

Abstract
Objective
Endostatin (EST) was found to initiate a redox signaling cascade associated with activation of NADPH oxidase in endothelial cells (ECs). The present study tested whether EST stimulate clustering of ceramide-enriched lipid rafts (LR), which assembles and activates NADPH oxidase to form redox signaling platforms.
Methods and Results
Using confocal microscopy, we first demonstrated a co-localization of LR clusters with NADPH oxidase subunits, gp91phox and p47phox in the ECs membrane upon EST stimulation. Immunoblot analysis of floated detergent-resistant membrane fractions found that in LR fractions NADPH oxidase subunits gp91phox and p47phox are enriched and that the activity of this enzyme increased dramatically, as measured by electron spin resonance (ESR) spectrometry. This EST-increased LR platform formation was shown to be attenuated by inhibition or RNA interference of acid sphingomyelinase (A-SMase). Functionally, EST pretreatment significantly impaired bradykinin or A23187-induced vasodilation in isolated small coronary arteries, which could be partially reversed by LR disruptors.
Conclusions
The early injury effect of EST on the vascular endothelium is associated with the formation of redox signaling platforms via lipid raft clustering.
Besides its proapototic effects, EST is also able to induce endothelial dysfunction. This early-stage action of EST is associated with LR clustering and consequent assembling and activation of NADPH oxidase.
Keywords: Endostatin, NADPH oxidase, coronary, endothelium, sphingolipid
Endostatin (EST), a ~20 kDa C-terminal fragment of collagen XVIII located in the basement membrane zones around blood vessels, is a naturally occurring peptide in the body 1, 2. In vivo, EST has been reported as one of the most potent endothelial cell (EC)-specific inhibitors of angiogenesis and tumor growth, and in vitro, extensive studies have demonstrated that EST specifically influences many cell processes such as proliferation and migration of ECs, ECs apoptosis and so on 3. Besides these generally known actions, recent studies from our laboratory demonstrated that EST reduced the NO bioavailability through enhanced O2.− production in the intact coronary endothelium, suggesting a potential role of EST in the impairment of endothelial function 4. This effect of EST was further demonstrated to be associated with the ceramide-mediated signaling pathway 4. However, it remains unknown how EST-stimulated ceramide leads to O2.− production and whether this peptide indeed produces endothelial dysfunction when it acts on intact vessels for a short time.
Recently, a growing body of evidence suggests that ceramide plays essential role in lipid raft (LR) clustering in a variety of cell types. When ceramide production is increased, LRs, the lipid microdomains that consist of cholesterol, sphingolipids, and some associated proteins 5, 6, can be clustered to form large ceramide-enriched macrodomains or platforms, mediating or amplifying transmembrane signals. There is increasing evidence that LRs clustering as a general mechanism participating in the initiation of receptor-mediated transmembrane cell signal transduction 7–10. Along with this line, we recently introduced a new concept regarding LR redox signaling platforms which is importantly involved in the redox regulation of endothelial function 11. We have demonstrated that these LR redox signaling platforms are characterized by gp91phox aggregation and p47phox translocation as well as by activation of acid sphingomyelinase (A- SMase) and subsequent production of ceramide 11. Given that EST stimulates ceramide production through A-SMase and that increased production of ceramide in the cell membrane is able to facilitate the formation of ceramide-enriched platforms 5, 6, the present study hypothesized that EST stimulates O2.− production through the formation of the LR redox signaling platforms in coronary arterials ECs (CAECs) and thereby leads to impairment of endothelium-dependent vasodilation in coronary arteries. We used a series of molecular and physiological approaches to test this hypothesis.
MATERIALS AND METHODS
Confocal analysis of LR clusters and its co-localization with NADPH oxidase subunits or A-SMase in CAECs
The primary cultures of bovine CAECs were obtained as we described previously4, 11, 12. All studies were performed using CAECs of 2–4 passages. For microscopic detection of LR platforms, CAECs were grown on glass coverslips and then treated with 100 nM EST (Upstate-Millipore, Billerica, MA) for 30 min to induce clustering of lipid rafts. In additional groups of cells, methyl-β-cyclodextrin, MCD (Sigma, St. Louis, MO, 1 mmol/L) and Nystatin (Nyst) (Sigma, St. Louis, MO, 10 μg/ml) were added to pre-treat the cells for 15 min before EST stimulation.
