable on the cancer cells in vivo (Figure 1B). We did find that VEGFR-2 was present on 89% of CD31 positive vessels, whereas VEGFR-3 was only present on 15% of CD31 positive vessels based on immunofluorescence (Supplementary Figure 1). Furthermore, we found that 93% of VEGFR-3 positive vessels were also LYVE-1 positive. Although PDGFR-β was not present on tumor cells, it was detected on less than one vessel per high power field (0.35 mm2 field size).
Figure 1.
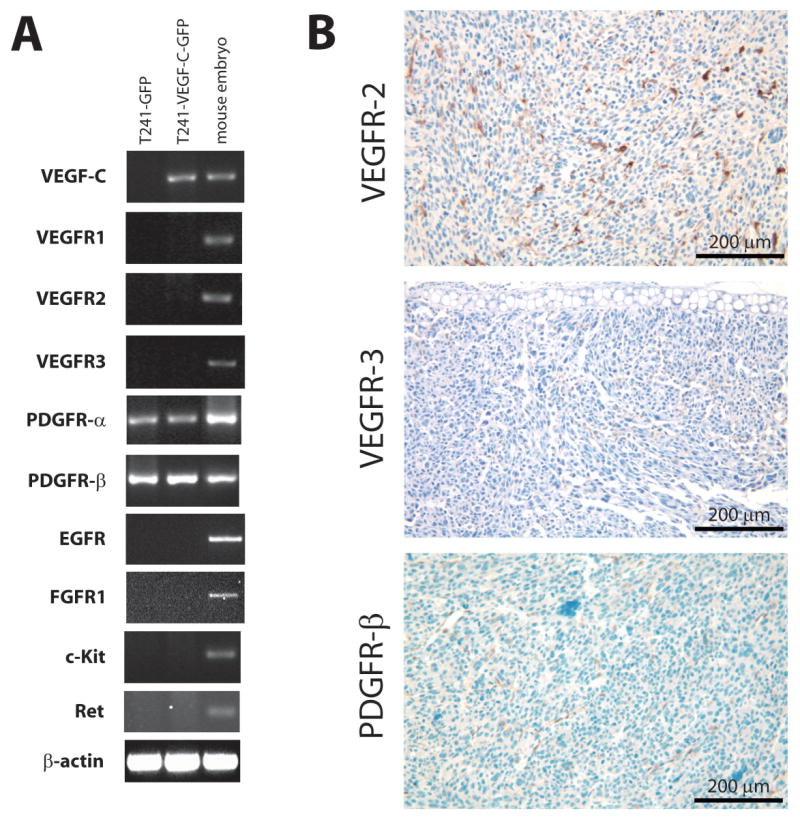
Analysis of molecular targets of cediranib and vandetanib in T241 tumor cell lines. A) Analysis of exogenous VEGF-C gene expression and other endogenous gene expression in T241 transfectants. The amplified cDNA products generated by RT-PCR analysis of total RNA from T241-GFP cells, T241-VEGF-C-GFP cells and murine embryo cells (as positive control) by oligonucleotide primers that recognize the specific genes indicated. B) Immunostaining of VEGFR-2, VEGFR-3 and PDGFR-β in T241-VEGF-C-GFP ear tumor. While VEGFR-2, VEGFR-3 and PDGFR-β expression were not detected in tumor cells, VEGFR-2 expression was detected on most tumor blood vessels and VEGFR-3 and PDGFR-β on a fraction of tumor blood vessels.
Effects of cediranib and vandetanib on endothelial cell and tumor cell proliferation
We then tested the effect of cediranib and vandetanib on proliferation of blood vascular endothelial cells (BECs), lymphatic endothelial cells (LECs), T241-GFP cells and T241-VEGF-C-GFP cells in vitro (Figure 2). The LEC and BEC proliferation IC50s for vandetanib were about 5 fold higher than those for cediranib, in concert with published data (13, 14). The T241-GFP and T241-VEGF-C-GFP cell proliferation IC50s for cediranib were at least 5 fold higher than for BECs and LECs (Figure 2). These data suggest that the effect of cediranib on the rate of tumor growth and metastasis is likely due to its effect on the vasculature and not the tumor cells, despite the presence of PDGFR-α and -β on the tumor cells. In contrast, the tumor cell proliferation IC50s for vandetanib were comparable to BECs and LECs (Figure 2B). The T241-GFP and T241-VEGF-C-GFP IC50s for vandetanib were comparable to that for cediranib.
Figure 2.
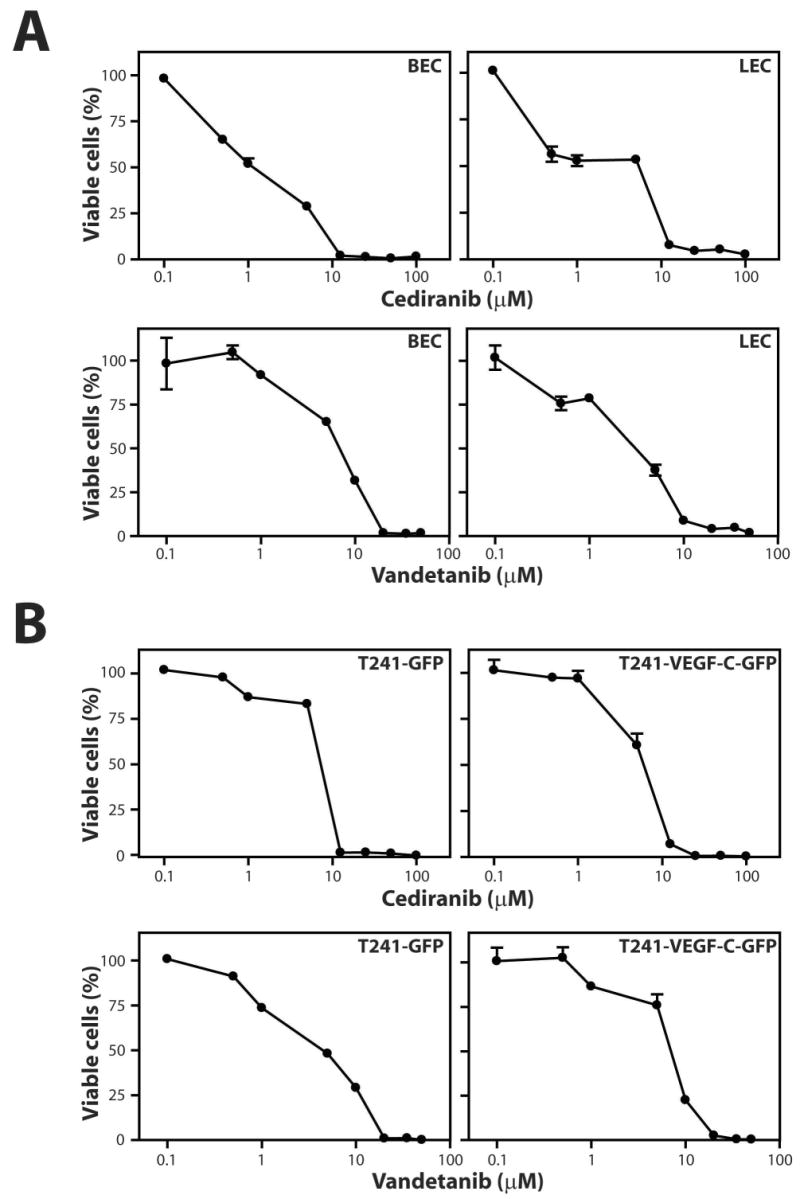
Tyrosine kinase inhibitors cediranib and vandetanib reduce the proliferation of blood and lymphatic endothelial cells more potently than T241 tumor cell lines. A) Dose response curves of cediranib and vandetanib on LECs and BECs. B) Dose response curves of cediranib and vandetanib on T241-GFP and T241-VEGF-C-GFP tumor cells. Proliferation was determined by WST-1 assay.