Detection of LR clusters were performed as we described previously 11–13. In brief, GM1 gangliosides enriched in LRs were stained by Alexa488-labeled cholera toxin B (alexa488-CTX, 1 μg/mL, 45 min) (Molecular Probes, Eugene, OR). For dual staining detection of the colocalization of LRs and NADPH oxidase subunits, gp91phox, or A-SMase, the CAECs were further incubated with mouse anti-gp91phox monoclonal antibody (BD Biosciences, San Jose, CA, USA, 1:200), or rabbit anti-A-SMase polyclonal antibodies (Santa Cruz, Santa Cruz, CA, USA, 1:200), separately, which was followed by Texas red-conjugated anti-mouse or anti-rabbit (Molecular Probes, Eugene, OR) secondary antibody as needed, respectively. Staining was visualized through sequentially scanning on an Olympus scanning confocal microscope (Olympus, Tokyo, Japan).
Isolation and detection of LR microdomains by gradient centrifugation
Since LR domains float in the membrane and are resistant to solubilization by non-ionic detergents such as Triton X-100 at low temperatures, they are also termed as floated detergent resistant membrane domains (DRMD). LR microdomains or DRMD were isolated as described previously 11, 14, 15. For immunodetection of LR-associated proteins, 50 μL of resuspended proteins were subjected to SDS-PAGE, transferred onto a nitrocellulose membrane, and blocked as described previously 11, 13. The membrane was probed with primary monoclonal antibodies against flotillin-1, anti-gp91phox, anti-p47phox (1:1000, BD Biosciences), respectively according to corresponding protocols, overnight at 4°C followed by incubation with horseradish peroxidase–labeled anti-mouse IgG (1:5000). The immunoreactive bands were detected by chemiluminescence methods (Pierce) and visualized on Kodak Omat film.
Eletromagnetic spin resonance (ESR) spectrometric detection of O2.−
ESR detection of O2.− was also performed as we described previously 13, 16. The ECs cell mixture loaded in glass capillaries was immediately analyzed by ESR (Noxygen Science Transfer & Diagnostics GmbH, Denzlingen, Germany) for production of O2.− at each minute for 10 min. The SOD-inhibitable signals were normalized by protein concentration and compared among different experimental groups. NADPH oxidase activity in both LRs fractions (mixture of fraction 3, 4) and non-raft fractions (mixture of fraction 7, 8) was measured by ESR. 50 μL of LR fractions or non-raft fractions were mixed with 1 mmol/L spin trap CMH was substituted for ECs for reactions. The relative increase of the ESR spectrum was used to represent NADPH oxidase activity.
RNA interference of A-SMase
Small interference RNAs (siRNAs) were purchased from QIAGEN. The DNA target sequence for A-SMase-siRNA is: 5′-AAGGCCGTGAGTTTCTACCT-3′. The scrambled small RNA (AATTCTCCGAA CGTGTCACGT) has been confirmed as non-silencing double stranded RNA and was used as control in the present study. Transfection of siRNA was performed using the QIAGEN TransMessengerTM transfection kit according to the manufacturer’s instructions.
Isolated perfused small coronary artery preparation
Fresh bovine hearts were obtained from a local abattoir. Small coronary arteries (~200 μm ID) were prepared and the vasodilator response measured as we described previously 11. After measurement of control vasodilator response, EST was added in the presence or absence of MCD or Nyst in the lumen of the artery and then the vasodilator response was redetermined.
Statistics
Data are presented as means ± SE. Significant differences between and within multiple groups were examined using ANOVA for repeated measures, followed by Duncan’s multiple-range test. A Students’ t-test was used to detect significant differences between two groups. P< 0.05 was considered statistically significant.
RESULTS
LRs clustering and aggregation or recruitment of signaling molecules in the membrane of CAECs
As shown in Figure 1, typical LR patches in a CAEC under resting condition and during endostatin stimulation were detected by confocal microscopy (left images). Under resting condition (control), there was only a diffuse fluorescent staining on the cell membrane by Alexa-CTX (Al488-CTX), indicating possible distribution of single LRs without clusters for fluorescent spots or patches. However, when these cells were incubated with EST, large fluorescent dots or patches were detected on the cell membrane. These fluorescent patches indicate the formation of LR clusters or macrodomains. In the presence of LR disruptors, either MCD or Nyst, EST-induced formation of LR clusters was substantially attenuated and in many cells there were no LR patches detectable (panel MCD+EST or NYST+EST).
Figure 1. Representative confocal images of LR clusters (Alexa488-CTX) and gp91phox(A), P47phox(B) labeling (Texas red-conjugated).