The comparable response of the T241 tumor cell lines and ECs to vandetanib is somewhat surprising as the tumor cells lack the primary targets of vandetanib. This result is consistent with published literature of IC50s for vandetanib on unstimulated HUVECs (> 3 μM) and 6 tumor cell lines (2.7 to 13.5 μM), showing that unstimulated ECs have similar or higher IC50s compared to tumor cells (13). Collectively, these in vitro data suggest that the in vivo biological effect of vandetanib on endothelial cells and tumor cells could be similar, whereas cediranib may be relatively more effective against endothelial cells in our tumor model.
Both cediranib and vandetanib treatments cause tumor growth delay
We next implanted T241-VEGF-C-GFP tumor cells in our ear model of lymphatic metastasis (11) and measured the effect of cediranib and vandetanib on the time required for these tumors to grow to 40 mm3. In both control groups, tumor growth took about 2 weeks (p > 0.05). As expected, both cediranib and vandetanib caused a significant delay in primary tumor growth (Table 1a, Supplementary Figure 2). However, at the doses used, the growth delay caused by vandetanib (about 2 weeks) was nearly twice as long as that caused by cediranib (about 1 week) (p < 0.05). Using MECA-32 immunohistochemistry, we found a reduction in the area density of intratumor blood vessels in both cediranib and vandetanib treated tumors when compared to controls (Figure 3), suggesting that the growth delay may be due to an anti-angiogenic effect of the TKIs. The equivalent reduction in vascular density suggests that the greater growth delay created by vandetanib may also be due in part to a direct tumor cell effect, as implied by our cell proliferation data (Figure 2), or through inhibition of EGFR and/or Ret on stromal cells.
Table 1.
| Table 1a: Cediranib and vandetanib treatments result in tumor growth delay in T241-VEGF-C-GFP tumors | |||
|---|---|---|---|
| Time for tumor to reach 40mm3 (days) | |||
| Treatment | Control | p-value | |
| cediranib | 23 ± 1.5 (n = 13) | 15 ± 1.4 (n = 10) | p < 0.001 |
| vandetanib | 30 ± 1.6 (n = 11) | 14 ± 2.0 (n = 7) | p < 0.001 |
| Table 1b: Cediranib, but not vandetanib, prevents the generation of lymphatic metastasis, but neither can stop metastasis after cancer cells have spread from T241-VEGF-C-GFP tumors. Data presented are the number of animals with lymphatic metastasis per the total number in its experimental group. | |||
|---|---|---|---|
| Intervention Protocol | |||
| Treatment | Control | p-value | |
| cediranib | 7/15 | 7/15 | 1.0 |
| vandetanib | 7/13 | 6/14 | 0.57 |
| Prevention Protocol | |||
| Treatment | Control | p-value | |
| cediranib | 10/19 | 15/17 | 0.031 |
| vandetanib | 11/11 | 11/14 | 0.23 |
Figure 3.
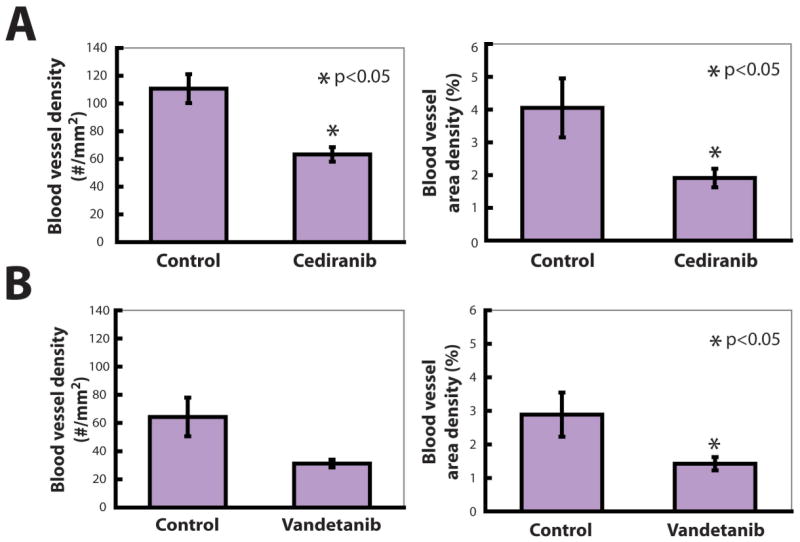
Cediranib and vandetanib reduce tumor angiogenesis. A) Cediranib treatment reduced the area and number density of intratumor blood vessels identified by MECA-32 immunohistochemistry. B) Vandetanib treatment also reduced the area density of intratumor blood vessels identified by MECA-32 immunohistochemistry.
Cediranib prevents formation of lymphatic metastasis
The ability of both TKIs to inhibit primary tumor growth suggested that these compounds may be able to inhibit both the formation of lymphatic metastasis and its subsequent growth in the lymph node. To test this hypothesis, we used two different treatment protocols, Intervention and Prevention. In the Intervention Protocol, which simulates the situation in which a patient is diagnosed with a primary tumor that may or may not have already spread, both cediranib and vandetanib were unable to reduce lymph node metastasis from T241-VEGF-C-GFP tumors (Table 1b). These data are consistent with our previous report using a blocking monoclonal antibody to VEGFR-3 (11).
In the Prevention Protocol, in which the TKIs were administered from the time of tumor implantation until the tumors reached a size of 40 mm3, cediranib was able to reduce the incidence of lymphatic metastasis from T241-VEGF-C-GFP tumors (Table 1b). In contrast, vandetanib was not able to prevent metastasis.
In spite of the ability of both TKIs to inhibit tumor growth, we found a marked difference in the ability of the two compounds to prevent the formation of lymphatic metastasis. This result was surprising; particularly in light of the longer growth delay induced by vandetanib. Vandetanib dose escalation was not possible as the animals were losing weight over the 30 days of treatment at a dose of 50 mg/kg (5 days per week). The group treated with vandetanib lost 7% of its body weight (p < 0.001) compared to the control group, which showed no weight change (p > 0.1). Animals treated with cediranib at a dose of 3 mg/kg (5 days per week) did not show any weight change (p > 0.1).
Cediranib inhibits lymphatic hyperplasia in the tumor margin and arrival of tumor cells in the draining lymph node
The difference in ability of cediranib and vandetanib to prevent lymphatic metastasis might be attributable to their differential effects on a specific step in the process of lymphatic metastasis. Using our previously described ear model and intravital microscopy (11), we tested the effect of both TKIs on peritumor lymphatic vessel size and tumor cell arrival in the lymph node. As both cediranib and vandetanib reduced primary tumor growth rate, size matched tumors (40 mm3) were used to compare treated tumors to controls. This mandated that treated tumors grew for a longer period of time prior to evaluation. Since a major correlate to the arrival of cells in the lymph node is time of primary tumor growth (11), using size matched tumors biases the experiments against the hypothesis that these TKIs can reduce tumor cell arrival and thus provides a rigorous test.
Cediranib reduced the diameter of peritumor lymphatic vessels in size matched T241-VEGF-C-GFP tumors (Figure 4A). This was accompanied by a decrease in the number of cells arriving in the draining lymph node (Figure 4B) quantified by MP-IVM. Cediranib treatment of T241-GFP tumors, which produce minimal levels of VEGF-C, showed no reduction in peritumor lymphatic diameter or lymph node cell arrival (data not shown). This result is likely due to the relatively normal peritumor lymphatic vessels and low rate of tumor cell arrival associated with T241-GFP (11). These data show that cediranib can inhibit lymphatic vessel hyperplasia and regional lymph node seeding induced by VEGF-C overexpression, but has little effect on quiescent or normal lymphatic vessels. Thus, cediranib can be used to target lymphangiogenic vessels stimulated by VEGF-C. Collectively, cediranib elicits an anti-lymphatic hyperplasia response (Figure 4A) in addition to its antiangiogenic effects (Figure 3A). This combination is attractive to prevent lymphatic metastasis.