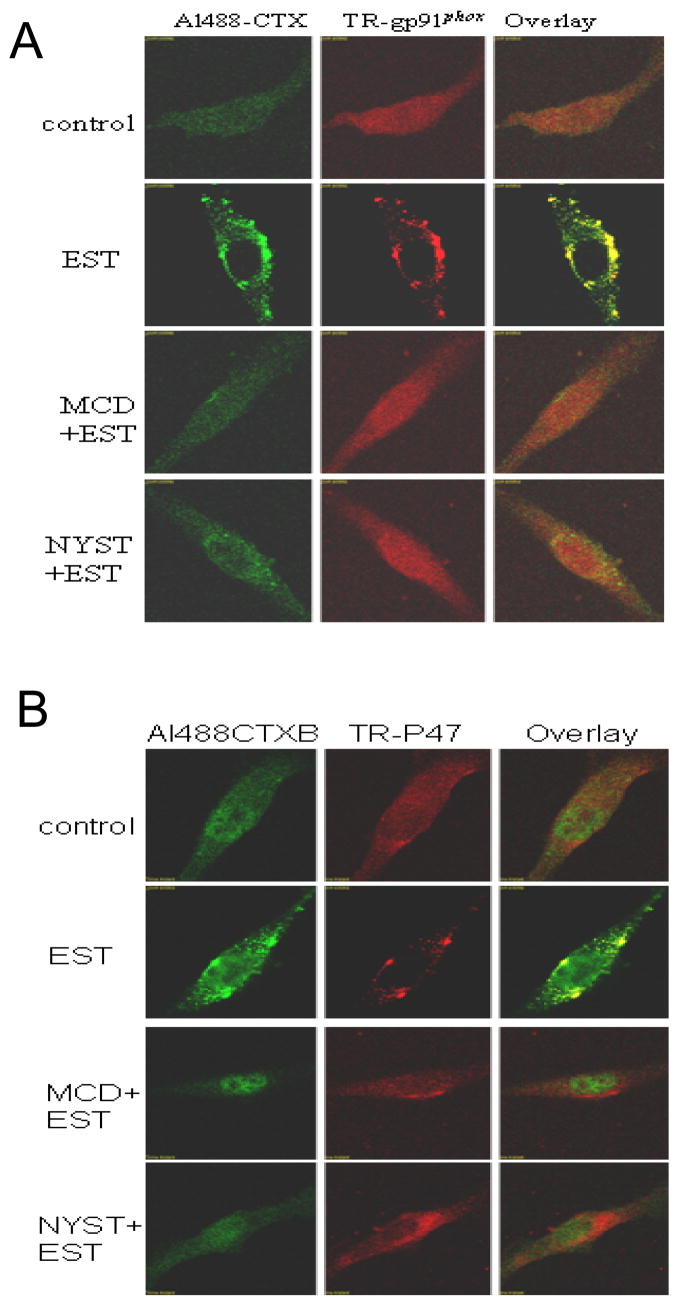
The overlay images exhibited yellow spots or patches (right), which represented co-localization of gp91phox or P47phox and LR component, ganglioside GM1.2. The figure presents representative images from experiments done using 6 batches of ECs cultures.
In the middle panel of Figure 1, the cell stimulated by EST was stained by a primary anti-gp91phox antibody and then by Texas red-conjugated secondary antibody. In addition to some diffuse signals under resting condition, red spots and patches were detected on the cell membrane when the cells were stimulated by EST (panel EST). In MCD or Nyst pretreatment groups, the red patches were not observed (panel MCD+EST or NYST+EST).
When two sequentially scanned images from the same cell with different wavelengths were merged together, there were a number of yellow areas either as dots or as patches resulting from green CTX and Texas red-antiboby (right images). These yellow patches were considered as a colocalization of LR clusters and NADPH oxidase subunit gp91phox. When the cells were pretreated by either MCD or Nyst, both CTX clusters and aggregated Texas red fluorescence were no longer observed. We also did similar experiments using p47phox antibody and found very similar distribution pattern to gp91phox. As shown in Figure 1B, LR clusters and colocalization of p47phox were confirmed in these ECs when they are stimulated by EST, while p47phox are diffusely distributed under control condition.
Enrichment of both gp91phox and p47phox in LR fractions of EC membrane
To further demonstrate an involvement of LRs in aggregations or translocation of NADPH subunits, we isolated membrane fractions by detergent treatments and gradient centrifugations. As shown in Figure 2A, Western blot analysis detected positive expression of flotillin-1 in fractions 3 to 5, which were referred to as LR fractions previously 23. The ratio of gp91phox or p47 phox between LRs fractions and other fractions was used to analyze their aggregation or translocation, because EST induces their clustering in LRs fractions of cell membrane, rather than increases expression. Figure 2B shows that gp91phox could be detected in most of the membrane fractions from CAECs; however, there was a marked increase in gp91phox protein in LR microdomains when CAECs were stimulated by EST. This increase was significantly inhibited by pretreatment of cells with Nyst. Similarly, as shown in Figure 2C, p47phox, a cytosolic NADPH oxidase subunit, was also found increased in LR fractions upon EST stimulation and this increase also inhibited by Nyst. Similarly, another LR disruptor, MCD blocked the enrichment of these two NADPH oxidase subunits in LR fractions.
Figure 2. Distribution and localization of gp91phox and p47phox in floated membrane fractions from ECs.

A: using LR marker protein flotillin-1, B: using anti-gp91phox, and C: using anti-p47phox (all were representative gels from 4 separated experiments).
NADPH oxidase activity in LR microdomains of EC
We also analyzed the NADPH oxidase activity present in both LR and non-raft fractions from control and EST-treated CAECs. We found that EST significantly increased NADPH oxidase activity by 147% in LR fractions, but only by 20% in non-raft fractions (Figure 3), which were both prevented by MCD pretreatment.
Figure 3. Effects of LRs disruption on EST-induced increase of NADPH oxidase activity in LR-enriched fractions and non-LR fractions.

*P < 0.05 vs. vehicle; & p<0.05 vs. EST; # P<0.05 vs. vehicle; n=6.
ESR assay of EST-induced O2.− production in CAECs
The direct consequence of activated NADPH oxidase is increased production of O2.− in CAECs. Figure 4 presented summarized data obtained by ESR assay using CAECs, showing the fold changes in O2.− production. After CAECs were treated with EST, O2.− levels detected by ESR increased significantly. When the cells were pretreated with MCD, this EST-induced increase in O2.− production was significantly reduced. MCD alone had no significant effects on the O2.− level.
Figure 4. Effects of LRs disruption on EST-induced O2.− production in CAECs.

Summarized data depicting changes in O2.− production in CAECs with different treatments. * indicates P<0.05 vs. control; # indicates P<0.05 vs. EST group; n = 5.
Association of A-SMase translocation and LR clustering
As shown in Figure 5, CAECs were double-stained with Alexa488 labeled CTX and anti-A-SMase primary antibody followed by Texas red-conjugated secondary antibody. Similar to that described in Figure 1, left and middle images represent fluorescence signals from Alexa488 and Texas Red, separately. Compared to control CAECs, EST induced significant LR patches on the cell membrane, accompanied by the aggregation of A-SMase. This was shown clearly in overlaid images, where many yellow dots or patches exhibited, indicating a colocalization of CTX-stained ganglioside M1 and A-SMase. When the CAECs were pretreated by a putative A-SMase inhibitor, desipramine, the EST-induced changes in LR clustering were blocked. In addition, when the CAECs were transfected with synthesized A-SMase siRNA (75% knocking down as shown in our preliminary experiments and previous studies 13), EST no longer induced LR clustering and colocalization with A-SMase with LR marker (siRNA+EST). However, CAECs transfected with scramble RNA still reserved LRs clustering responses to EST (scrambleRNA+EST).
Figure 5. Confocal microscopic analysis of A-SMase in LR clusters in ECs stimulated with EST.

Texas-red conjugated anti-A-SMase in the left and LRs markers (Al488-CTX in the middle) were used to stain cells. Desip = desipramine; A-SMase siRNA was A-SMase siRNA transfection. The images are representatives from 6 separated experiments.
LR disruptors reversed EST-induced impairment of endothelium-dependent vasodilation to BK or A23187
Incubation of small coronary arteries with EST (100 nM, perfused into the lumen) had no significant effect on the basal arterial diameters, but markedly attenuated the BK-induced increase in arterial diameters. In the presence of Nyst (for 15 minutes), however, EST-induced inhibition of vasodilator response to BK was reversed. Treatment of the arteries with Nyst alone had no significant effect on basal arterial diameters or BK-induced vasodilation at the same doses used (Figure 6A). As shown in Figure 6D, the effects of Nyst to reverse the action of EST were also observed in arterial preparations that use A23187 as endothelium-dependent vasodilator. Similar results were obtained when we used another LR disruptor, MCD. EST-induced impairment of endothelium-dependent vasodilator response to BK and A23187 were also reversed by MCD treatment (Figure 6B and E).