Figure 4.
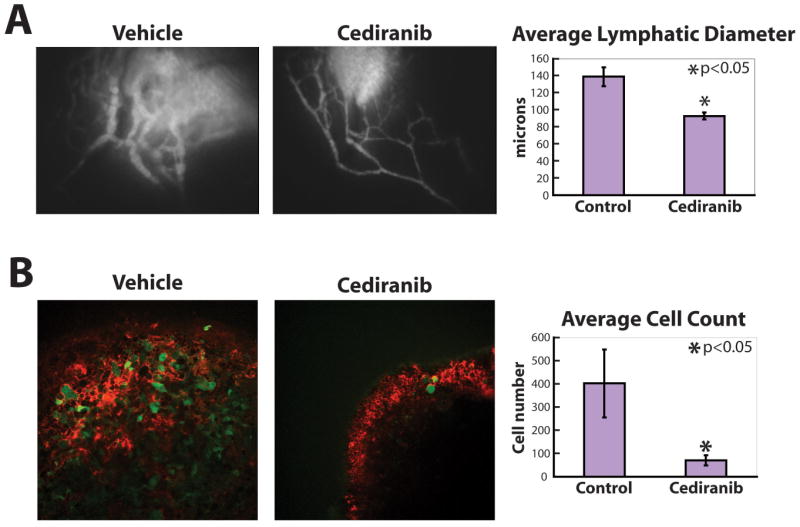
Cediranib reduced lymphatic hyperplasia and tumor cell arrival in lymph nodes. A) Tumor margin lymphatic hyperplasia induced by T241-VEGF-C-GFP tumors (at 40 mm3) was suppressed by daily treatment with cediranib (n = 12) compared with vehicle treated animals (n = 10). B) The number of T241-VEGF-C-GFP cells (green) in the lymph node when the tumor size was 40 mm3 was greatly reduced by daily treatment with cediranib (n = 12) compared with vehicle treated animals (n = 8). Red signal from TMR-dextran lymphangiography shows lymph entering the lymph node from the efferent lymphatic vessel.
Vandetanib does not inhibit tumor margin lymphatic hyperplasia and arrival of tumor cells in the draining lymph node
Vandetanib did not cause a reduction in peritumor lymphatic diameter or tumor cell arrival from T241-VEGF-C-GFP tumors (Figure 5). These data explain the lack of a reduction in lymphatic metastasis using vandetanib in the Prevention Protocol. Since there was no anti-lymphatic hyperplasia effect, there was no reduction in tumor cell arrival in the draining lymph node and thus no reduction in lymphatic metastasis in our model.
Figure 5.
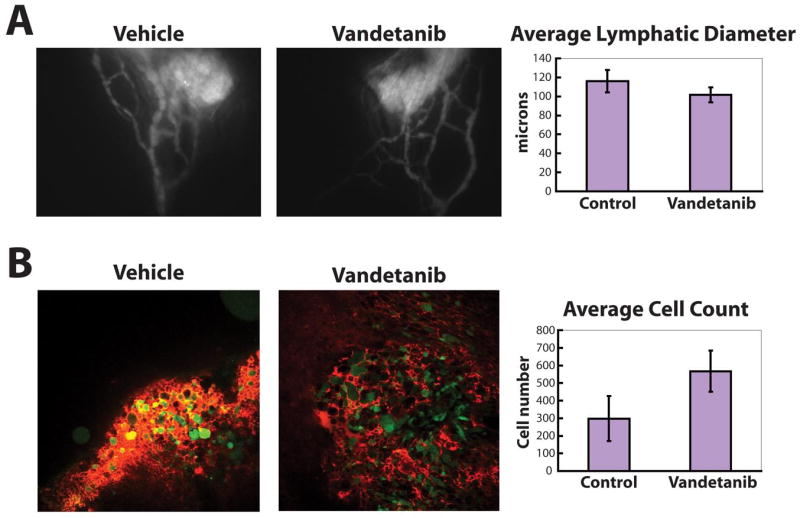
Vandetanib did not reduce lymphatic hyperplasia and tumor cell arrival in lymph nodes. A) Tumor margin lymphatic hyperplasia induced by T241-VEGF-C-GFP tumors (at 40 mm3) was not influenced by daily treatment with vandetanib (n = 11) compared with vehicle treated animals (n = 9). B) The number of T241-VEGF-C-GFP cells (green) in the lymph node when the tumor size was 40 mm3 was not changed by daily treatment with vandetanib (n = 10) compared with vehicle treated animals (n = 9). Red signal from TMR-dextran lymphangiography shows lymph entering the lymph node from the efferent lymphatic vessel.
Many potential explanations exist for the differential effect of the two TKIs on lymphatic hyperplasia in our model. Although, both TKIs caused reductions in tumor growth and blood vessel density (Figure 3), the pharmacokinetics of targeting blood vessels differs from that of lymphatic vessels. To cause a lymphatic effect, a drug has to be transported across the blood vessel wall and through the intervening tissue to the lymphatic vessels in sufficient quantity. Vandetanib may be transported through the tissue less efficiently than cediranib, resulting in the lack of a lymphatic effect. As the animals receiving vandetanib at 50 mg/kg/day 5 times a week were losing body mass, a higher dose could not be administered to overcome transport barriers. Thus, the transvascular and interstitial transport properties of cediranib and vandetanib may account for the differential efficacy of the two drugs.
Our previous findings in the same tumor model show a reduction in peritumor lymphatic hyperplasia, a reduction in cell arrival and a reduction in lymph node metastasis when only VEGFR-3 is blocked using a monoclonal antibody against VEGFR-3 (11). If vandetanib was effectively blocking VEGFR-3 signaling in LECs in our model, we would expect a similar result. Thus, the data presented here suggest that vandetanib did not effectively block VEGFR-3 signaling in tumor associated lymphatic vessels in our model. This may be a result of insufficient concentrations of vandetanib in lymphatic vessels or limited effectiveness of vandetanib in blocking VEGFR-3 signaling in vivo.
Another possibility is that subtle differences in the pharmacodynamic effects of the two TKIs, particularly against targets with higher in vitro IC50's than VEGFR-2 and -3 (13, 14), may account for the differential response in lymphatic vessel hyperplasia and lymphatic metastasis. For instance, VEGFR-1 signaling is important in pathological angiogenesis (18) and macrophage activation (19). Macrophages have been implicated as playing an important role in lymphangiogenesis (20-23). We found no significant difference in the density of macrophages in tumor tissue after treatment with cediranib (data not shown), arguing against this possibility. PDGFR-β is involved in tumor lymphangiogenesis in animal models and is present on lymphatic vessels (24). Both of these targets have an in vitro IC50 (relative to VEGFR-2) for cediranib lower than vandetanib and could explain the differences in the efficacy of the two molecules. Using imatinib mesylate, a TKI that inhibits PDGFR-β, bcr-abl, c-Kit and stem cell factor, we observed no reduction in lymphatic metastasis when treatment was administered during tumor growth (data not shown). Other possible targets for inhibition include EGFR or Ret signaling in the stromal cells of T241-VEGF-C-GFP tumors, which may be important for tumor growth. Vandetanib activity against these molecules could account for the greater primary tumor growth inhibition when compared to cediranib without affecting lymphatic morphology or metastasis.
Our data shows that cediranib, which blocks signaling from VEGFR-2 and -3, can inhibit lymphatic hyperplasia and lymphatic metastasis induced by VEGF-C overexpressing T241 fibrosarcomas. This result is consistent with our previous report that used a blocking VEGFR-3 monoclonal antibody under the same conditions (11). Cediranib also had the benefit of inhibiting primary tumor growth through an antiangiogenic mechanism. Thus, the TKI cediranib appears to be a compelling compound for use in anti-tumor and anti-lymphatic metastatic strategies.