Figure 6. Effects of LR disruptors on EST-induced impairment of the vasodilator responses.

A: Nyst, responses to BK; D: Nyst, responses to A23187. B: MCD, responses to BK; E: MCD, responses to A23187. C: DPI, apocynin, responses to BK; F: DPI, apocynin, responses to A23187. n=7 or 6, *P<0.05 vs. control.
In Figure 6C and 6F, two NADPH oxidase inhibitors, DPI and apocynin were used to preincubate the vessels and the vasodilation response to BK and A23187 were determined. Both inhibitors significantly blocked the EST-induced impairment of vasodilation response.
DISCUSSION
In previous studies from our laboratory and by others, EST was demonstrated to produce O2.− via enhanced NADPH oxidase activity and thereby regulate cell function or activities 4, 28. However, the mechanism how this peptide activates NADPH oxidase is still poorly understood. The present study tested a hypothesis that EST induces or enhances NADPH oxidase activation through the formation of LR signaling platforms. To test this hypothesis, we first determined whether EST induces the formation of this LR redox signaling platform associated with NADPH oxidase. By using confocal co-localization analysis of LR marker, Alexa488-labeled CTXB and Texas red conjugated gp91phox and p47phox, two typical NADPH oxidase subunits, we demonstrated that EST stimulated LR clustering and enhanced enrichment of NADPH oxidase subunits in LR fractions. These results suggest that gp91phox could be aggregated in response to EST stimulation. This aggregation of NADPH oxidase subunits may be of importance in the response of this enzyme to agonists in vascular ECs. Although there are many studies demonstrating an assembling or aggregation of NADPH oxidase subunits to form a enzyme complex and to produce O2− 29. This assembling or aggregation is usually centered by its membrane subunits such as gp91phox or other Nox. However, little is known whether these subunits themselves are able to aggregate or be clustered. The present studies provide evidence that in response to EST the membrane NADPH oxidase subunits are able to be aggregated and that this aggregation may form an NADPH oxidase-mediated redox platform, which may be a prerequisite or driving force to initiate assembling of other subunits and consequent activating cascade of this enzyme 11.
In addition to aggregation of gp91phox, we also found that a cytosolic subunit, p47phox was enriched in membrane LR fractions upon EST stimulation. This p47phox enrichment in LR fractions may represent its translocation into the cell membrane. In this regard, p47phox has been reported to be translocated and then interacts with membrane-bound NADPH oxidase subunits in response to agonists such as Ang II, TNF-α, growth factors, and thrombin 30–34. It is now known that this translocation of p47phox is governed by serine phosphorylation and that p47phox protein contains a number of phosphorylation sites for PKC, PKA, and MAPK 32, 33. The findings of the present study further demonstrate that this p47phox translocation occurs in LR microdomains, in particular, in LR platforms during agonist stimulations. LR clustering may provide a driving force to promote this p47phox translocation in response to agonist stimulations such as EST used in the present study.
To provide direct evidence that aggregation or assembling of NADPH oxidase components via LR clustering is involved in the activation of this enzyme, we isolated the whole membrane fractions as well as its LR fractions from CAECs and determined NADPH oxidase–derived O2.− production. It was found that EST indeed enhanced NADPH oxidase activity in the whole membrane fraction, which was inhibited by MCD, a lipid raft disruptor that could extract cholesterol from the cell membrane 5. In isolated LR fractions, EST more dramatically increased NADPH oxidase activity, which was also blocked by treatment of cells with MCD. We also demonstrated that EST significantly increased O2.− production in intact CAECs, which was blocked by MCD. These results confirm that aggregated NADPH oxidase subunits in LR platforms are functioning as an active enzyme complex.
Next, we explored the mechanism by which EST stimulates LR clustering and thereby form LR redox signaling platforms. Given a major contributing role of ceramide to the formation of ceramide-enriched signaling platforms, it is very possible that A-SMase translocation and activation are importantly implicated in EST-induced LR clustering and redox signaling platforms formation. To test this hypothesis, we double-stained the CAECs with Al488-labeled CTXB and Texas red-conjugated anti-A-SMase and found that EST significantly increased the enrichment of A-SMase in the LR platforms. In the presence of desipramine, an inhibitor of A-SMase, this EST-stimulated enrichment of A-SMase in the LR platforms was almost completely blocked. However, although desipramine is often used as ASMase inhibitor, its action on ceramidase may be involved and thus interpretation of the results obtained by using this compound should be cautious. For this reason, we also used A-SMase specific siRNA to knock down this gene and found that clustering of LRs with A-SMase was blocked. These results together confirm that aggregation in LRs and consequent activation of A-SMase is essential to the process of LR clustering. This role of A-SMase in mediating LR clustering in ECs has been also found in response to other agonists such as FasL and TNF-α 13, 36. It seems that the A-SMase activation is a common mechanism mediating LR clustering and corresponding signaling platforms formation 37.