Supplementary Material
Acknowledgments
The authors thank Emmanuelle di Tomaso, Johanna Lahdenranta, James Tyrrell, Juliane M. Jürgensmeier and Anderson Ryan for their scientific and technical input, and Sylvie Roberge and Carolyn Smith for their outstanding technical support. This work was supported by NCI Grants R01CA85140 (RKJ) and P01CA80124 (RKJ), and in part by an unrestricted gift from AstraZeneca.
Financial support provided by NCI Grants R01CA85140 (RKJ) and P01CA80124 (RKJ), and in part by an unrestricted gift from AstraZeneca.
Abbreviations
- BECs
blood vessel endothelial cells
- E-IVM
epifluorescence intravital microscopy
- GFP
green fluorescent protein
- LECs
lymphatic endothelial cells
- MP-IVM
multiphoton intravital microscopy
- PDGFR
platelet derived growth factor receptor
- TKI
tyrosine kinase inhibitor
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
References
- 1.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–53. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 2.Pepper MS. Lymphangiogenesis and tumor metastasis: myth or reality? Clin Cancer Res. 2001;7:462–8. [PubMed] [Google Scholar]
- 3.Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer. 2002;2:573–83. doi: 10.1038/nrc863. [DOI] [PubMed] [Google Scholar]
- 4.Wissmann C, Detmar M. Pathways targeting tumor lymphangiogenesis. Clin Cancer Res. 2006;12:6865–8. doi: 10.1158/1078-0432.CCR-06-1800. [DOI] [PubMed] [Google Scholar]
- 5.Padera TP, Kadambi A, di Tomaso E, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296:1883–6. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- 6.Padera TP, Stoll BR, Tooredman JB, et al. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 7.Jain RK, Tong RT, Munn LL. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007;67:2729–35. doi: 10.1158/0008-5472.CAN-06-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Y, Kozaki K, Karpanen T, et al. Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J Natl Cancer Inst. 2002;94:819–25. doi: 10.1093/jnci/94.11.819. [DOI] [PubMed] [Google Scholar]
- 9.He Y, Rajantie I, Pajusola K, et al. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer Res. 2005;65:4739–46. doi: 10.1158/0008-5472.CAN-04-4576. [DOI] [PubMed] [Google Scholar]
- 10.Stacker SA, Caesar C, Baldwin ME, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 11.Hoshida T, Isaka N, Hagendoorn J, et al. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res. 2006;66:8065–75. doi: 10.1158/0008-5472.CAN-06-1392. [DOI] [PubMed] [Google Scholar]
- 12.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 13.Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–55. [PubMed] [Google Scholar]
- 14.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 15.Brown EB, Campbell RB, Tsuzuki Y, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nature Medicine. 2001;7:864–8. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
- 16.Padera TP, Stoll BS, So PTC, Jain RK. High-speed intravital multiphoton scanning laser microscopy of microvasculature, lymphatics, and leukocyte-endothelial interactions. Molecular Imaging. 2002;1:9–15. doi: 10.1162/15353500200200004. [DOI] [PubMed] [Google Scholar]
- 17.Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284–90. [PubMed] [Google Scholar]
- 18.Carmeliet P, Moons L, Luttun A, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–83. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 19.Pipp F, Heil M, Issbrucker K, et al. VEGFR-1-selective VEGF homologue PlGF is arteriogenic: evidence for a monocyte-mediated mechanism. Circ Res. 2003;92:378–85. doi: 10.1161/01.RES.0000057997.77714.72. [DOI] [PubMed] [Google Scholar]
- 20.Schoppmann SF, Birner P, Stockl J, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002;161:947–56. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skobe M, Hamberg LM, Hawighorst T, et al. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. Am J Pathol. 2001;159:893–903. doi: 10.1016/S0002-9440(10)61765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerjaschki D. The crucial role of macrophages in lymphangiogenesis. J Clin Invest. 2005;115:2316–9. doi: 10.1172/JCI26354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–75. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 24.Cao R, Bjorndahl MA, Religa P, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004;6:333–45. doi: 10.1016/j.ccr.2004.08.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.