The other important findings of the present study are that EST impaired the endothelium-dependent vasodilator responses and that this action of EST is associated with enhanced formation of LR clusters. This EST-induced endothelial dysfunction occurred no matter whether a receptor-mediated endothelium-dependent vasodilator, BK or a non-receptor-mediated endothelial NO stimulatory agonist, A23187 was used. Although A23187 bypasses the receptor step and activates eNOS through calcium, endostatin-induced O2.− production may interact with NO to generate ONOO− impairing the A23187-induced vasodilator responses. Our results provide direct evidence that EST produces an early action to induce endothelial dysfunction prior to detectable apoptotic effect. In this regard, EST has been reported to induce a detectable apoptosis only when it was incubated with ECs at least for 6 hours 38, 39. Given a relative resistance of ECs to apoptosis 40, the functional impairment may represent one of the most important pathological actions of this apoptotic peptide in ECs. Since EST-impaired vasodilator responses could be restored in the presence of LR disruptors or NADPH oxidase inhibitors or silencers, it seems that EST-induced endothelial dysfunction is due to increased O2.− production from LR platforms with aggregated NADPH oxidase subunits. Increased O2.− could reduce the bioavailability of NO, resulting in impairment of endothelium-dependent vasodilation, as shown in our previous studies and by others 4, 41–44.
In summary, the present study demonstrated that EST is able to induce endothelial dysfunction as an early-stage acute action, which is associated with assembling and activation of NADPH oxidase in LR clusters. The clustered LRs together with activated NADPH oxidase constitute a redox signaling platform mediating the pathological actions of EST on the vascular endothelium.
Acknowledgments
This study was supported by grants from the National Institute of Health (HL-57244, HL-75316, and DK54927).
Footnotes
Publisher's Disclaimer: This is an un-copyedited author manuscript that was accepted for publication in Arteriosclerosis, Thrombosis, and Vascular Biology, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at Arteriosclerosis, Thrombosis, and Vascular Biology. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
References
- 1.OReilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 2.Rehn M, Pihlajaniemi T. Alpha-1(Xviii), a Collagen Chain with Frequent Interruptions in the Collagenous Sequence, a Distinct Tissue Distribution, and Homology with Type-Xv Collagen. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4234–4238. doi: 10.1073/pnas.91.10.4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkman J. Antiangiogenesis in cancer therapy - endostatin and its mechanisms of action. Experimental Cell Research. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Zhang AY, Teggatz EG, Zou AP, Campbell WB, Li PL. Endostatin uncouples NO and Ca2+ response to bradykinin through enhanced O-2(-center dot) production in the intact coronary endothelium. American Journal of Physiology-Heart and Circulatory Physiology. 2005;288:H686–H694. doi: 10.1152/ajpheart.00174.2004. [DOI] [PubMed] [Google Scholar]
- 5.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochimica Et Biophysica Acta-Molecular Cell Research. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Gulbins E, Grassme H. Ceramide-enriched membrane domains in infectious biology. Chemistry and Physics of Lipids. 2006;143:53–53. [Google Scholar]
- 7.Furne C, Corset V, Herincs Z, Cahuzac N, Hueber AO, Mehlen P. The dependence receptor DCC requires lipid raft localization for cell death signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4128–4133. doi: 10.1073/pnas.0507864103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta N, DeFranco AL. Visualizing lipid raft dynamics and early signaling events during antigen receptor-mediated B lymphocyte activation. Faseb Journal. 2003;17:C203–C203. doi: 10.1091/mbc.02-05-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jury EC, Kabouridis PS. T-lymphocyte signalling in systemic lupus erythematosus: a lipid raft perspective. Lupus. 2004;13:413–422. doi: 10.1191/0961203304lu1045rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sprenger RR, Fontijn RD, van Marle J, Pannekoek H, Horrevoets AJG. Spatial segregation of transport and signalling functions between human endothelial caveolae and lipid raft proteomes. Biochemical Journal. 2006;400:401–410. doi: 10.1042/BJ20060355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006;47:74–80. doi: 10.1161/10.1161/01.HYP.0000196727.53300.62. [DOI] [PubMed] [Google Scholar]
- 12.Jin S, Yi F, Li PL. Contribution of Lysosomal Vesicles to the Formation of Lipid Raft Redox Signaling Platforms in Endothelial Cells. Antioxid Redox Signal. 2007 doi: 10.1089/ars.2007.1660. [DOI] [PubMed] [Google Scholar]
- 13.Zhang AY, Yi F, Jin S, Xia M, Chen QZ, Gulbins E, Li PL. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal. 2007;9:817–828. doi: 10.1089/ars.2007.1509. [DOI] [PubMed] [Google Scholar]
- 14.Parkin ET, Hussain I, Turner AJ, Hooper NM. The amyloid precursor protein is not enriched in caveolae-like, detergent-insoluble membrane microdomains. J Neurochem. 1997;69:2179–2188. doi: 10.1046/j.1471-4159.1997.69052179.x. [DOI] [PubMed] [Google Scholar]
- 15.Radeva G, Sharom FJ. Isolation and characterization of lipid rafts with different properties from RBL-2H3 (rat basophilic leukaemia) cells. Biochemical Journal. 2004;380:219–230. doi: 10.1042/BJ20031348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang G, Zhang F, Muh R, Yi F, Chalupsky K, Cai H, Li PL. Autocrine/paracrine pattern of superoxide production through NAD(P)H oxidase in coronary arterial myocytes. American Journal of Physiology-Heart and Circulatory Physiology. 2007;292:H483–H495. doi: 10.1152/ajpheart.00632.2006. [DOI] [PubMed] [Google Scholar]
- 17.Geiger J, Zou AP, Campbell WB, Li PL. Inhibition of cADP-ribose formation produces vasodilation in bovine coronary arteries. Hypertension. 2000;35:397–402. doi: 10.1161/01.hyp.35.1.397. [DOI] [PubMed] [Google Scholar]
- 18.Campbell WB, Deeter C, Gauthier KM, Ingraham RH, Falck JR, Li PL. 14,15-Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of K(Ca) channels. Am J Physiol Heart Circ Physiol. 2002;282:H1656–1664. doi: 10.1152/ajpheart.00597.2001. [DOI] [PubMed] [Google Scholar]
- 19.Fukuhara M, Neves LA, Li P, Diz DI, Ferrario CM, Brosnihan KB. The angiotensin II AT1 receptor antagonist irbesartan prevents thromboxane A2-induced vasoconstriction in the rat hind-limb vascular bed in vivo. J Hypertens. 2001;19:561–566. doi: 10.1097/00004872-200103001-00007. [DOI] [PubMed] [Google Scholar]
- 20.Pratt PF, Li P, Hillard CJ, Kurian J, Campbell WB. Endothelium-independent, ouabain-sensitive relaxation of bovine coronary arteries by EETs. Am J Physiol Heart Circ Physiol. 2001;280:H1113–1121. doi: 10.1152/ajpheart.2001.280.3.H1113. [DOI] [PubMed] [Google Scholar]
- 21.Li P, Fukuhara M, Diz DI, Ferrario CM, Brosnihan KB. Novel angiotensin II AT(1) receptor antagonist irbesartan prevents thromboxane A(2)-induced vasoconstriction in canine coronary arteries and human platelet aggregation. J Pharmacol Exp Ther. 2000;292:238–246. [PubMed] [Google Scholar]
- 22.Li PL, Jin MW, Campbell WB. Effect of selective inhibition of soluble guanylyl cyclase on the K(Ca) channel activity in coronary artery smooth muscle. Hypertension. 1998;31:303–308. doi: 10.1161/01.hyp.31.1.303. [DOI] [PubMed] [Google Scholar]
- 23.Shao DM, Segal AW, Dekker LV. Lipid rafts determine efficiency of NADPH oxidase activation in neutrophils. Febs Letters. 2003;550:101–106. doi: 10.1016/s0014-5793(03)00845-7. [DOI] [PubMed] [Google Scholar]
- 24.Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond) 2005;109:217–226. doi: 10.1042/CS20050067. [DOI] [PubMed] [Google Scholar]
- 25.Rueckschloss U, Duerrschmidt N, Morawietz H. NADPH oxidase in endothelial cells: impact on atherosclerosis. Antioxid Redox Signal. 2003;5:171–180. doi: 10.1089/152308603764816532. [DOI] [PubMed] [Google Scholar]
- 26.Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life. 2000;50:267–269. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 27.Meyer JW, Schmitt ME. A central role for the endothelial NADPH oxidase in atherosclerosis. FEBS Lett. 2000;472:1–4. doi: 10.1016/s0014-5793(00)01397-1. [DOI] [PubMed] [Google Scholar]
- 28.Clamp AR, Jayson GC. The clinical potential of antiangiogenic fragments of extracellular matrix proteins. Br J Cancer. 2005;93:967–972. doi: 10.1038/sj.bjc.6602820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 30.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lassegue B, Griendling KK. Out phoxing the endothelium: what’s left without p47? Circ Res. 2002;90:123–124. [PubMed] [Google Scholar]
- 32.Dewas C, Dang PM, Gougerot-Pocidalo MA, El-Benna J. TNF-alpha induces phosphorylation of p47(phox) in human neutrophils: partial phosphorylation of p47phox is a common event of priming of human neutrophils by TNF-alpha and granulocyte-macrophage colony-stimulating factor. J Immunol. 2003;171:4392–4398. doi: 10.4049/jimmunol.171.8.4392. [DOI] [PubMed] [Google Scholar]
- 33.Mitsui K, Iwashita S. Examination of the role of serine phosphorylation in phospholipase C-gamma and its related P47 in cAMP-mediated depression of epidermal growth factor signal transduction. FEBS Lett. 1990;268:157–160. doi: 10.1016/0014-5793(90)80997-w. [DOI] [PubMed] [Google Scholar]
- 34.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J Biol Chem. 1999;274:19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 35.Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K, Kolesnick R, Gulbins E. CD95 signaling via ceramide-rich membrane rafts. J Biol Chem. 2001;276:20589–20596. doi: 10.1074/jbc.M101207200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang DX, Zou AP, Li PL. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. American Journal of Physiology-Heart and Circulatory Physiology. 2003;284:H605–H612. doi: 10.1152/ajpheart.00697.2002. [DOI] [PubMed] [Google Scholar]
- 37.Gulbins E, Li PL. Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol. 2006;290:R11–26. doi: 10.1152/ajpregu.00416.2005. [DOI] [PubMed] [Google Scholar]
- 38.Dhanabal M, Ramchandran R, Waterman MJ, Lu H, Knebelmann B, Segal M, Sukhatme VP. Endostatin induces endothelial cell apoptosis. J Biol Chem. 1999;274:11721–11726. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- 39.Ren B, Wang Y, Ndebele K, Zhi Q, Chen FH, Wang YZ, Parangi S. Multiple signaling is involved in endostatin-mediated apoptosis in ECV 304 endothelial cells. Front Biosci. 2005;10:1089–1097. doi: 10.2741/1602. [DOI] [PubMed] [Google Scholar]
- 40.Duval H, Harris M, Li J, Johnson N, Print C. New insights into the function and regulation of endothelial cell apoptosis. Angiogenesis. 2003;6:171–183. doi: 10.1023/B:AGEN.0000021390.09275.bc. [DOI] [PubMed] [Google Scholar]
- 41.Sheng JZ, Braun AP. Small- and intermediate-conductance Ca2+-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. Am J Physiol Cell Physiol. 2007;293:C458–467. doi: 10.1152/ajpcell.00036.2007. [DOI] [PubMed] [Google Scholar]
- 42.Zhu J, Drenjancevic-Peric I, McEwen S, Friesema J, Schulta D, Yu M, Roman RJ, Lombard JH. Role of superoxide and angiotensin II suppression in salt-induced changes in endothelial Ca2+ signaling and NO production in rat aorta. Am J Physiol Heart Circ Physiol. 2006;291:H929–938. doi: 10.1152/ajpheart.00692.2005. [DOI] [PubMed] [Google Scholar]
- 43.Church JE, Fulton D. Differences in eNOS activity because of subcellular localization are dictated by phosphorylation state rather than the local calcium environment. J Biol Chem. 2006;281:1477–1488. doi: 10.1074/jbc.M505968200. [DOI] [PubMed] [Google Scholar]
- 44.Zhu L, He P. Platelet-activating factor increases endothelial [Ca2+]i and NO production in individually perfused intact microvessels. Am J Physiol Heart Circ Physiol. 2005;288:H2869–2877. doi: 10.1152/ajpheart.01080.2004. [DOI] [PubMed] [Google Scholar]